Abstract
We investigate termination effects in aluminosilicate (AlSi) and aluminogermanate (AlGe) imogolite nanotubes (NTs) by means of semi-local and range-corrected hybrid Density Functional Theory (DFT) simulations. Following screening and identification of the smallest finite model capable of accommodating full relaxation of the NT terminations around an otherwise geometrically and electrostatically unperturbed core region, we quantify and discuss the effects of physical truncation on the structure, relative energy, electrostatics and electronic properties of differently terminated, finite-size models of the NTs. In addition to composition-dependent changes in the valence (VB) and conduction band (CB) edges and resultant band gap (BG), the DFT simulations uncover longitudinal band bending and separation in the finite AlSi and AlGe models. Depending on the given termination of the NTs, such longitudinal effects manifest in conjunction with the radial band separation typical of fully periodic AlSi and AlGe NTs. The strong composition dependence of the longitudinal and radial band bending in AlSi and AlGe NTs suggests different mechanisms for the generation, relaxation and separation of photo-generated holes in AlSi and AlGe NTs, inviting further research in the untapped potential of imogolite compositional and structural flexibility for photo-catalytic applications.
1. Introduction
Termination-induced structural relaxation and reconstruction can alter, and thus be used to tailor, the mechanical, electronic and optical properties of nanostructured materials [1,2,3,4,5]. Physical truncation of a solid-state material leaves the terminal atoms under-coordinated and incompletely bonded, disrupting the crystalline periodicity by introducing dangling bonds on the medium-exposed surfaces of the material. These dangling bonds and their additional interactions with the surrounding medium can strongly alter the properties of finite-sized materials leading to structural reconstruction as well as the emergence of unexpected optical, charge-carrier and chemical properties [6,7,8,9,10].
In spite of tremendous progress in the quantitative characterization and understanding of termination effects in 2D (crystalline) materials [1,2,3,4,5,6,7,8,9,10], these kinds of studies remain extremely rare for 1D nanostructure materials such as nanotubes. This is mostly due to the currently unsolved challenges in the direct quantitative resolution of the terminations of 1D materials, as well as in the unambiguous detection, separation and interpretation of termination and “bulk-like” fingerprints in structural or spectroscopic characterization experiments. The limitations of existing first principles or force fields simulation methods in accurately simulating realistic models of 1D nanomaterials, ideally with inclusion of medium effects, has further slowed down progress in this field. To contribute to progress in this area, here we investigate modern, favorably scaling density functional theory (DFT) implementations to explore, in silico, termination effects in open-ended aluminosilicate (AlSi) and aluminogermanate (AlGe) imogolite nanotubes (NTs).
AlSi and AlGe NTs are structurally analogous to the naturally occurring hydrous-aluminosilicate imogolite [11,12,13]. Their walls consist of a single layer of octahedrally coordinated aluminum hydroxide (gibbsite) with pendant tetrahedral silanol (Si-OH) groups facing the tube cavity (Figure 1). The only difference between AlSi and AlGe NTs is the substitution of silanol groups with germanol (Ge–OH) groups. The chemical formulas of the AlSi and AlGe NTs are (Al2SiO7H4)N and (Al2GeO7H4)N, respectively. N defines the number of radially non-equivalent aluminum atoms along the NT circumference, and is necessarily an even number due to the zig-zag rolling of the nanotubes [14].
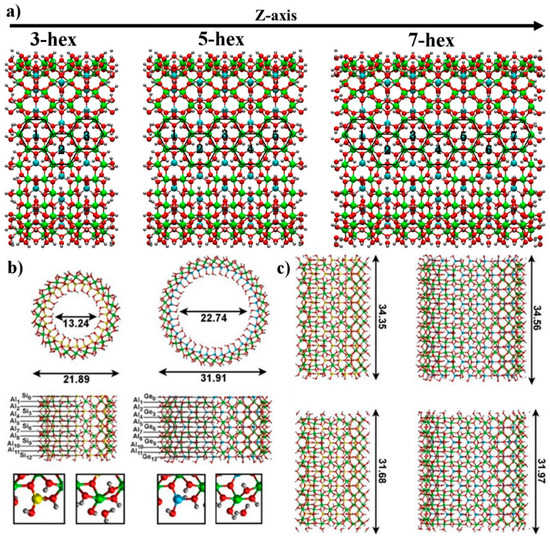
Figure 1.
(a) Optimized geometry of the finite AlSi/AlGe NT models. The hex-3, 5 and 7 labeling reflects the number of complete Al hexagons along the (zigzag) NT axis. Al hexagons have been highlighted in black. (b) Front and side view of half of the optimized AlSi and AlGe finite models. The position of the tube-rings (R = 0–12) is marked by the black horizontal lines. R = 0 corresponds to the innermost Si(Ge) ring. The small squared panels contain a close up of the optimized atomic structure for the OH (left) and H2O (right) terminations of the AlSi and AlGe NT models. The optimized diameter of the finite NT models is reported in Å. (c) Side view of the full hex-7 models for the AlSi (left), and AlGe (right) NTs. The top and bottom rows reports the OH- and H2O-terminated models, respectively. The optimized length of the NT models is also marked in Å. The color labeling for the atoms is as follows: H (gray), O (red), C (brown), Al (green), Si (yellow), Ge (cyan).
Synthetic imogolite NTs can be cheaply synthesized in water solution [11,15,16,17,18,19,20] with extremely high structural and compositional flexibility [21,22,23,24]. Both sides of the imogolite NTs’ cavities can be easily functionalized [25,26,27,28]. The synthetic flexibility of imogolite NTs leads to widely tunable properties, with exciting reports as regards the performance of imogolite NT for chemical separation [17,20,21,22,23,24,26,29], co-photo-catalyst [30], hybrid nanocomposites [31] and colloidal, liquid-crystal applications [32,33,34,35]. In addition to these examples, the potential range of applicability for AlSi AlGe NTs has been recently expanded via synthesis of Fe-doped imogolite NTs, capable of absorbing visible (and UV) light. These advances have already opened up experimental research in the potential of imogolite NTs for UV–visible light photo-catalytic applications [36,37,38,39,40,41]. For the interested reader, Ref. [42,43] provide recent reviews of imogolite NTs.
In spite of the substantial and rapidly increasing rate of progress in the synthesis, characterization and application of imogolite NTs, the available atomistic insights into their open-ended terminations is limited to (not strictly atomically resolved) transmission electron microscopy images [11,19,44]. As a result, no truly atomic understanding is currently available regarding the role of NT terminations in the emerging electronic, optical and chemical properties of these systems. In analogy to the truncation effects in metal-oxides [11,12,13], terminal relaxation in imogolite NTs may also induce properties markedly different from the innermost, bulk-like regions of the systems.
To start exploring this subject, here we quantify the effects that physical truncation and the ensuing structural rearrangements bring about in finite models of AlSi and AlGe NTs, using computational methods and set-ups previously benchmarked and applied to imogolite NTs [45,46,47,48,49,50]. To the best of our knowledge, this manuscript reports the first ever DFT characterization of the structure and electronic properties for different models of the AlSi and AlGe NT terminations. The present results should accordingly be a useful and timely contribution to research in the field of imogolite NTs, especially in terms of the quantification of the extent to which the electronic properties of the peripheral and innermost regions of imogolite nanotubes can differ, potentially leading to profoundly diverse photo-chemical reactivity. Quantification of the structure and steric hindrance of the terminations of imogolite nanotubes should also be a valuable addition to ongoing research into the potential of this class of materials for nanofluidic and chemical separation applications [17,20,21,22,23,24,26,27,29], as well as a necessary reference for future research in termination effects for double-walled imogolite nanotubes [51,52,53,54,55]. Last but not least, the present results should also provide a valuable, currently missing, first principles benchmark reference for ongoing work in the development of accurate tight-binding DFT methods [56,57,58] or force fields parameterizations [59,60,61,62] towards the simulation of more extended NT models and time-scales than are practically viable at the DFT level.
2. Models and Methods
2.1. Finite Models of the NTs Terminations
A suitable model of the NT terminations needs to be necessarily finite, in contrast to the 1D periodic models that have been routinely used in the literature for Imo-NTs so far [45,47,48,49,50,63]). It should also be sufficiently extended to accommodate full relaxation of the NT terminations around an otherwise unperturbed, bulk-like core region. Even though, depending on the composition and synthetic protocol used, real imogolite NTs can be from 20 nm [15] to 1 μm long [54], the relaxation of the tube terminations should affect only the most peripheral regions a few nm apart from the physical truncation of the NT, as happens for 2D oxide substrates [11,12,13]. On this basis, instead of simulating > 20 nm long NTs, we target the simulation-assisted identification of the smallest model that can accommodate the full, unconstrained relaxation of the terminations around a “bulk-like” core region unaffected by the structural and electronic rearrangements that the physical truncation of the NT induces.
To this end, we created differently sized finite models starting from the periodic structures of the AlGe (36 Al atoms in the NT, zigzag circumference) and AlSi (24 Al atoms) NTs optimized with the use of periodic boundary conditions. To define and label the longitudinal size of these models, we count the number of complete Al-hexagons present along the NT axis. Our models are accordingly labeled hex-3, hex-5 and hex-7 (Figure 1). The overall stoichiometry and number of atoms of these (electro-neutral) models is reported in Table 1. To eliminate electrostatic dipoles parallel to the NT axis, which may bias the geometry optimization and electronic structure characterization, all the finite NT models were set up with a symmetry plane passing through the central Si0 ring (Figure 1).

Table 1.
Full stoichiometry for the hex-3, 5 and 7 models of the OH- and H2O-terminatied AlSi and AlGe NTs. The total number of atoms of the model is shown in brackets.
The composition of the imogolite (zig-zag) structural unit, (Al2Si(Ge)O7H4)N, leads to two alternative neutral terminations for the AlSi/Ge NTs. One, labeled as the H2O termination, sees the aluminum hydroxide network of the NT truncated at the level of the Al-O bonds. The terminal (under-coordinated) Al atoms are then saturated with H2O molecules (instead of hydroxyl groups, –OH), which preserves the overall electro-neutrality of the system. The other, labeled as the OH termination, sees the NT truncated at the level of the Si(Ge)-O bonds, with one extra hydroxyl (OH) group saturating the terminal (Si)Ge atoms and maintaining the system electro-neutral. It is doubtless that the real terminations of the NTs may be substantially more complex than the highly idealized scenarios considered here, with defects and/or counter-ions contributing to the overall electro-neutrality of the finite NTs [19]. Nonetheless, in the absence of any experimental or first principles characterization of the NT terminations, the present models, although undoubtedly highly idealized, are expected to provide original and valuable insights, stimulating further computational and experimental research in the field.
2.2. Computational Methods
2.2.1. Geometry Optimization
Due to the computational challenging dimension of the systems studied, with a number of atoms ranging from 744 to 2232 (Table 1), their structure was relaxed by means of the linear-scaling DFT program ONETEP [64,65,66,67], using the same computational set-ups benchmarked and applied in earlier studies of imogolite NTs [47,48,49,50,51]. We used the PBE exchange-correlation (XC) functional [68], Kleinman–Bylander norm-conserving pseudopotentials [69], and a 1000 eV kinetic energy cutoff. The valence states of the O (Al, Si, Ge) atoms were described by means of four (nine) non-orthogonal Wannier functions (NGWFs) localized within an 8 Bohr radius from the atomic sites. The valence states of the H atoms were modeled using one NGWF only, also of 8 Bohr localization radius. For all the simulations, the density kernel (Kαβ) was optimized without any truncation. In all cases we used periodic boundary conditions ensuring a vacuum-buffer of at least 15 Å in each direction. The rationale beyond the selection of the different NTs dimensions is presented and discussed in Section 3.1. Geometry optimizations were carried out at the quasi-Newton level, via the Broyden–Fletcher–Goldfarb–Shanno (BFGS) approach [70]. In these simulations, the positions of all of the atoms were optimized without constraint, within a 0.05 eV/Å threshold for convergence on the atomic forces. Due to the absence of any dangling bonds in the systems (Section 2.1), spin-singlet solutions were found to have lower energy than alternative spin-polarized ones.
2.2.2. Hybrid, Range-Corrected DFT Simulations
The PBE-optimized structures were used for single-point DFT and non-local hybrid DFT simulations, carried out by means of the CP2K package [71]. We used Goedecker–Teter–Hutter (GTH) norm-conserving pseudopotentials [72], and the molecularly optimized (MOLOPT) DZVP-SR basis set [73]. We also used the auxiliary density matrix method (ADMM) and the cpFIT3 basis set to compute the exchange integrals. Hybrid DFT simulations were run using the PBE0-TC-LRC XC-functional [74] with an exact-exchange contribution of 25% and a real-space cut-off of 8 Å, complemented by a long-range (PBE exchange hole derived) correction [75].
3. Results and Discussion
3.1. Structural Relaxation of the Finite Alsi and AlGe NT models
To quantify the geometrical relaxation of the finite AlSi and AlGe NT models we resorted to two structural descriptors, namely the ring-resolved average displacement (⟨∆r⟩), and the layer-resolved average ring-diameter (⟨D⟩). ⟨∆r⟩ is defined as:
ri defines the position of the ith atom of the Rth ring (Figure 1b) in the finite NT model. Conversely, indicates the position of the same atom in the periodically repeated NT structure. N refers to the number of atoms in a given ring. A given ring contains all the longitudinally equivalent Si (Al) atoms and bridging hydroxyls (OH), in addition to the closest OH groups pointing to the NT ends. Only longitudinally equivalent Si atoms and bridging OH groups were included in the evaluation of ⟨∆r (R = 0)⟩ for the central ring (see also Figure 1b). Terminal H2O molecules in the H2O-terminated NTs were discarded for the evaluation of the average ring-displacement ⟨∆r⟩. The calculated ⟨∆r⟩ values define the level of relaxation of the ring by comparison to its analog in the optimized geometry for the fully periodic model. In the following, we rely on the value of ⟨∆r⟩ to separate between core and terminal regions of the finite NT model.
While ⟨∆r⟩ allows analysis of the longitudinal distortion in the NT finite models, the average radial-resolved ring-diameter, ⟨D(j,R)⟩, quantifies the cross-section relaxation. ⟨D(j,R)⟩ is computed as:
di(j,R) refers to the diameter of the ith pair of atoms in the jth NT layer (j: H1, O2,…H7, see Figure 2) in the Rth ring. As per Equation (2), ⟨D(j,R)⟩ is defined from the average of the calculated di(j,R) over the M = N/2 layer-equivalent atom-pairs of species j in a given ring R.
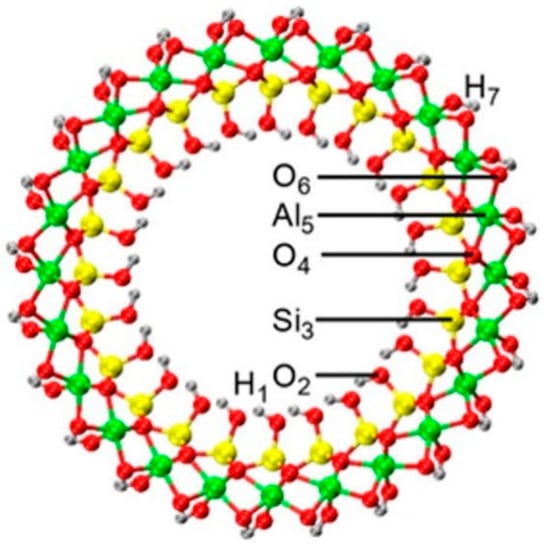
Figure 2.
The atomic structure of the AlSi NT (N = 24) with the labeling used for the atomic radial layers.
Figure 3 reports the values of ⟨∆r(R)⟩ and ⟨D(j,R)⟩ calculated for the hex-3, hex-5 and hex-7 models of the AlSi and AlGe NTs. We recall that the radial layers of the NTs are labeled from the inside to the outside: H1, O2, Si3 (Ge3), O4, Al5, O6, H7 (Figure 2).
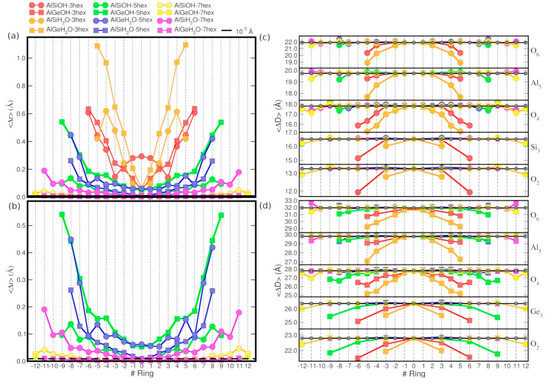
Figure 3.
(a) Calculated ⟨∆r⟩ values (Å) for the OH- and H2O-terminated AlSi and AlGe models as a function of the longitudinal (hex-3, hex-5, hex-7) extension. Panel (b) reports a close up of hex-5 and hex-7 results in comparison with the adopted 10−3 Å displacement threshold (horizontal black line). Panels (c,d) show the layer-resolved (O2,4,6, Si3 (Ge3), and Al5, see Figure 2) average ring-diameter (⟨D(j,R)⟩, Å) for the differently sized (hex-3, hex-5, and hex-7), OH- and H2O-terminated models of the AlSi and AlGe NTs, respectively. Results for the periodically optimized NTs are shown as black points.
As seen in Figure 3a,b, with an average ring displacement (⟨∆r⟩) for the innermost rings larger than 0.1 Å, the size of the AlSi and AlGe hex-3 models is evidently insufficient to prevent the relaxation of the tube-extremities from affecting the core region, regardless of the H2O or OH termination.
At odds with the AlSi results (⟨∆r⟩ ≤ 10−3 Å for the R = 0 ring), the computed ⟨∆r⟩ values for the core-rings of the AlGe hex-5 models are larger than 0.05 Å. Thus, the latter systems too turns out to be insufficiently extended to prevent the relaxation at the NT ends from altering the structure of the NT core region. For this condition to be fulfilled, the simulations indicate that use of the hex-7 models is necessary, as demonstrated by the ⟨∆r⟩ ≤ 10−3 Å values for their innermost core rings.
The results for the layer-resolved average ring-diameter (⟨D⟩) for the periodic and finite systems in Figure 3c,d reveals strong inward relaxation (up to ~3 Å) for the smallest hex-3 models, regardless of the (AlSi or AlGe) composition and the OH or H2O termination. The structural relaxation of the terminations becomes progressively smaller (down to ~0.25 Å) as the longitudinal size of the model is increased from hex-3 to hex-5, and to hex-7. Notably, whereas the inward relaxation of the innermost O2, Si3 (Ge3), and O4 atoms is systematically found for all the NT compositions and terminations studied, the radial relaxation of the O6 atoms is not. As shown in Figure 3c,d, depending on the AlSi or AlGe composition (thence diameter of the NT, Figure 1) and on the OH or H2O termination, the terminal O6 atoms contribute to the relaxation of the NT ends in a qualitatively different way. Specifically, the relaxation of the H2O-terminated AlGe hex-7 model is observed to take place via the substantial (~0.5 Å) increase of the O6-diameter.
Overall, these results indicate that depending on the NT composition and diameter, different (H-bonding-mediated) relaxation mechanisms can be present at the terminations of imogolite NTs. These results invite further research in the subject, especially for less idealized models of the NT terminations, ideally in the presence of solvent.
3.2. Relative Energy of the Finite AlSi and AlGe NT models
Calculation of the grand canonical formation energies (Eform) for the finite models of the AlSi and AlGe NTs enables comparison of their relative energies per imogolite unit. Eform were computed as:
where Etot is the total energy calculated for the finite NT model. N indicates the (integer) number of imogolite building blocks, the energy of which (EN) was obtained from the periodically optimized analog. ni indicates the number of atoms of species i in the system that needs to be added to the (N) imogolite units to recover the full stoichiometry of the finite NT model (Table 1). μi is the chemical potential of the atoms of species i. μi was taken as the DFT energy of the bulk or the molecular reference for the given atom. These were as follows: bulk Ge and Si in the diamond structure, bulk HPC Al, isolated singlet H2 molecule, and isolated triplet O2 molecule.
Based on the conclusion that hex-7 models are needed to accommodate the full relaxation of the NT terminations around an unperturbed core-region, we calculated Eform only for this size of system. The results are displayed in Table 2. Owing to the more contained distortion of the Al(OH)3 layer (Figure 3), the H2O termination turns out to be energetically favored over the OH one, regardless of the AlSI or AlGe composition. The substantially increased Eform difference between the H2O and OH termination of the (wider) AlGe NT with respect to the (narrower) AlSi indicates that as the NT diameter increases, the possibility of stabilizing the OH termination via hydrogen-bonding at the NT rim is progressively lost. The lower formation energy for AlGe–H2O with respect to AlSi–H2O in Table 2 suggests a more favorable hydration (per imogolite unit). This result stems from the different H-bonding connectivity of the terminal H2O molecules with the hydroxyl (OH) groups present at the NT rim for the AlGe and AlSi NTs (Figure 1). We reiterate that these results have been obtained for the NT models in vacuo, neglecting solvent-induced effects, which may be substantial and majorly alter the present calculated trends. The present result (in vacuo) should nevertheless provide a useful first principles reference for further study of the role of solvation for the NT terminations.
Table 2.
Computed grand-canonical formation energy (Eform, eV) for the the OH and H2O termination of the AlSi and AlGe hex-7 models.
3.3. Electronic Properties of the Finite AlSi and AlGe NT Models
In this section we quantify the effects of the termination relaxation on the electronic properties of the finite AlSi and AlGe models. Given the neglect of solvent effects in the simulations, which may alter the relative energies of the H2O- and OH-terminated models (Table 2), we characterized both the OH- and H2O-terminated hex-7 models, which have been shown (Section 3.1) to accommodate the full relaxation of the terminations around an otherwise structurally unperturbed core-region.
The PBE0-TC-LRC XC vacuum-aligned density of states (DOS) and layer-resolved projected DOS (LDOS) for the 7-hex models are displayed in Figure 4 (AlSi) and Figure 5 (AlGe). For a more compact visualization, the calculated valence band (VB) and conduction band (CB) edges, and the resulting band gaps (BGs) for all the models at both the PBE and PBE0-TC-LRC levels, are combined in Figure 6.
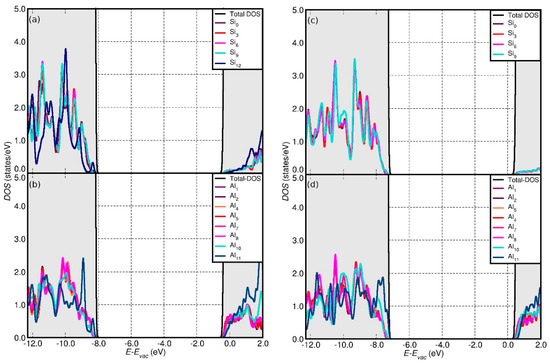
Figure 4.
Calculated (vacuum-aligned) total density of states (DOS), together with the Al and Si ring resolved (Figure 1) local density of states (LDOS) for the OH- (a,b) and H2O- (c,d) terminated AlSi hex-7 model.
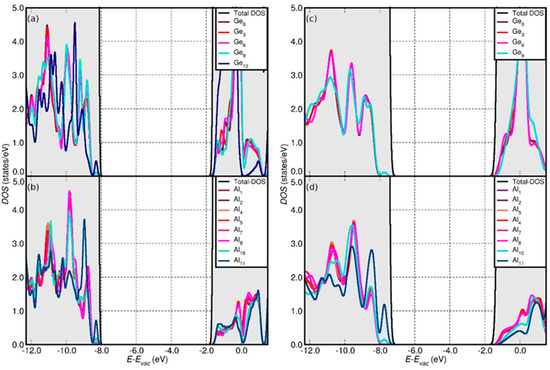
Figure 5.
Calculated (vacuum-aligned) DOS, Al ring and Si ring LDOS for the OH- (a,b) and H2O (c,d)-terminated AlGe hex-7 model.
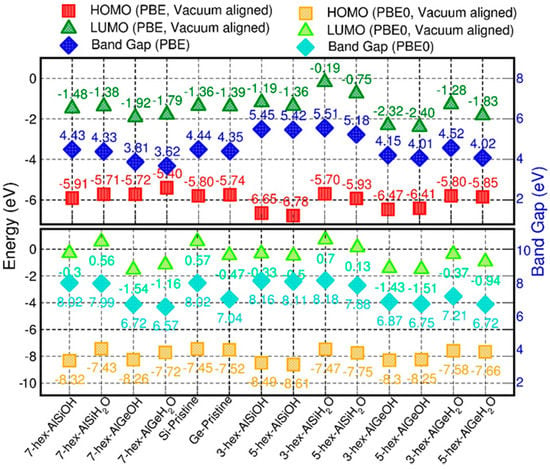
Figure 6.
PBE and PBE0-TC-LRC (shorthand notation: PBE0) vacuum-aligned VB and CB edges, and resulting BGs, for the OH- and H2O-terminated finite models of the AlSi and AlGe NTs, together with the results for their fully periodic counterparts.
Regardless of the NT composition and the XC-functional considered, the OH termination results in noticeably lower VB edges by comparison to the H2O-terminated models. The OH-induced downshift of the VB edge at PBE0-TC-LRC level is larger (0.78 eV on average) than for PBE (0.59 eV on average). The OH termination also induces the downshift of the CB-edge of the NTs by comparison to the results for the H2O-terminated models, with the calculated differences semi-quantitatively following the results for the VB edge.
The simulations indicate that the finite models of the AlSi NTs result in slightly larger (up to ~0.2 eV) BGs than for the periodic AlSi NTs, especially in the case of the smaller hex-3 and hex-5 systems. The opposite trend is observed for the finite AlGe models, which are found to have narrower (up to ~0.5 eV) BGs with respect to their periodic counterpart, the only exception being the 3-hex, H2O-terminated AlGe NT. Comparison of the vacuum-aligned VB and CB edges for the finite AlGe models (Figure 6) reveals that the BG reduction, with respect to the periodic model, is due to a larger downshift of the CB edges (up to ~1.1 eV) with respect to the VB ones (up to ~0.7 eV). Conversely, the BG for the smaller hex-3 and hex-5 AlSi models increases as a consequence of the termination-induced downshift (upshift) of the VB (CB) edges.
Previous computational studies [31,33,34] of periodically repeated AlSi and AlGe NTs have shown a clear correlation between the presence of a permanent polarization at the NT wall and the occurrence of a separation in real-space for the VB and CB edges of the NTs. For pristine hydroxylated NTs, this separation leads to the CB and VB edges being localized outside and inside the NT, respectively. The availability of optimized finite models allows us to investigate whether such a separation is maintained or modified due to the relaxation of the NT terminations. Figure 7 reports the computed real-space distribution of the VB and CB edges for the optimized hex-7 models of the OH- and H2O-terminated AlSi and AlGe NTs.
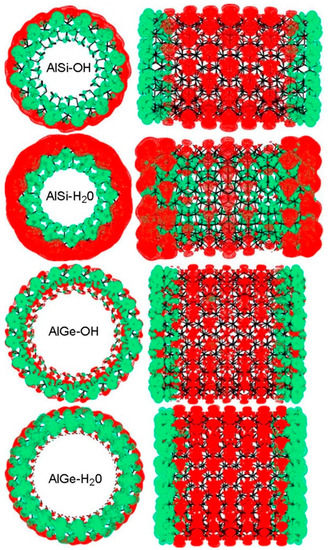
Figure 7.
Real-space localization of the VB (green) and CB (red) edges for the OH- and H2O-terminated hex-7 models of the AlSi and AlGe NTs. The band edges have been plotted considering Kohn States within 0.5 eV from the Highest Occupied Molecular Orbital (HOMO) and Lowest Unoccupied Molecular Orbital (LUMO) levels.
The calculated results indicate that the truncation of the AlSi NTs does not alter the pristine radial separation of the VB and CB edges. This conclusion holds regardless of the OH or H2O termination of the NT model. However, and owing to the calculated spilling of the CB edge inside the NT cavity, this conclusion does not hold for the AlGe NTs, regardless of the H2O or OH termination.
Analysis of the real-space localization of the VB and CB edges along the NT axis indicates that, apart from the H2O-terminated AlSi NTs, the VB and CB edges tend to accumulate at the NT ends and the innermost regions, respectively. This result reflects the calculated upward band-bending induced by the relaxation of the NT terminations (Figure 4 and Figure 5). As noted above, the H2O-terminated hex-7 AlSi NT deviates from this trend with accumulation of the CB edge at the NT ends, and the spreading of the VB edge across the whole NTs. These results reveal that control over the NT’s composition, diameter and specific termination could enable the tuning of the radial and longitudinal separation of the VB and CB edges in imogolite NTs. Such flexibility may in turn translate to synthetic control over the potentially strongly different optical properties and mechanisms for the relaxation and/or separation of photo-generated electron–hole (e–h) pairs.
Based on the calculated results, and neglecting solvent effects as well as the known presence of point-defects in imogolite NTs [19], the photo-generated holes in AlGe and OH-terminated AlSi NTs should accumulate at the NT ends, potentially favoring photo-oxidative processes therein. The marked longitudinal and radial separation of the VB and CB edges in the OH-terminated AlSi NTs also suggests that the solvent-mediated stabilization of this system may result in the beneficial separation of e–h pairs, with e- and h-accumulation at the NT core (outer surface) and termination (inner surface), respectively. We hope the present results, although on necessarily idealized models, will stimulate further interest in and research into the potential of imogolite NTs for photo-catalytic applications. The markedly different separation of band-edges calculated for periodic models of single- and double-walled imogolite AlGe NTs [47,56] invites future research also into the quantification of the extent to which the termination effects in double-walled AlGe NTs can be different from the single-walled analogs considered in this study.
3.4. Atomic Charges and Electrostatic Properties of the Finite AlSi and AlGe NT Models
In order to further characterize the termination effects in the AlSi and AlGe NTs, and to sustain ongoing efforts in the development of continuously improved, atomic charge-based force fields for imogolite NTs, we performed a charge analysis on all the finite models studied and their 1D periodic counterparts. Charge analysis was carried out on the basis of three different charge decompositions schemes: Hirshfield [76], Mulliken [77] and Bloechel (DDAPC) [78]. In spite of quantitative differences, all the charge analysis schemes used resulted in the same qualitative trends in terms of the longitudinal changes of atomic charges due to relaxation of the NT ends. For the sake of compactness, we therefore discuss here only the results for the DDAPC charges. Hirshfield and Mulliken’s results are available upon request to the authors.
Figures S1–S4 (Supplementary Material) report ring-averaged, atom-resolved charges for all the systems studied. For all the compositions and terminations modeled, the atomic charges for the central rings of the hex-7 models are very similar to those for the periodic analogs, with effectively negligible differences smaller than 0.01/0.02 a.u. This result reiterates the coexistence of fully relaxed terminations around an unperturbed core for the hex-7 models.
Our analysis indicates that the Ge3(Si3) and Al5 atoms become less positive moving from the core to the termination regions, with differences reaching up to 0.3 a.u. in the H2O-terminated models and 0.2 a.u. in the OH-terminated ones. Owing to the reduced positive charge for the terminal Ge3(Si3) and Al5 atoms, the O6 and O4 atoms at the NT end lose some of their electronic density, leading to smaller negative charges.
The terminal O4 atoms in the OH-ended models systematically present a less negative charge with respect to the O6 ones, indicative of an accumulation of electronic density at the outer wall of the NT termination. Conversely, the O4 atoms at NT rim in the larger hex-5 and hex-7 H2O-terminated models are more negatively charged than the O6 ones. This behavior is particularly marked in the hex-5 AlSiH2O case (0.05 a.u. average difference between the O4 and O6 atoms).
In combination with the corresponding structural relaxation (Figure 3), the calculated composition- and termination-dependent redistribution of the atomic charges at the NT termination (Figures S1–S4) results in different changes in the electrostatic potential along the NT axis (Figure 8) and in the accumulation of the VB and CB edges in the finite NTs (Figure 7). As noted above, these changes suggest, at a very qualitative level, potentially different mechanisms for the relaxation of photo-generated charge-carriers in the differently terminated AlSi and AlGe NTs.
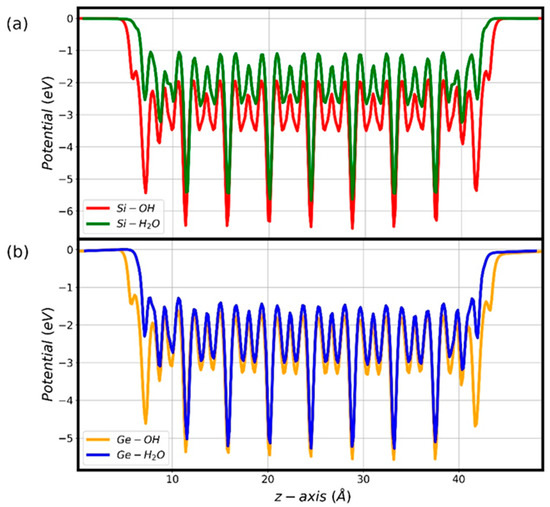
Figure 8.
Radially averaged PBE0-TC-LRC electrostatic potential along the hex-7 AlSi (a) and AlGe (b) NT (z) axis as a function of the OH or H2O termination.
We finally note that almost identical longitudinal trends are found for the atomic charges calculated at both the PBE and the PBE0-TC-LRC level. Thus, the XC-functional does not seem to majorly alter ground-state charge redistribution in these systems. Qualitatively, the most notable difference is that the PBE0-TC-LRC functional tends to accumulate more electronic density on the oxygen atoms, thus leading to larger positive charges on the electropositive atoms (H, Si, Ge, and Al).
4. Conclusions
To summarize, the PBE and PBE0-TC-LRC DFT simulations of differently sized, OH- and H2O-terminated finite models of AlSi and AlGe NTs indicate that:
- Models longer than 30 Å (1416-2232 atoms for the composition and termination considered here) are required to accommodate the relaxation of the NT ends around an unperturbed core region.
- In vacuo, NT termination by Al atoms coordinated to an additional H2O molecule is energetically favored over the truncation of the NT at the terminal Si atoms in the presence of additional hydroxyls (OH).
- For the largest and most converged models studied, the physical truncation of the AlSi NTs negligibly alters the calculated BG in comparison to what was obtained at the same level of theory for the periodic systems, owing to the quantitatively similar shifts for the VB and CB edges. Whereas H2O termination of the AlSi NTs is found to very weakly alter the energy of the VB and CB edges with respect to the vacuum level, the truncation-induced downward shifts of both band edges for the OH-terminated AlSi NTs is substantial (~0.9 eV).
- In contrast to the results for the AlSi NTs, the terminations effects in the AlGe NTs narrow the BG by ~0.3 eV (OH termination) and ~0.5 eV (H2O termination), owing to larger (up to ~1.1 eV) downshifting of the CB edge by comparison to the VB one (up to ~0.7 eV).
- Regardless of the OH or H2O termination, the accumulation of the VB and CB edges on opposite sides of the NT cavity typical of the periodic models (VB: inside, CB: outside) is maintained for AlSi NTs. OH termination of the AlSi NTs leads also to the accumulation of the VB and CB edges at the NT ends and core, respectively. Conversely, H2O termination suppresses such longitudinal separation of the VB and CB edges. For AlGe NTs, both the OH and H2O terminations result in longitudinal separation of the VB and CB edges (VB: end, CB: core), albeit with partial removal of their radial separation on different sides of the NT cavity. These changes are rationalized on the basis of the changes that structural relaxation at the NT ends induce in the atomic charges and electrostatics along the NT-axis.
- The calculated composition- and termination-dependent radial and longitudinal band separation and bending suggest potentially different optical excitations and mechanisms for the generation, relaxation and separation of photo-generated electron–hole (e-h) pairs.
In spite of the clear limitations due to the strongly idealized nature of the models for the NT terminations and the neglect of solvents, we hope the present results and considerations will stimulate further research in this subject, especially considering the current lack of computational or experiment-derived insights into the role of the NT terminations for the (photo-chemical) reactivity of imogolite NTs. More specifically, we anticipate these results will provide valuable tools towards future conclusive understanding of the inevitably different (photo-)chemical reactivity of the innermost and termination regions in single- and double-walled imogolite nanotubes of tunable composition and length. The results presented provide also a hitherto missing first principles reference for the development and benchmarking of future tight-binding or force fields descriptions of the termination effects in imogolite nanotubes.
Supplementary Materials
Supplementary Materials can be found at https://www.mdpi.com/2073-4352/10/11/1051/s1. Figures S1 to S4: calculated atomic charges for all the considered models.
Author Contributions
E.P. and G.T. designed the research. E.P. executed and analyzed the simulations, whose results were discussed with J.D.E., Z.C. and G.T., E.P. and G.T. wrote the initial draft of the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
E.P.: J.D.E., and G.T. acknowledge support from EPSRC UK (EP/I004483/1, EP/K013610/1, EP/P022189/1 and EP/P022189/2. E.P. acknowledges support from CINECA (ICT-CMSP-2018). Via membership of UKCP Consortium (EP/K013610/1, EP/P022189/1, and EP/P022189/2), this work made use of the ARCHER UK and UK Materials and Molecular Modeling Hub (EP/P020194/1) High-Performance Computing facilities. Z.C. and G.T. acknowledge also support by the Royal Society Newton Advanced Fellowship scheme (grant No. NAF\R1\180242).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Diebold, U. The surface science of titanium dioxide. Surf. Sci. Rep. 2003, 48, 53–229. [Google Scholar] [CrossRef]
- Bliem, R.; McDermott, E.; Ferstl, P.; Setvin, M.; Gamba, O.; Pavelec, J.; Schneider, M.A.; Schmid, M.; Diebold, U.; Blaha, P.; et al. Subsurface cation vacancy stabilization of the magnetite (001). Surf. Sci. 2014, 346, 1215–1218. [Google Scholar] [CrossRef] [PubMed]
- Goniakowski, J.; Finocchi, F.; Noguera, C. Polarity of oxide surfaces and nanostructures. Rep. Prog. Phys. 2008, 71, 016501. [Google Scholar] [CrossRef]
- Ray, P.C. Size and Shape Dependent Second Order Nonlinear Optical Properties of Nanomaterials and Their Application in Biological and Chemical Sensing. Chem. Rev. 2010, 110, 5332–5365. [Google Scholar] [CrossRef]
- Puzder, A.; Williamson, A.; Reboredo, F.A.; Galli, G. Structural Stability and Optical Properties of Nanomaterials with Reconstructed Surfaces. Phys. Rev. Lett. 2003, 91, 157405. [Google Scholar] [CrossRef] [PubMed]
- Naydenov, B.; Boland, J.J. Engineering the electronic structure of surface dangling bond nanowires of different size and dimensionality. Nanotechnology 2013, 24, 275202. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.-J.; Li, S.-L.; Gong, P.; Li, Y.-L.; Fang, X.-Y.; Jia, Y.-H.; Caob, M. Effect of surface dangling bonds on transport properties of phosphorous doped SiC nanowires. Phys. E Low-Dimens. Syst. Nanostruct. 2018, 104, 247–253. [Google Scholar] [CrossRef]
- Asaduzzaman, A.M.; Springborg, M. Structural and electronic properties of Si?Ge nanoparticles. Phys. Rev. B 2006, 74, 165406. [Google Scholar] [CrossRef]
- Wu, H.; Dey, R.; Siretanu, I.; Van den Ende, D.; Shui, L.; Zhou, G.; Mugele, F. Electrically Controlled Localized Charge Trapping at Amorphous Fluoropolymer–Electrolyte Interfaces. Small 2020, 16, 1905726. [Google Scholar] [CrossRef]
- Brawand, N.P.; Vörösab, M.; Galli, G. Surface dangling bonds are a cause of B-type blinking in Si nanoparticles. Nanoscale 2015, 7, 3737–3744. [Google Scholar] [CrossRef]
- Mukherjee, S.; Bartlow, V.M.; Nair, S. Phenomenology of the Growth of Single-Walled Aluminosilicate and Aluminogermanate Nanotubes of Precise Dimensions. Chem. Mater. 2005, 17, 4900. [Google Scholar] [CrossRef]
- Wada, S.-I.; Eto, A.; Wada, K. Synthetic allophane and imogolite. J. Soil Sci. 1979, 30, 347–355. [Google Scholar] [CrossRef]
- Barron, P.F.; Wilson, M.A.; Campbell, A.S.; Frost, R.L. Detection of imogolite in soils using solid state 29Si NMR. Nat. Cell Biol. 1982, 299, 616–618. [Google Scholar] [CrossRef]
- Monet, G.; Amara, M.S.; Rouzière, S.; Paineau, E.; Elkaim, E.; Chai, Z.; Elliott, J.D.; Poli, E.; Liu, L.M.; Teobaldi, G.; et al. Structural resolution of inorganic nanotubes with complex stoichiometry. Nat. Commun. 2018, 9, 2033–2042. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S.; Kim, K.; Nair, S. Short, Highly Ordered, Single-Walled Mixed-Oxide Nanotubes Assemble from Amorphous Nanoparticles. J. Am. Chem. Soc. 2007, 129, 6820–6826. [Google Scholar] [CrossRef]
- Levard, C.; Rose, J.; Masion, A.; Doelsch, E.; Borschneck, D.; Olivi, L.; Dominici, C.; Grauby, O.; Woicik, J.C.; Bottero, J.-Y. Synthesis of Large Quantities of Single-Walled Aluminogermanate Nanotube. J. Am. Chem. Soc. 2008, 130, 5862–5863. [Google Scholar] [CrossRef]
- Kang, D.-Y.; Zang, J.; Wright, E.R.; McCanna, A.L.; Jones, C.W.; Nair, S. Dehydration, Dehydroxylation, and Rehydroxylation of Single-Walled Aluminosilicate Nanotubes. ACS Nano 2010, 4, 4897–4907. [Google Scholar] [CrossRef]
- Yucelen, G.I.; Choudhury, R.P.; Vyalikh, A.; Scheler, U.; Beckham, H.W.; Nair, S. Formation of Single-Walled Aluminosilicate Nanotubes from Molecular Precursors and Curved Nanoscale Intermediates. J. Am. Chem. Soc. 2011, 133, 5397–5412. [Google Scholar] [CrossRef]
- Yucelen, G.I.; Choudhury, R.P.; Leisen, J.; Nair, S.; Beckham, H.W. Defect Structures in Aluminosilicate Single-Walled Nanotubes: A Solid-State Nuclear Magnetic Resonance Investigation. J. Phys. Chem. C 2012, 116, 17149–17157. [Google Scholar] [CrossRef]
- Levard, C.; Rose, J.; Thill, A.; Masion, A.; Doelsch, E.; Maillet, P.; Spalla, O.; Olivi, L.; Cognigni, A.; Ziarelli, F.; et al. Formation and Growth Mechanisms of Imogolite-Like Aluminogermanate Nanotubes. Chem. Mater. 2010, 22, 2466–2473. [Google Scholar] [CrossRef]
- Bottero, I.; Bonelli, B.; Ashbrook, S.E.M.; Wright, P.A.; Zhou, W.; Tagliabue, M.; Armandi, M.; Garrone, E. Synthesis and characterization of hybrid organic/inorganic nanotubes of the imogolite type and their behaviour towards methane adsorption. Phys. Chem. Chem. Phys. 2011, 13, 744–750. [Google Scholar] [CrossRef] [PubMed]
- Bonelli, B.; Armandi, M.; Garrone, E. Surface properties of alumino-silicate single-walled nanotubes of the imogolite type. Phys. Chem. Chem. Phys. 2013, 15, 13381–13390. [Google Scholar] [CrossRef] [PubMed]
- Amara, M.S.; Paineau, E.; Rouzière, S.; Guiose, B.; Krapf, M.-E.M.; Taché, O.; Launois, P.; Thill, A. Hybrid, Tunable-Diameter, Metal Oxide Nanotubes for Trapping of Organic Molecules. Chem. Mater. 2015, 27, 1488–1494. [Google Scholar] [CrossRef]
- Kang, D.-Y.; Brunelli, N.A.; Yucelen, G.I.; Venkatasubramanian, A.; Zang, J.; Leisen, J.; Hesketh, P.J.; Jones, C.W.; Nair, S. Direct synthesis of single-walled aminoaluminosilicate nanotubes with enhanced molecular adsorption selectivity. Nat. Commun. 2014, 5, 3342. [Google Scholar] [CrossRef] [PubMed]
- Kang, D.-Y.; Zang, J.; Jones, C.W.; Nair, S. Single-Walled Aluminosilicate Nanotubes with Organic-Modified Interiors. J. Phys. Chem. C 2011, 115, 7676–7685. [Google Scholar] [CrossRef]
- Zanzottera, C.; Armandi, M.; Esposito, S.; Gorrone, E.; Bonelli, B. Physico-chemical properties of imogolite nanotubes functionalized on both external and internal surfaces. J. Phys. Chem. C. 2012, 116, 20417. [Google Scholar] [CrossRef]
- Picot, P.; Taché, O.; Coradin, T.; Thill, A.; Malloggi, F. Behaviour of hybrid inside/out Janus nanotubes at an oil/water interface. A route to self-assembled nanofluidics? Faraday Discuss 2016, 191, 391–406. [Google Scholar] [CrossRef]
- Picot, P.; Gobeaux, F.; Coradin, T.; Thill, A. Dual internal functionalization of imogolite nanotubes as evidenced by optical properties of Nile red. Appl. Clay Sci. 2019, 178, 105133. [Google Scholar] [CrossRef]
- Nasi, R.; Sannino, F.; Picot, P.; Thill, A.; Oliviero, O.; Esposito, S.; Armandi, M.; Bonelli, B. Hybrid organic-inorganic nanotubes effectively adsorb some organic pollutants in aqueous phase. Appl. Clay Sci. 2020, 186, 105449. [Google Scholar] [CrossRef]
- Katsumata, K.-I.; Hou, X.; Sakai, M.; Nakajima, A.; Fujishima, A.; Matsushita, N.; MacKenzie, K.; Okada, K. Visible-light-driven photodegradation of acetaldehyde gas catalyzed by aluminosilicate nanotubes and Cu(II)-grafted TiO2 composites. Appl. Catal. B Environ. 2013, 138, 243–252. [Google Scholar] [CrossRef]
- Marzán, L.L.; Philipse, A.P. Synthesis of platinum nanoparticles in aqueous host dispersions of inorganic (imogolite) rods. Colloids Surf. A Physicochem. Eng. Asp. 1994, 90, 95–109. [Google Scholar] [CrossRef]
- Paineau, E.; Krapf, M.-E.M.; Amara, M.-S.; Matskova, N.V.; Dozov, I.; Rouzière, S.; Thill, A.; Launois, P.; Davidson, P. A liquid-crystalline hexagonal columnar phase in highly-dilute suspensions of imogolite nanotubes. Nat. Commun. 2016, 7, 10271. [Google Scholar] [CrossRef] [PubMed]
- Paineau, E.; Monet, G.; Peyre, V.; Goldmann, C.; Rouzière, S.; Launois, P. Colloidal Stability of Imogolite Nanotube Dispersions: A Phase Diagram Study. Langmuir 2019, 35, 12451–12459. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.J.; Paineau, E.; Anthony, D.B.; Gao, Y.; Leese, H.; Rouzière, S.; Launois, P.; Shaffer, M.S.P. Inorganic Nanotube Mesophases Enable Strong Self-Healing Fibers. ACS Nano 2020, 14, 5570–5580. [Google Scholar] [CrossRef]
- Paineau, E.; Rouzière, S.; Monet, G.; Diogo, C.C.; Morfin, I.; Launois, P. Role of initial precursors on the liquid-crystalline phase behavior of synthetic aluminogermanate imogolite nanotubes. J. Colloid Interface Sci. 2020, 580, 275–285. [Google Scholar] [CrossRef]
- Ma, W.; Yah, W.O.; Otsuka, H.; Takahara, A. Application of imogolite clay nanotubes in organic–inorganic nanohybrid materials. J. Mater. Chem. 2012, 22, 11887. [Google Scholar] [CrossRef]
- Arancibia-Miranda, N.; Escudey, M.; Pizarro, C.; DeNardin, J.C.; García-González, M.T.; Fabris, J.D.; Charlet, L. Preparation and characterization of a single-walled aluminosilicate nanotube-iron oxide composite: Its applications to removal of aqueous arsenate. Mater. Res. Bull. 2014, 51, 145–152. [Google Scholar] [CrossRef]
- Avellan, A.; Levard, C.; Kumar, N.P.; Rose, J.; Olivi, L.; Thill, A.; Chaurand, P.; Borschneck, D.; Masion, A. Structural incorporation of iron into Ge–imogolite nanotubes: A promising step for innovative nanomaterials. RSC Adv. 2014, 4, 49827–49830. [Google Scholar] [CrossRef]
- Castro, C.; Arancibia-Miranda, N.; Acuña-Rougier, C.; Escudey, M.; Tasca, F. Spectroscopic and Electrochemical Studies of Imogolite and Fe-Modified Imogolite Nanotubes. Nanomaterials 2016, 6, 28. [Google Scholar] [CrossRef]
- Shafia, E.; Esposito, S.; Armandi, M.; Manzoli, M.; Garrone, E.; Bonelli, B. Isomorphic substitution of aluminium by iron into single-walled alumino-silicate nanotubes: A physico-chemical insight into the structural and adsorption properties of Fe-doped imogolite. Microporous Mesoporous Mater. 2016, 224, 229–238. [Google Scholar] [CrossRef]
- Bahadori, E.; Vaiano, V.; Esposito, S.; Armandi, M.; Sannino, D.; Bonelli, B. Photo-activated degradation of tartrazine by H2O2 as catalyzed by both bare and Fe-doped methyl-imogolite nanotubes. Catal. Today 2018, 304, 199–207. [Google Scholar] [CrossRef]
- Paineau, E. Imogolite Nanotubes: A Flexible Nanoplatform with Multipurpose Applications. Appl. Sci. 2018, 8, 1921. [Google Scholar] [CrossRef]
- Paineau, E.; Launois, P. Nanomaterials from Imogolite: Structure, Properties, and Functional Materials. Nanomater. Clay Miner. 2019, 257–284. [Google Scholar] [CrossRef]
- Thill, A. Characterization of Imogolite by Microscopic and Spectroscopic Methods. Dev. Clay Sci. 2016, 7, 223. [Google Scholar]
- Teobaldi, G.; Beglitis, N.S.; Fisher, A.J.; Zerbetto, F.; Hofer, W.A. Hydroxyl vacancies in single-walled aluminosilicate and aluminogermanate nanotubes. J. Phys. Condens. Matter 2009, 21, 195301. [Google Scholar] [CrossRef]
- Poli, E.; Elliott, J.D.; Hine, N.D.M.; Mostofi, A.A.; Teobaldi, G. Large-scale density functional theory simulation of inorganic nanotubes: A case study on Imogolite nanotubes. Mater. Res. Innov. 2015, 19, S272–S282. [Google Scholar] [CrossRef]
- Poli, E.; Elliott, J.D.; Ratcliff, L.E.; Andrinopoulos, L.; Dziedzic, J.; Hine, N.D.M.; Mostofi, A.A.; Skylaris, C.-K.; Haynes, P.D.; Teobaldi, G. The potential of imogolite nanotubes as (co-)photocatalysts: A linear-scaling density functional theory study. J. Phys. Condens. Matter 2016, 28, 74003. [Google Scholar] [CrossRef]
- Elliott, J.D.; Poli, E.; Scivetti, I.; Ratcliff, L.E.; Andrinopoulos, L.; Teobaldi, G. Chemically Selective Alternatives to Photoferroelectrics for Polarization-Enhanced Photocatalysis: The Untapped Potential of Hybrid Inorganic Nanotubes. Adv. Sci. 2016, 4, 1600153. [Google Scholar] [CrossRef]
- Poli, E.; Elliott, J.D.; Chulkov, S.K.; Watkins, M.B.; Teobaldi, G. The Role of Cation-Vacancies for the Electronic and Optical Properties of Aluminosilicate Imogolite Nanotubes: A Non-local, Linear-Response TDDFT Study. Front. Chem. 2019, 7, 210. [Google Scholar] [CrossRef]
- Monet, G.; Paineau, E.; Chai, Z.; Amara, M.S.; Orecchini, A.; Jimenez-Ruiz, M.; Ruiz-Caridad, A.; Fine, L.; Rouzière, S.; Liu, L.; et al. Solid wetting-layers in inorganic nano-reactors: The water in imogolite nanotube case. Nanoscale Adv. 2020, 2, 1869–1877. [Google Scholar] [CrossRef]
- Maillet, P.; Levard, C.; Larquet, E.; Mariet, C.; Spalla, O.; Menguy, N.; Masion, A.; Doelsch, E.; Rose, J.; Thill, A. Evidence of Double-Walled Al−Ge Imogolite-Like Nanotubes. A Cryo-TEM and SAXS Investigation. J. Am. Chem. Soc. 2010, 132, 1208–1209. [Google Scholar] [CrossRef] [PubMed]
- Maillet, P.; Levard, C.; Spalla, O.; Masion, A.; Rose, J.; Thill, A. Growth kinetic of single and double-walled aluminogermanate imogolite-like nanotubes: An experimental and modeling approach. Phys. Chem. Chem. Phys. 2011, 13, 2682–2689. [Google Scholar] [CrossRef] [PubMed]
- Thill, A.; Maillet, P.; Guiose, B.; Spalla, O.; Belloni, L.; Chaurand, P.; Auffan, M.; Olivi, L.; Rose, J. Physico-chemical Control over the Single- or Double-Wall Structure of Aluminogermanate Imogolite-like Nanotubes. J. Am. Chem. Soc. 2012, 134, 3780–3786. [Google Scholar] [CrossRef] [PubMed]
- Amara, M.-S.; Paineau, E.; Bacia-Verloop, M.; Krapf, M.-E.M.; Davidson, P.; Belloni, L.; Levard, C.; Rose, J.; Launois, P.; Thill, A. Single-step formation of micron long (OH)3Al2O3Ge(OH) imogolite-like nanotubes. Chem. Commun. 2013, 49, 11284–11286. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.Y.; Picot, P.; Brubach, J.-B.; Roy, P.; Thill, A.; Le Caer, S. Water Adsorption in Single- and Double-Walled Inorganic Nanotubes. J. Phys. Chem. C 2019, 123, 19768–19777. [Google Scholar] [CrossRef]
- Lourenço, M.P.; Guimarães, L.; Da Silva, M.C.; De Oliveira, C.; Heine, T.; Duarte, H.A. Nanotubes With Well-Defined Structure: Single- and Double-Walled Imogolites. J. Phys. Chem. C 2014, 118, 5945–5953. [Google Scholar] [CrossRef]
- Guimarães, L.; Lourenço, M.P.; Duarte, H.A. Nanotubes with well-defined structure: Imogolites. Chem. Model. 2015, 12, 151–183. [Google Scholar] [CrossRef]
- Da Silva, M.C.; Dos Santos, E.C.; Lourenço, M.P.; Gouvea, M.P.; Duarte, H.A. Structural, Electronic, and Mechanical Properties of Inner Surface Modified Imogolite Nanotubes. Front. Mater. 2015, 2, 16. [Google Scholar] [CrossRef][Green Version]
- Pouvreau, M.; Greathouse, J.A.; Cygan, R.T.; Kalinichev, A.G. Structure of Hydrated Gibbsite and Brucite Edge Surfaces: DFT Results and Further Development of the ClayFF Classical Force Field with Metal–O–H Angle Bending Terms. J. Phys. Chem. C 2017, 121, 14757–14771. [Google Scholar] [CrossRef]
- Scalfi, L.; Fraux, G.; Boutin, A.; Coudert, F.-X. Structure and Dynamics of Water Confined in Imogolite Nanotubes. Langmuir 2018, 34, 6748–6756. [Google Scholar] [CrossRef]
- González, R.I.; Rojas-Nunez, J.; Valencia, F.J.; Munoz, F.; Baltazar, S.E.; Allende, S.; Rogan, J.; Valdivia, J.A.; Kiwi, M.; Ramírez, R.; et al. Imogolite in water: Simulating the effects of nanotube curvature on structure and dynamics. Appl. Clay Sci. 2020, 191, 105582. [Google Scholar] [CrossRef]
- Fernandez-Martinez, A.; Tao, J.; Wallace, A.F.; Bourg, I.C.; Johnson, M.R.; De Yoreo, J.J.; Charlet, L. Cuelloe and Laurent Charlet. Curvature-induced hydrophobicity at imogolite–water interfaces. Environ. Sci. Nano 2020, 7, 2759. [Google Scholar] [CrossRef]
- Fan, Y.; Xia, Y.; Mei, L. Energetic Minimum Structures of Imogolite Nanotubes: A First-Principles Prediction. J. Phys. Chem. C 2009, 113, 14834–14837. [Google Scholar] [CrossRef]
- Skylaris, C.-K.; Haynes, P.D.; Mostofi, A.A.; Payne, M.C. IntroducingONETEP: Linear-scaling density functional simulations on parallel computers. J. Chem. Phys. 2005, 122, 084119. [Google Scholar] [CrossRef] [PubMed]
- Haynes, P.D.; Skylaris, C.-K.; Mostofi, A.A.; Payne, M.C. ONETEP: Linear-scaling density-functional theory with local orbitals and plane waves. Phys. Status Solidi 2006, 243, 2489–2499. [Google Scholar] [CrossRef][Green Version]
- Hine, N.D.M.; Haynes, P.D.; Mostofi, A.A.; Skylaris, C.-K.; Payne, M. Linear-scaling density-functional theory with tens of thousands of atoms: Expanding the scope and scale of calculations with ONETEP. Comput. Phys. Commun. 2009, 180, 1041–1053. [Google Scholar] [CrossRef]
- Hine, N.D.M.; Robinson, M.; Haynes, P.D.; Skylaris, C.-K.; Payne, M.C.; Mostofi, A.A. Accurate ionic forces and geometry optimization in linear-scaling density-functional theory with local orbitals. Phys. Rev. B 2011, 83, 195102. [Google Scholar] [CrossRef]
- Perdew, J.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef]
- Gonze, X.; Stumpf, R.; Scheffler, M. Analysis of separable potentials. Phys. Rev. B 1991, 44, 8503–8513. [Google Scholar] [CrossRef]
- Pfrommer, B.G.; Côté, M.; Louie, S.G.; Cohen, M.L. Relaxation of Crystals with the Quasi-Newton Method. J. Comput. Phys. 1997, 131, 233–240. [Google Scholar] [CrossRef]
- Vandevondele, J.; Krack, M.; Mohamed, F.; Parrinello, M.; Chassaing, T.; Hutter, J. Quickstep: Fast and accurate density functional calculations using a mixed Gaussian and plane waves approach. Comput. Phys. Commun. 2005, 167, 103–128. [Google Scholar] [CrossRef]
- Goedecker, S.; Teter, M.A.; Hutter, J. Separable dual-space Gaussian pseudopotentials. Phys. Rev. B 1996, 54, 1703–1710. [Google Scholar] [CrossRef] [PubMed]
- Vandevondele, J.; Hutter, J. Gaussian basis sets for accurate calculations on molecular systems in gas and condensed phases. J. Chem. Phys. 2007, 127, 114105. [Google Scholar] [CrossRef] [PubMed]
- Strand, J.; Chulkov, S.K.; Watkins, M.; Shluger, A. First principles calculations of optical properties for oxygen vacancies in binary metal oxides. J. Chem. Phys. 2019, 150, 044702. [Google Scholar] [CrossRef] [PubMed]
- Ernzerhof, M.; Perdew, J.P. Generalized gradient approximation to the angle- and system-averaged exchange hole. J. Chem. Phys. 1998, 109, 3313–3320. [Google Scholar] [CrossRef]
- Hirshfeld, F.L. Bonded-atom fragments for describing molecular charge densities. Theor. Chem. Acc. 1977, 44, 129–138. [Google Scholar] [CrossRef]
- Mulliken, R.S. Electronic Population Analysis on LCAO–MO Molecular Wave Functions. I. J. Chem. Phys. 1955, 23, 1833–1840. [Google Scholar] [CrossRef]
- Blochl, P.E. Electrostatic decoupling of periodic images of plane-wave-expanded densities and derived atomic point charges. J. Chem. Phys. 1995, 103, 7422. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).