
New Insights into the Genome Organization of Yeast Killer Viruses Based on “Atypical” Killer Strains Characterized by High-Throughput Sequencing

 ,
,
Abstract
:1. Introduction
2. Results
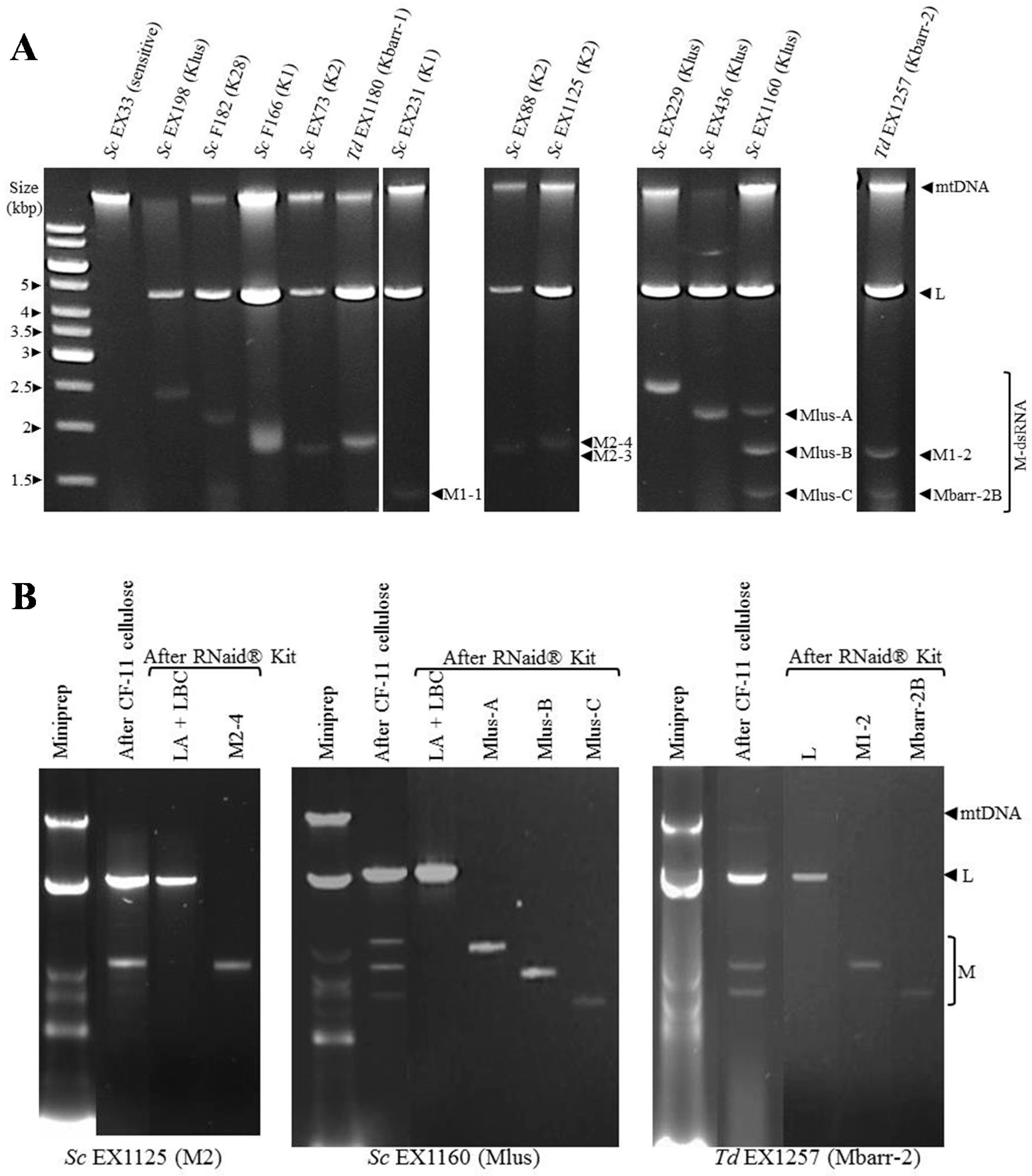
2.1. Phenotypic and Genotypic Characterization of S. cerevisiae and T. delbrueckii Killer Yeasts
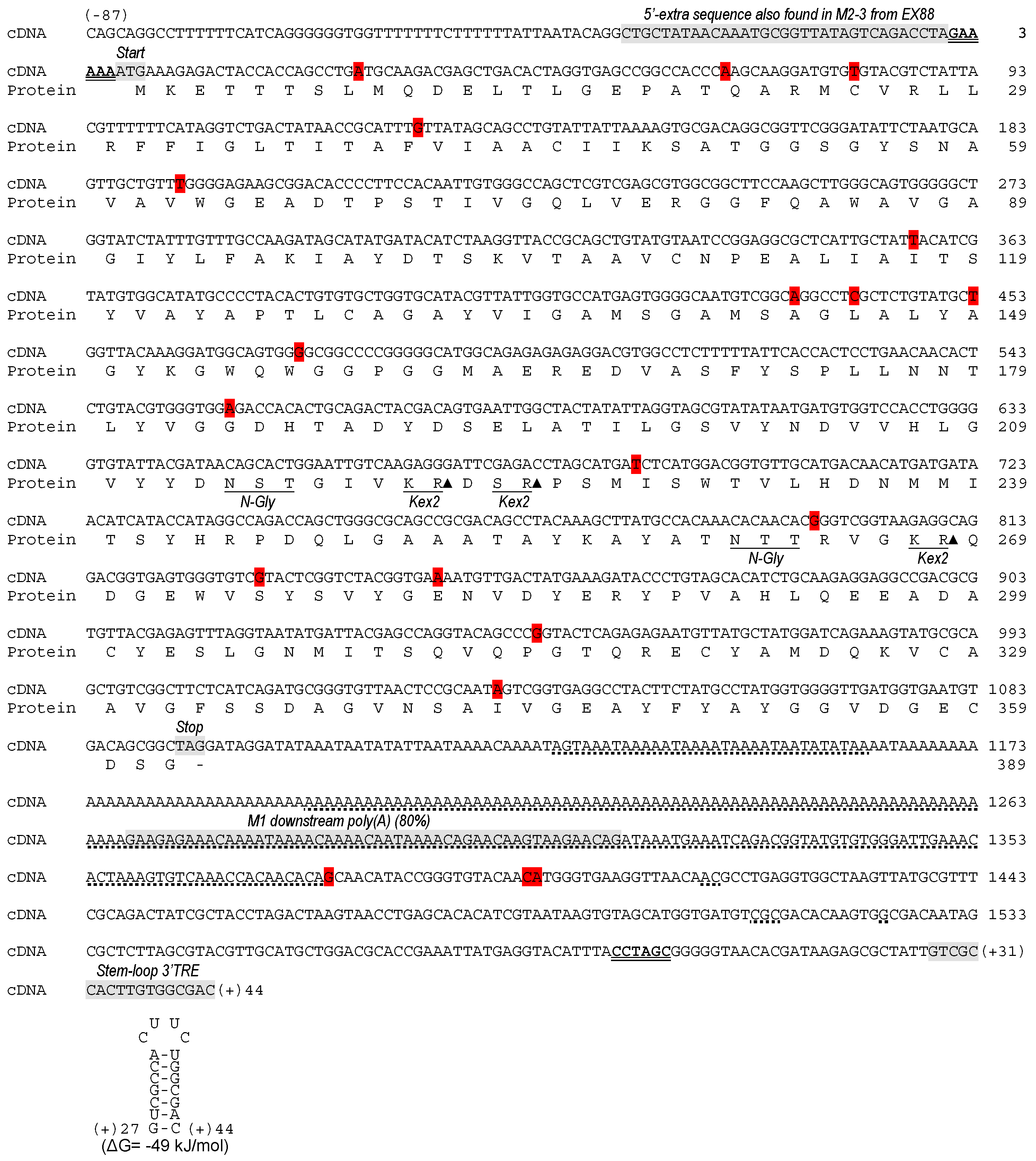
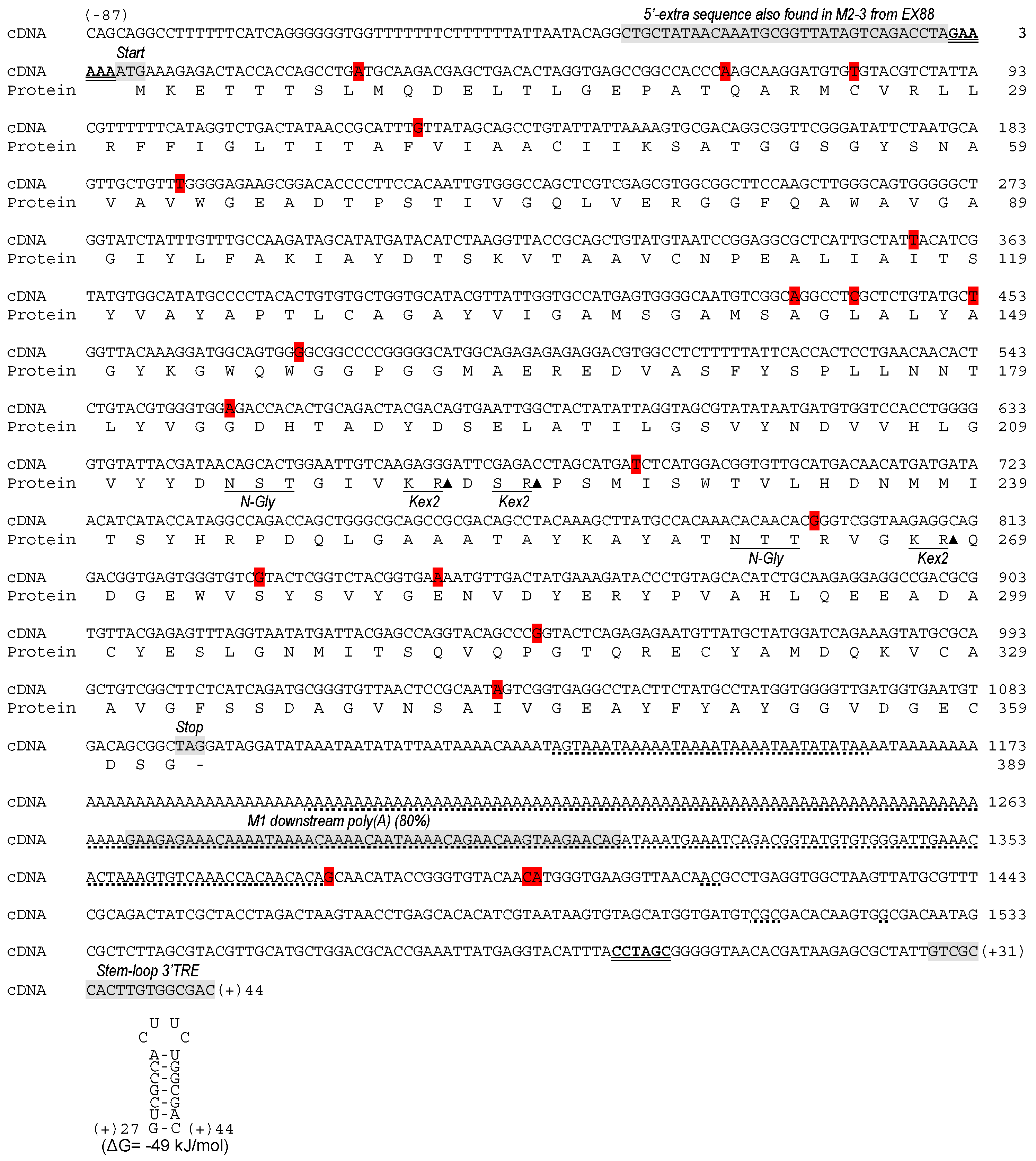
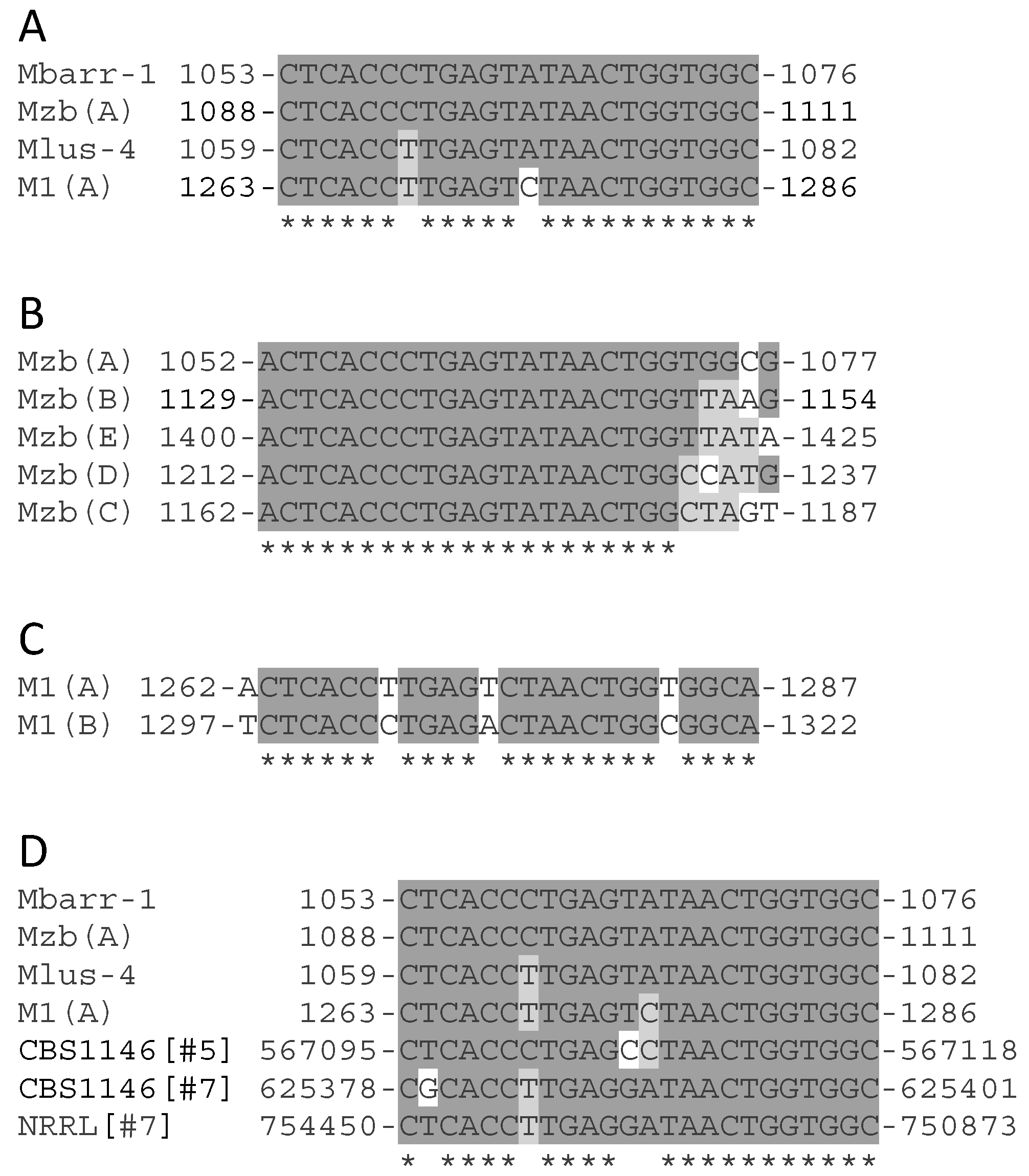
2.2. Analysis of the dsRNA Sequence from ScV-M and TdV-M Viruses
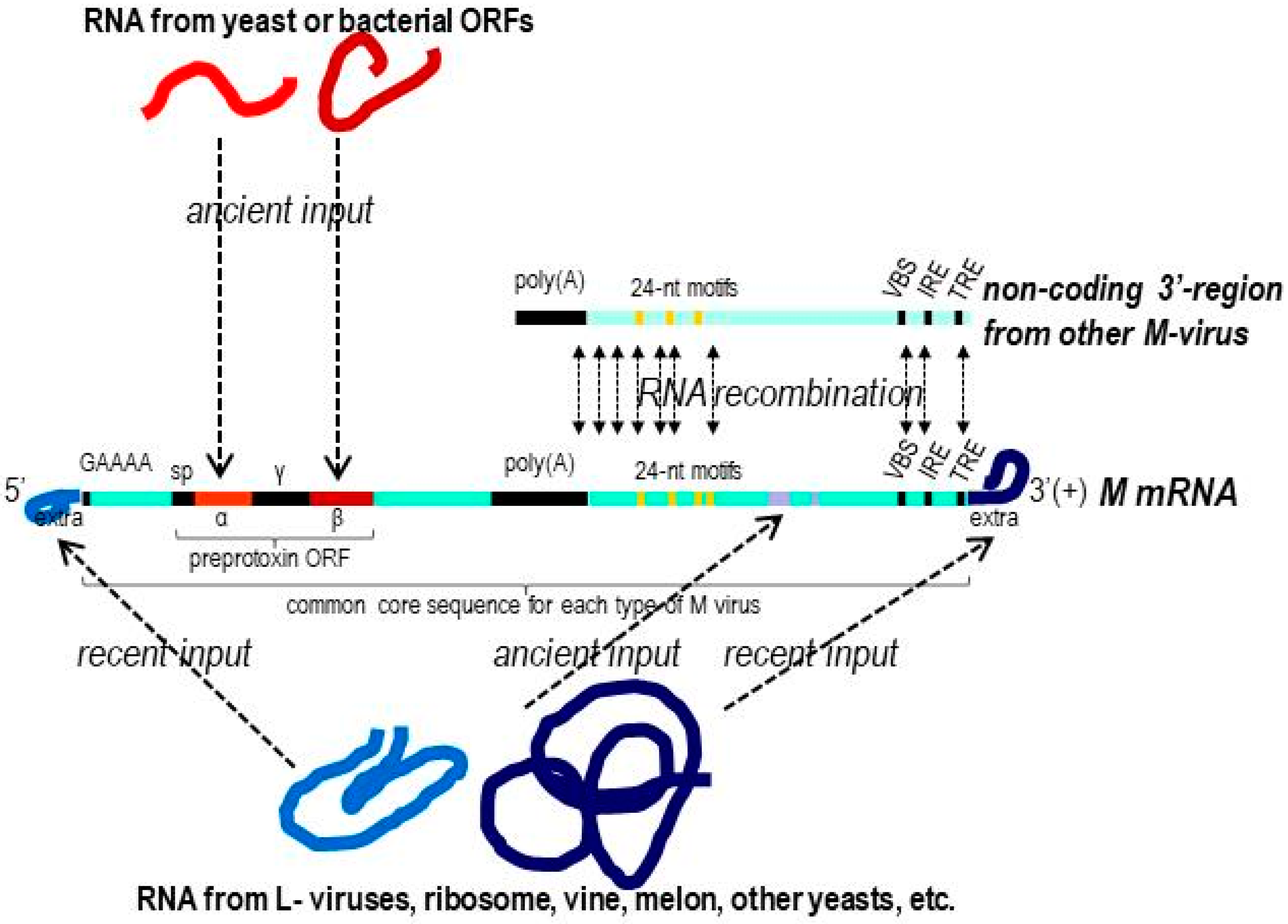
2.3. Analysis of 5′- and 3′-Extra Sequences of M-Genomes
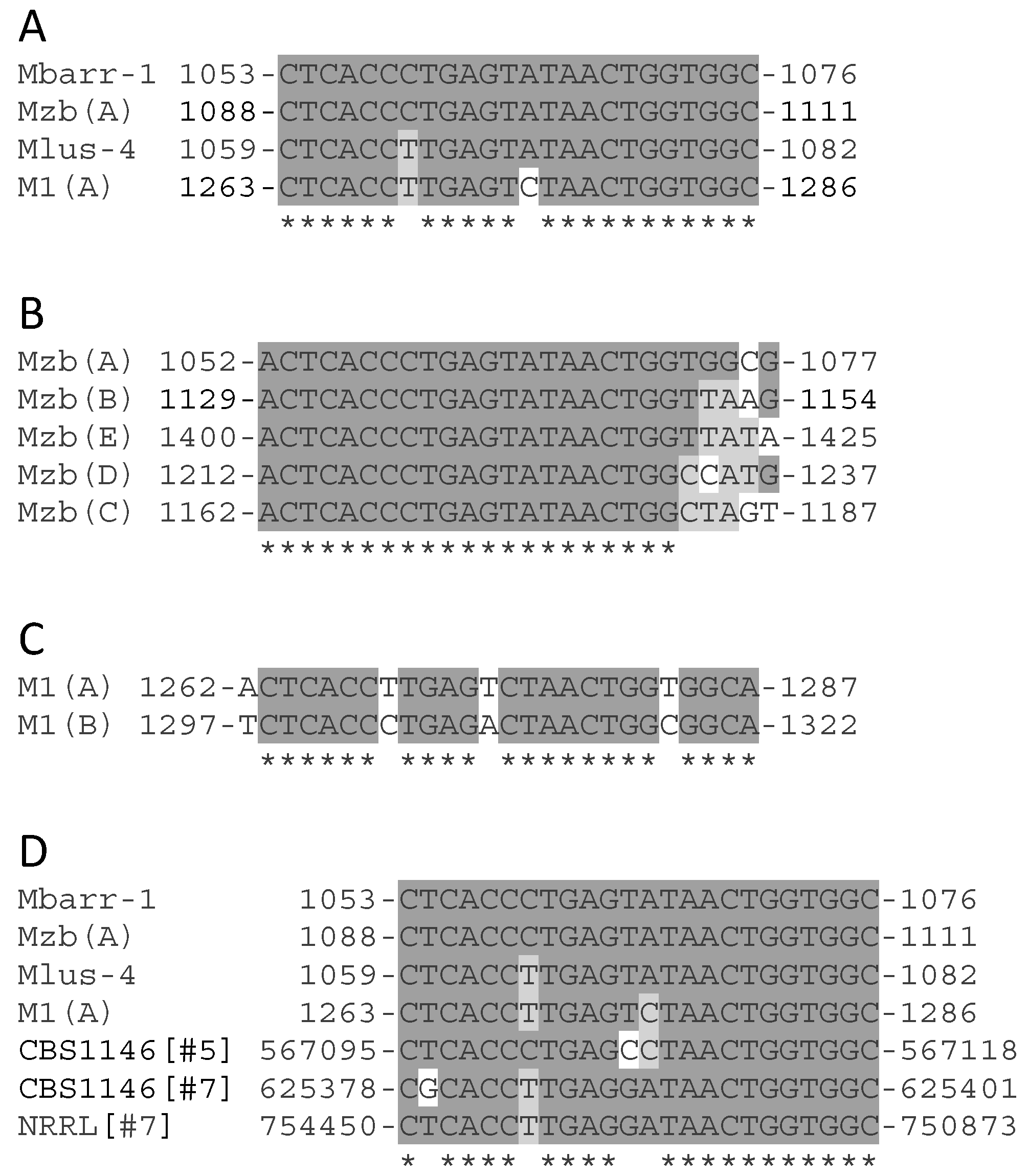
2.4. Analysis of Common Core Sequence of M-Genomes
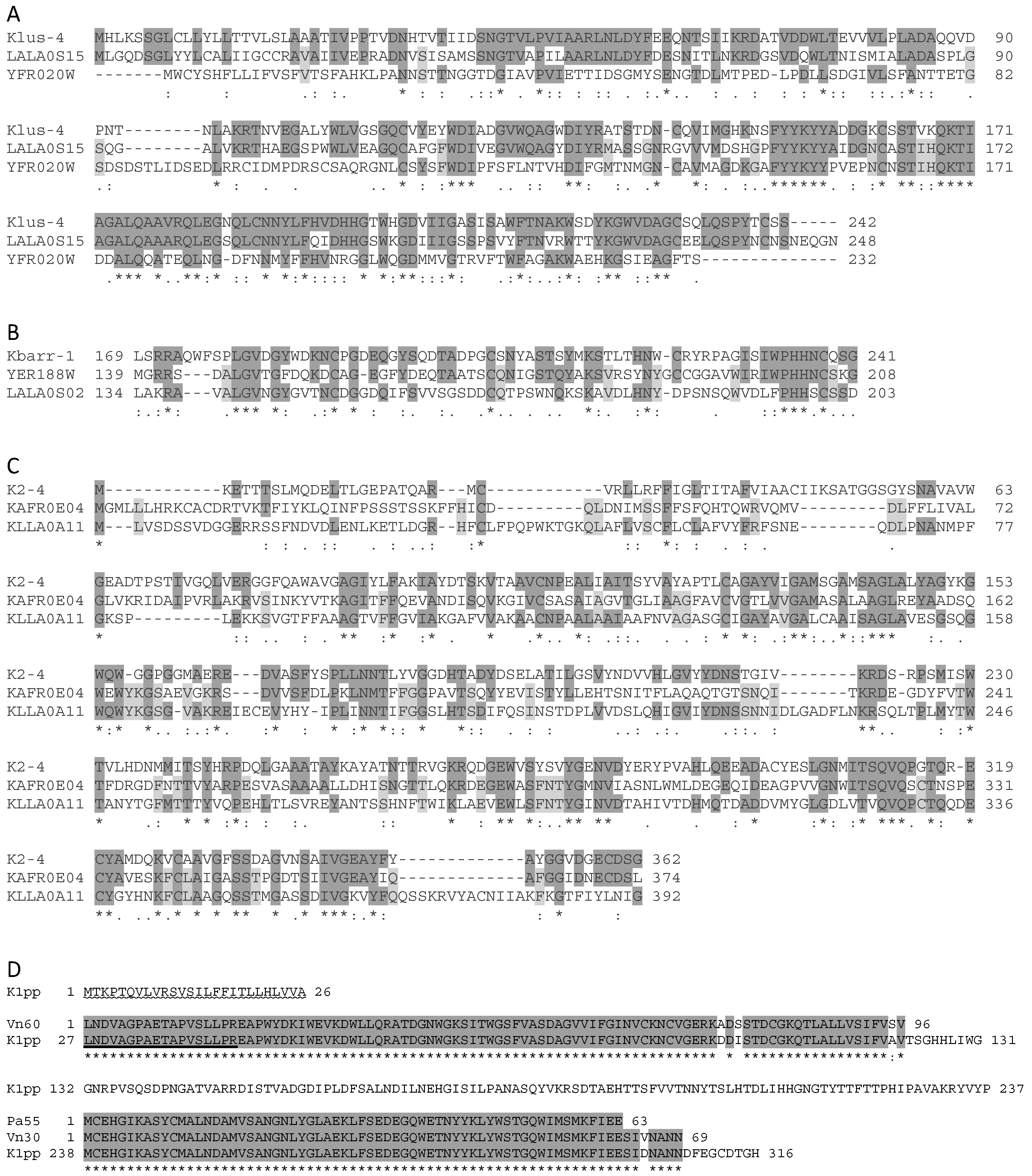
2.5. Analysis of ScV-M and TdV-M Preprotoxin ORF Sequences
3. Discussion
3.1. Phenotypic and Genotypic Characterization of Atypical Killer Yeasts
3.2. Analysis of dsRNA Sequences from M Viruses
3.3. Sequence Comparison of the Killer Preprotoxins
4. Conclusions
5. Materials and Methods
5.1. Yeast Strains and Media
5.2. Determination of Yeast Killer Activity
5.3. Total Nucleic Acid Preparation and Nuclease Digestion
5.4. Nucleic Acid Analysis for Killer Yeast Typing
5.5. Viral dsRNA Purification
5.6. Preparation and Sequencing of cDNA Libraries from Purified Viral dsRNA
5.7. Viral dsRNA Sequence Assembly
5.8. Real-Time Quantitative PCR (qPCR) Conditions and Analysis
5.9. Miscellaneous
5.10. Nucleotide Sequence Accession Number
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Ramírez, M.; Velázquez, R.; Maqueda, M.; López-Piñeiro, A.; Ribas, J.C. A new wine Torulaspora delbrueckii killer strain with broad antifungal activity and its toxin-encoding double-stranded RNA virus. Front. Microbiol. 2015, 6, 983. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Cousiño, N.; Maqueda, M.; Ambrona, J.; Zamora, E.; Esteban, E.; Ramírez, M. A new wine Saccharomyces cerevisiae double-stranded RNA virus encoded killer toxin (Klus) with broad antifungal activity is evolutionarily related to a chromosomal host gene. Appl. Environ. Microbiol. 2011, 77, 1822–1832. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, M.J.; Breinig, F. Yeast viral killer toxins: Lethality and self-protection. Nat. Rev. Microbiol. 2006, 4, 212–221. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, M.J.; Tipper, D.J. Sequence of the M28 dsRNA: Preprotoxin is processed to an α/β heterodimeric protein. Virology 1995, 213, 341–351. [Google Scholar] [CrossRef] [PubMed]
- Magliani, W.; Conti, S.; Gerloni, M.; Bertolotti, D.; Polonelli, L. Yeast killer systems. Clin. Microbiol. Rev. 1997, 10, 369–400. [Google Scholar] [PubMed]
- Belda, I.; Ruiz, J.; Alonso, A.; Marquina, D.; Santos, A. The biology of Pichia membranifaciens killer toxins. Toxins 2017, 9, 112. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, M.J.; Neuhausen, F. Killer toxin-secreting double-stranded RNA mycoviruses in the yeasts Hanseniaspora uvarum and Zygosaccharomyces bailii. J. Virol. 1994, 68, 1765–1772. [Google Scholar] [PubMed]
- Weiler, F.; Rehfeldt, K.; Bautz, F.; Schmitt, M.J. The Zygosaccharomyces bailii antifungal virus toxin zygocin: Cloning and expression in a heterologous fungal host. Mol. Microbiol. 2002, 46, 1095–1105. [Google Scholar] [CrossRef] [PubMed]
- Weiler, F.; Schmitt, J.M. Zygocin, a secreted antifungal toxin of the yeast Zygosaccharomyces bailii, and its effect on sensitive fungal cells. FEMS Yeast Res. 2003, 3, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Bussey, H.; Vernet, T.; Sdicu, A.-M. Mutual antagonism among killer yeasts: Competition between Kl and K2 killers and a novel cDNA-based K1-K2 killer strain of Saccharomyces cerevisiae. Can. J. Microbiol. 1988, 34, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Ball, S.G.; Tirtiaux, C.; Wickner, R.B. Genetic control of L-A and L-(BC) dsRNA copy number in killer systems of Saccharomyces cerevisiae. Genetics 1984, 107, 217. [Google Scholar]
- Sommer, S.S.; Wickner, R.B. Yeast L dsRNA consists of at least three distinct RNA’s; evidence that the non-mendelian genes [hok], [nex] and [exl] are on one of these dsRNA’s. Cell 1982, 31, 429–441. [Google Scholar] [CrossRef]
- Dinman, J.D.; Wickner, R.B. Ribosomal frameshifting efficiency and gag/gag-pol ratio are critical for yeast M1 double-stranded RNA virus propagation. J. Virol. 1992, 66, 3669–3676. [Google Scholar] [PubMed]
- Fujimura, T.; Ribas, J.C.; Makhov, A.M.; Wickner, R.B. Pol of gag-pol fusion protein required for encapsidation of viral RNA of yeast L-A virus. Nature 1992, 359, 746–749. [Google Scholar] [CrossRef] [PubMed]
- Icho, T.; Wickner, R.B. The double-stranded RNA genome of yeast virus L-A encodes its own putative RNA polymerase by fusing two open reading frames. J. Biol. Chem. 1989, 264, 6716–6723. [Google Scholar] [PubMed]
- Park, C.M.; Lopinski, J.D.; Masuda, J.; Tzeng, T.H.; Bruen, J.A. A second double-stranded RNA virus from yeast. Virology 1996, 216, 451–454. [Google Scholar] [CrossRef] [PubMed]
- Wickner, R.B.; Bussey, H.; Fujimura, T.; Esteban, R. Viral RNA and the killer phenomenon of Saccharomyces. In The Mycota: Genetics and Biotechnology; Kück, U., Ed.; Springer: Berlin, Germany, 1995; Volume 2, pp. 211–226. [Google Scholar]
- Fujimura, T.; Esteban, R.; Esteban, L.M.; Wickner, R.B. Portable encapsidation signal of the L-A double-stranded-RNA virus of Saccharomyces cerevisiae. Cell 1990, 62, 819–828. [Google Scholar] [CrossRef]
- Thiele, D.J.; Leibowitz, M.J. Structural and functional analysis of separated strands of killer double-stranded RNA of yeast. Nucleic Acids Res. 1982, 10, 6903–6918. [Google Scholar] [CrossRef] [PubMed]
- Thiele, D.J.; Wang, R.W.; Leibowitz, M.J. Separation and sequence of the 3′ termini of M double-stranded RNA from killer yeast. Nucleic Acids Res. 1982, 10, 1661–1678. [Google Scholar] [CrossRef] [PubMed]
- Hannig, E.M.; Leibowitz, M.J. Structure and expression of the M2 genomic segment of a type 2 killer virus of yeast. Nucleic Acids Res. 1985, 13, 4379–4400. [Google Scholar] [CrossRef] [PubMed]
- Wickner, R.B.; Fujimura, T.; Esteban, R. Viruses and prions of Saccharomyces cerevisiae. Adv. Virus Res. 2013, 86, 1–36. [Google Scholar] [PubMed]
- Berrye, A.; Bevane, A. A new species of double-stranded RNA from yeast. Nature 1972, 239, 279–280. [Google Scholar] [CrossRef]
- Cansado, J.; Barros Velázquez, J.; Sieiro, C.; Gacto, M.; Villa, T.G. Presence of non-suppressive, M2-related dsrnas molecules in Saccharomyces cerevisiae strains isolated from spontaneous fermentations. FEMS Microbiol. Lett. 1999, 181, 211–215. [Google Scholar] [CrossRef] [PubMed]
- Vodkin, M.H.; Fink, G.R. A nucleic acid associated with a killer strain of yeast. Proc. Natl. Acad. Sci. USA 1973, 70, 1069–1072. [Google Scholar] [CrossRef] [PubMed]
- Sangorrín, M.P.; Lopes, C.A.; Jofré, V.; Querol, A.; Caballero, A.C. Spoilage yeasts from patagonian cellars: Characterization and potential biocontrol based on killer interactions. World J. Microbl. Biotechnol. 2007, 24, 945–953. [Google Scholar] [CrossRef]
- Wickner, R.B. Yeast RNA virology: The killer systems. In The Molecular and Cellular Biology of the Yeast Saccharomyces: Genome Dynamics, Protein Synthesis, and Energetics; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 1991; pp. 263–296. [Google Scholar]
- Fujimura, T.; Wickner, R.B. Reconstitution of template-dependent in vitro transcriptase activity of a yeast double-stranded RNA virus. J. Biol. Chem. 1989, 264, 10872–10877. [Google Scholar] [PubMed]
- Hannig, E.M.; Leibowitz, M.J. Yeast Type 2 Killer Virus M2 RNA 3′-Terminus (Positive Strand); GenBank: X02609.1. Available online: www.ncbi.nlm.nih.gov/nuccore/X02609.1 (accessed on 7 August 2017).
- Meskaukas, A. Nucleotide sequence of cDNA to yeast M2-1 dsRNA segment. Nucleic Acids Res. 1990, 18, 6720. [Google Scholar] [CrossRef]
- Fried, H.M.; Fink, G.R. Electron microscopic heteroduplex analysis of killer double-stranded RNA species from yeast. Proc. Natl. Acad. Sci. USA 1978, 75, 4224–4228. [Google Scholar] [CrossRef] [PubMed]
- Gmyl, A.P.; Korshenko, S.A.; Belousov, E.V.; Khitrina, E.V.; Agol, V.I. Nonreplicative homologous RNA recombination: Promiscuous joining of RNA pieces? RNA 2003, 9, 1221–1231. [Google Scholar] [CrossRef] [PubMed]
- Sztuba-Solińska, J.; Urbanowicz, A.; Figlerowicz, M.; Bujarski, J.J. RNA-RNA recombination in plant virus replication and evolution. Annu. Rev. Phytopathol. 2011, 49, 415–443. [Google Scholar] [CrossRef] [PubMed]
- Holland, J.; Spindler, K.; Horodyski, F.; Grabau, E.; Nichol, S.; VandePol, S. Rapid evolution of RNA genomes. Science 1982, 215, 1577–1585. [Google Scholar] [CrossRef] [PubMed]
- Steinhauer, D.A.; Holland, J.J. Direct method for quantitation of extreme polymerase error frequencies at selected single base sites in viral RNA. J. Virol. 1986, 57, 219–228. [Google Scholar] [PubMed]
- Lai, M.M. RNA recombination in animal and plant viruses. Microbiol. Rev. 1992, 56, 61–79. [Google Scholar] [PubMed]
- Rodriguez-Cousino, N.; Esteban, R. Relationships and evolution of double-stranded RNA totiviruses of yeasts inferred from analysis of L-A-2 and L-BC variants in wine yeast strain populations. Appl. Environ. Microbiol. 2017, 83, e02991-16. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Cousiño, N.; Gómez, P.; Esteban, R. L-A-lus, a new variant of the L-A totivirus found in wine yeasts with Klus killer toxin-encoding mlus double-stranded RNA: Possible role of killer toxin-encoding satellite RNAs in the evolution of their helper viruses. Appl. Environ. Microbiol. 2013, 79, 4661–4674. [Google Scholar] [CrossRef] [PubMed]
- Guthrie, C.; Fink, G.R. Guide to yeast genetics and molecular biology. Methods Enzymol. 1991, 194, 3–57. [Google Scholar]
- Kaiser, C.; Michaelis, S.; Mitchell, A. Methods in Yeast Genetics; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 1994. [Google Scholar]
- Ramírez, M.; Vinagre, A.; Ambrona, J.; Molina, F.; Maqueda, M.; Rebollo, J.E. Genetic instability of heterozygous hybrid populations of natural wine yeasts. Appl. Environ. Microbiol. 2004, 70, 4686–4691. [Google Scholar] [CrossRef] [PubMed]
- Maqueda, M.; Zamora, E.; Rodríguez-Cousiño, N.; Ramírez, M. Wine yeast molecular typing using a simplified method for simultaneously extracting mtDNA, nuclear DNA and virus dsRNA. Food Microbiol. 2010, 27, 205–209. [Google Scholar] [CrossRef] [PubMed]
- Toh-e, A.; Guerry, P.; Wickner, R.B. Chromosomal superkiller mutants of Saccharomyces cerevisiae. J. Bacteriol. 1978, 136, 1002–1007. [Google Scholar]
- Schmieder, R.; Edwards, R. Quality control and preprocessing of metagenomic datasets. Bioinformatics 2011, 27, 863–864. [Google Scholar] [CrossRef] [PubMed]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. Spades: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed]
- Futami, R.; Muñoz-Pomer, L.; Dominguez-Escriba, L.; Covelli, L.; Bernet, G.P.; Sempere, J.M. GPRO: The professional tool for annotation, management and functional analysis of omic databases. Biotechvana Bioinform. 2011. Available online: http://biotechvana.uv.es/bioinformatics/article_files/35/pdf/GPRO.pdf (accessed on 1 August 2017).
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Thorvaldsdottir, H.; Robinson, J.T.; Mesirov, J.P. Integrative Genomics Viewer (IGV): High-performance genomics data visualization and exploration. Brief. Bioinform. 2013, 14, 178–192. [Google Scholar] [CrossRef] [PubMed]
- Sambrook, J.; Fritsch, E.F.; Maniatis, T. Molecular Cloning: A Laboratory Manual; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 1989. [Google Scholar]
- Thompson, J.D.; Gibson, T.J.; Higgins, D.G. Unit 2.3: Multiple sequence alignment using ClustalW and ClustalX. In Current Protocols in Bioinformatics; John Wiley & Sons, Inc.: New York, NY, USA, 2003; pp. 2.3.1–2.3.22. [Google Scholar]
- Zuker, M.; Mathews, D.H.; Turner, D.H.; Barciszewski, J.; Clark, B.F.C. Algorithms and thermodynamics for RNA secondary structure prediction: A practical guide. In RNA Biochemistry and Biotechnology; Kluwer Academic Publishers: Dordrecht, The Netherlands, 1999. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Virus | Previous Estimated Size/Sequenced Length (bp)/Yeast Strain/ΔG | Newly Analysed Yeast Strain | Killer Phenotype/dsRNA Isotype | Size Estimated/Length (bp) Sequenced/ΔG | 5′-Extra Sequence (bp)/% Identity with: Size [Position] | 3′-Extra Sequence (bp)/% Identity with: Size [Position] | Stem-Loop Involving 5′-Extra Sequence [Position] (ΔG) | Stem-Loop Involving 3′-Extra Sequence [position] (ΔG) |
|---|---|---|---|---|---|---|---|---|
| M1 | 1830/1801/S.c. TF325/−1807 | S.c. EX231 | K1*/M1-1 | 1300/1933/−1937 | 66/94% S.c. LBC-2 virus, 66 nt [A(−)66 to T(−)1] | 66 /no identity found | Not found | Not found |
| T.d. EX1257 | Kbarr-2/M1-2 | 1700/1933/−1916 | 66/no identity found | 65/94% S.c. 26S rRNA, 77 nt [C1783 to G(+)57] | Not found | Not found | ||
| M2 | 1700/1163 + 209/S.c. 1384/−1861 | S.c. EX88 | K2/M2-3 | 1650/1625/−2020 | 33/no identity found | None | [A(−)23 to T126] (−69.5) | Not found |
| S.c. EX1125 | K2/M2-4 | 1750/1723/−2297 | 87/no identity found | 44 /no identity found | [T(−)42 to A145] (−205) | [C1603 to G(+)39] (−137) | ||
| Mlus | 2300/2033/S.c. EX229/−1970 | S.c. EX229 | Klus/Mlus-4 | 2300/2314/−2242 | 50/no identity found | 230/98% S.c. 16S mit rRNA, 242 nt [C2022 to G(+)203] | [C(−)50 to G107] (−30.1) | Not found |
| S.c. EX436 | Klus/Mlus-1 | 2000/2346/−2409 | 51/93% Vitis vinifera, 47 nt [A(−)30 to T17], and 85% Saccharomycopsis fibuligera, 45 nt [A(−)30 to A15] | 198/100% S.c. 18S rRNA, 198 nt [A(+)1 to G(+)198] | [C(−)51 to G107] (−46.0) | Not found | ||
| S.c. EX1160 | Klus*/Mlus-A | 2100/2268/−2723 | 218/no identity found | 17/no identity found | [T(−)125 to A97] (−246), or [A(−)67 to T129] (−62.8) | Not found | ||
| S.c. EX1160 | Klus*/Mlus-B | 1700/1937/−2066 | 77/no identity found | None | Not found | Not found | ||
| S.c. EX1160 | Klus*/Mlus-C | 1350/2055/−1962 | 22/no identity found | None | Not found | Not found | ||
| Mbarr-1 | 1700/1705/T.d. EX1180/−1727 | T.d. EX1257 | Kbarr-2*/Mbarr-2B | 1300/1835/−1878 | 64/88% Cucumis melo, 41 nt [A(−)18 to G23] | 66/97% T.d. (95% S.c.) 26S rRNA, 76 nt [G1759 to C(+)65] | [T(−)2 to A53] (−40.6) | Not found |
| Strain | Genotype [Relevant Phenotype] | Origin |
|---|---|---|
| Sc EX88 * | MAT a/α HO/HO cyhS/cyhS M2-3 [K2+] | M. Ramírez a (from wine) |
| Sc EX85 * | MAT a/α HO/HO cyhS/cyhS LA M2-3 [K2+] | M. Ramírez a (from wine) |
| Sc EX85R * | MAT a/α HO/HO CYHR/cyhS M20 [cyhR K20] | M. Ramírez a (from EX85) |
| Sc EX229 * | MAT a/α HO/HO cyhS/cyhS LA LBC Mlus-4 [Klus+] | M. Ramírez a (from wine) |
| Sc EX229-R1 * | MAT a/α HO/HO CYHR/cyhS [cyhR Klus0] | M. Ramírez a (from EX229) |
| Sc EX33 * | MAT a/α HO/HO [LA0 K10 K20 K280 Klus0] | M. Ramírez a (from wine) |
| Sc EX73 * | MAT a/α HO/HO LA M2-3 [K2+] | M. Ramírez a (from wine) |
| Sc EX198 * | MAT a/α HO/HO LA LBC Mlus-3 [Klus+] | M. Ramírez a (from wine) |
| Sc EX231 | MAT a/α HO/HO LA LBC M1-1 [K1+] | This study (from wine) |
| Sc EX436 | MAT a/α HO/HO LA Mlus-1 [Klus+] | This study (from wine) |
| Sc EX1125 | MAT a/α HO/HO LA LBC M2-4 [K2+] | This study (from wine) |
| Sc EX1160 | MAT a/α HO/HO LA LBC Mlus-A Mlus-B Mlus-C[Klus+] | This study (from wine) |
| Sc F166 * | MAT α leu1 kar1 LA-HNB M1 [K1+] | J.C. Ribas b (from R. Wickner) |
| Sc F182 * | MAT α his2 ade1 leu2-2 ura3-52 ski2-2 LA M28 [K28+] | J. C. Ribas b (from M. Schmitt) |
| Td EX1180 * | wt LAbarr-1 Mbarr-1 [Kbarr-1+] | M. Ramírez a (from wine) |
| Td EX1180-11C4 * | cyhR LAbarr-1 Mbarr-1 [cyhR Kbarr-1+] | M. Ramírez a (from EX1180) |
| Td EX1180-2K− * | cyhR LAbarr-1 Mbarr-10 [cyhR Kbarr0] | M. Ramírez a (from EX1180) |
| Td EX1257 | wt LAbarr-2 M1-2 Mbarr-2B [Kbarr-2+] | This study (from wine) |
| Td EX1257-CYH5 | cyhR LAbarr-2 M1-2 Mbarr-2B [cyhR Kbarr-2+] | M. Ramírez a (from EX1257) |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ramírez, M.; Velázquez, R.; López-Piñeiro, A.; Naranjo, B.; Roig, F.; Llorens, C. New Insights into the Genome Organization of Yeast Killer Viruses Based on “Atypical” Killer Strains Characterized by High-Throughput Sequencing. Toxins 2017, 9, 292. https://doi.org/10.3390/toxins9090292
Ramírez M, Velázquez R, López-Piñeiro A, Naranjo B, Roig F, Llorens C. New Insights into the Genome Organization of Yeast Killer Viruses Based on “Atypical” Killer Strains Characterized by High-Throughput Sequencing. Toxins. 2017; 9(9):292. https://doi.org/10.3390/toxins9090292
Chicago/Turabian StyleRamírez, Manuel, Rocío Velázquez, Antonio López-Piñeiro, Belén Naranjo, Francisco Roig, and Carlos Llorens. 2017. "New Insights into the Genome Organization of Yeast Killer Viruses Based on “Atypical” Killer Strains Characterized by High-Throughput Sequencing" Toxins 9, no. 9: 292. https://doi.org/10.3390/toxins9090292
APA StyleRamírez, M., Velázquez, R., López-Piñeiro, A., Naranjo, B., Roig, F., & Llorens, C. (2017). New Insights into the Genome Organization of Yeast Killer Viruses Based on “Atypical” Killer Strains Characterized by High-Throughput Sequencing. Toxins, 9(9), 292. https://doi.org/10.3390/toxins9090292