
Different Metabolic Pathways Are Involved in Response of Saccharomyces cerevisiae to L-A and M Viruses


 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
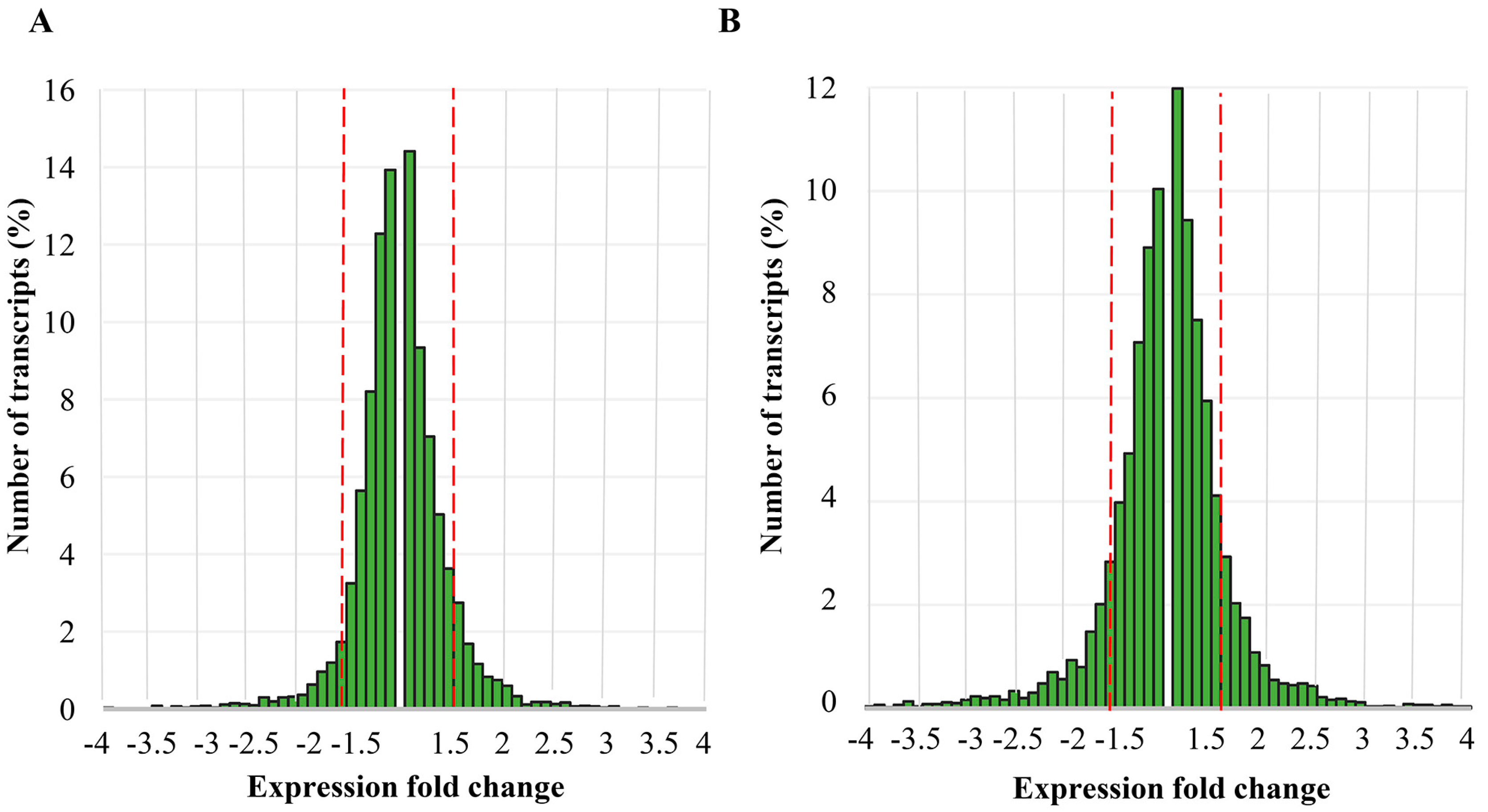
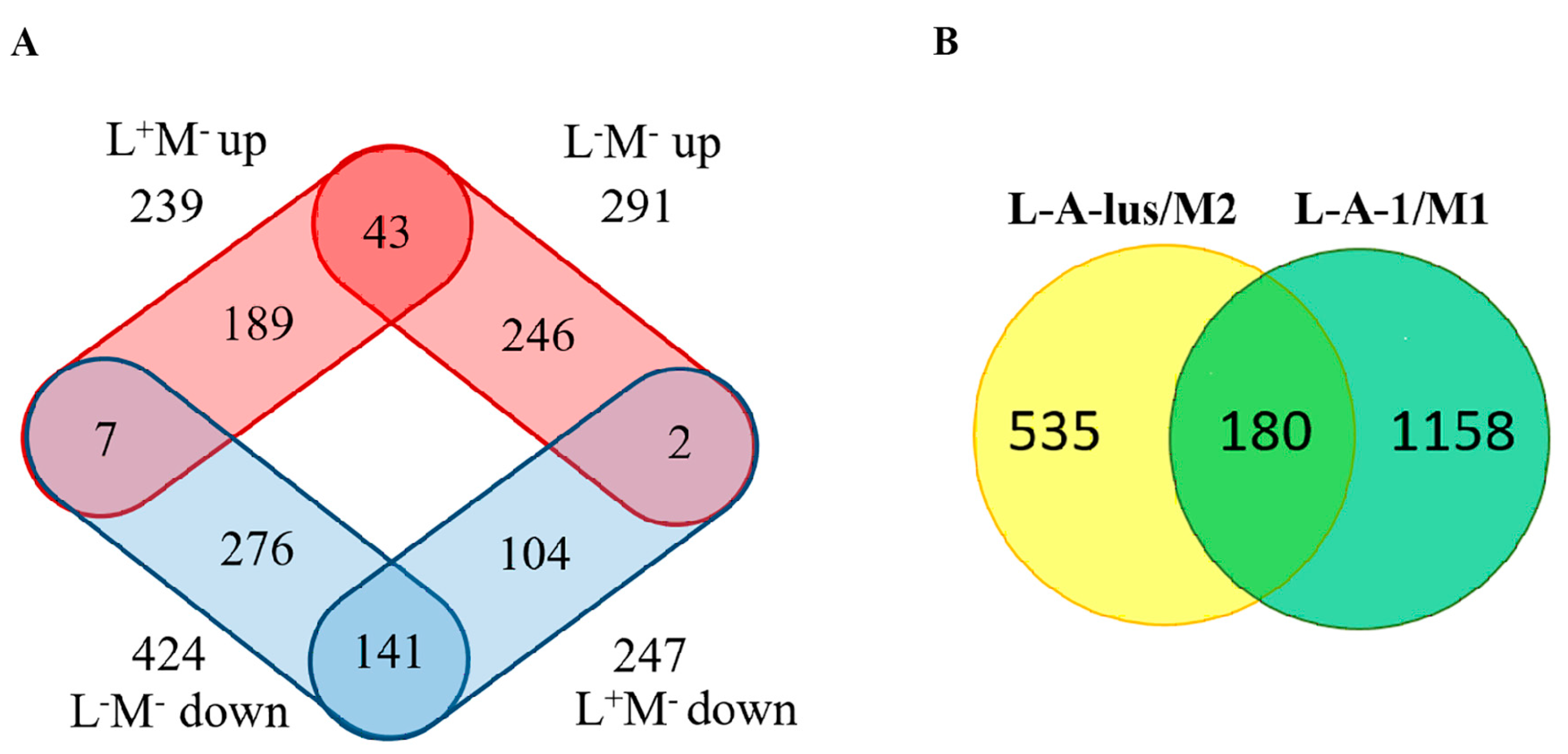
2.1. Differential Gene Expression Induced by Elimination of Viral DsRNA(s)
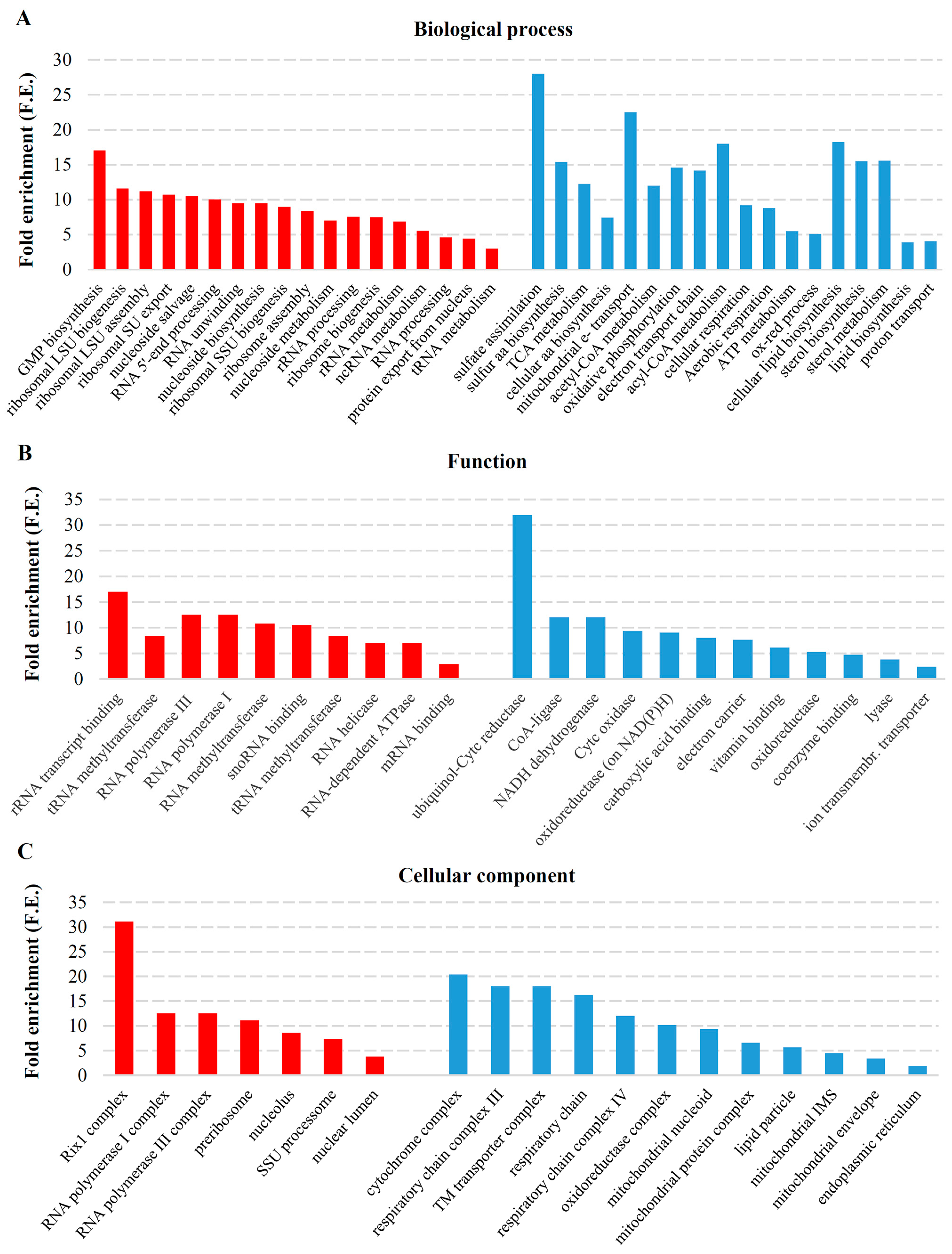
2.2. Transcriptional Response to M-2 DsRNA Elimination
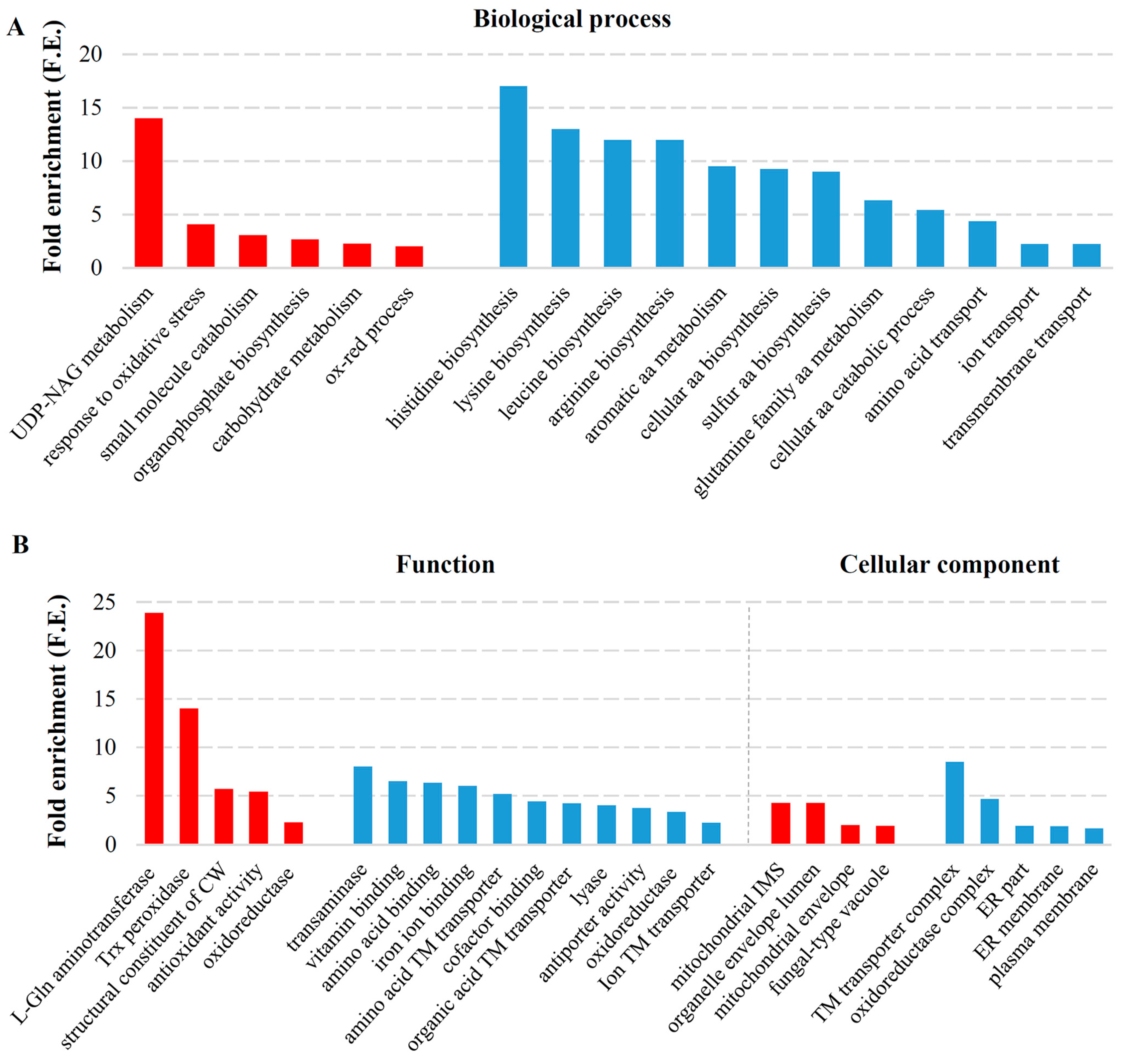
2.3. Transcriptional Response to Elimination of Both M-2 and L-A-lus
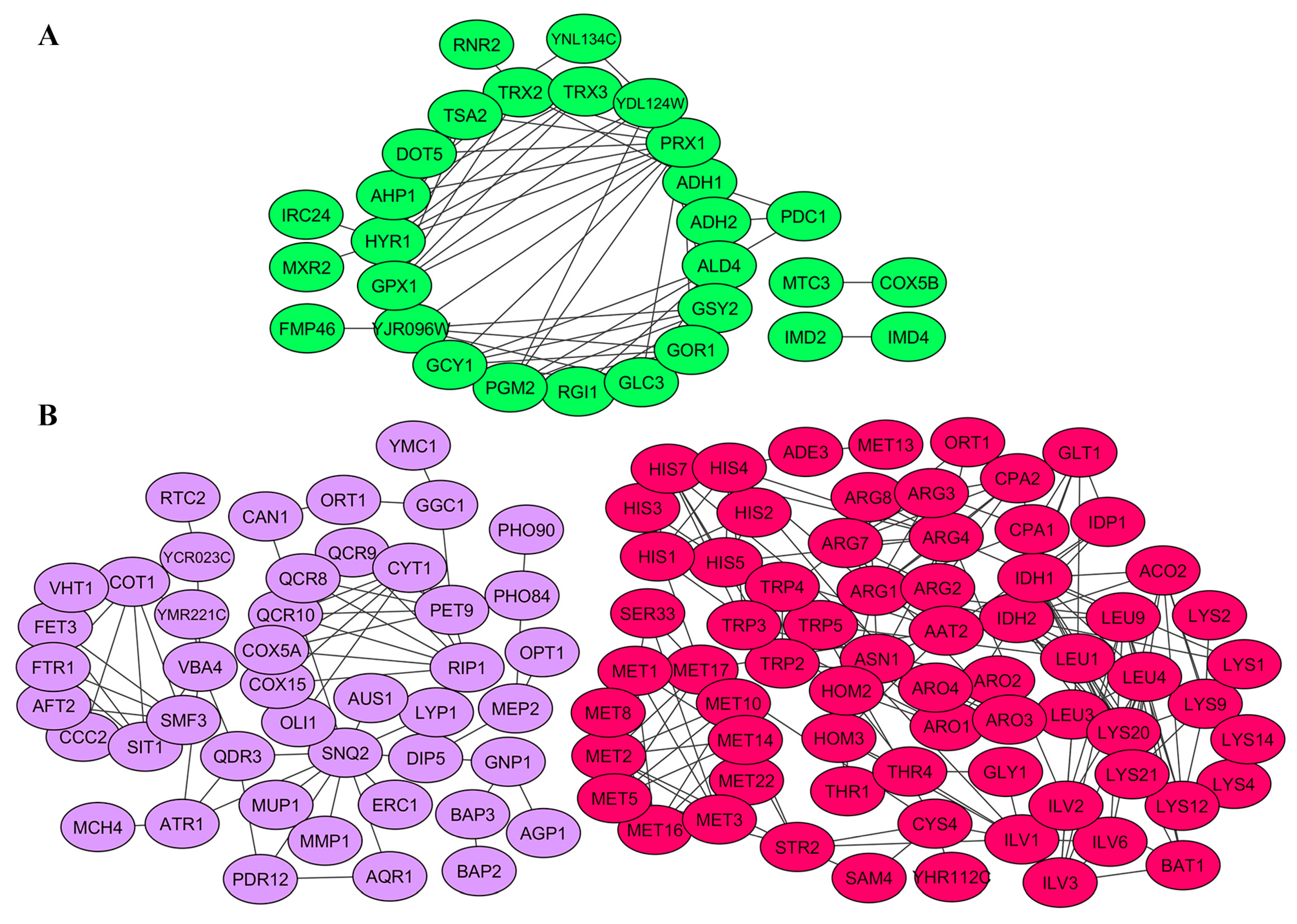
2.4. Gene Products Involved in M-2 and L-A-lus Virus Biology Are Physically and Functionally Highly Interconnected In Vivo
2.5. Link between Host Gene Expression Altered by Viral DsRNA and Cell Sensitivity to K2 Toxin
3. Discussion
4. Materials and Methods
4.1. Yeast Strains and Culture Media
4.2. Curing Yeast Strain from M-2 DsRNA
4.3. Curing Yeast Strain from L-A-lus DsRNA
4.4. Detection of Killing Phenotype
4.5. Total RNA and DsRNA Extraction
4.6. Detection of L-A-lus and M-2 DsRNAs by 2-step RT-PCR
4.7. Preparation of Total RNA for Next-Generation Sequencing
4.8. RNA Sequencing and Data Analysis
4.9. QRT-PCR Analysis
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Ghabrial, S.A. Origin, adaptation and evolutionary pathways of fungal viruses. Virus Genes 1998, 16, 119–131. [Google Scholar] [CrossRef] [PubMed]
- Wickner, R.B. Double-stranded and single-stranded RNA viruses of Saccharomyces cerevisiae. Annu. Rev. Microbiol. 1992, 46, 347–375. [Google Scholar] [CrossRef]
- Wickner, R.B. Prions and RNA viruses of Saccharomyces cerevisiae. Annu. Rev. Genet. 1996, 30, 109–139. [Google Scholar] [CrossRef]
- Liu, H.; Fu, Y.; Jiang, D.; Li, G.; Xie, J.; Cheng, J.; Peng, Y.; Ghabrial, S.A.; Yi, X. Widespread horizontal gene transfer from double-stranded RNA viruses to eukaryotic nuclear genomes. J. Virol. 2010, 84, 11876–11887. [Google Scholar] [CrossRef]
- Ahn, I.-P.; Lee, Y.-H. A viral double-stranded RNA up regulates the fungal virulence of Nectria radicicola. Mol. Plant-Microbe Interact. 2001, 14, 496–507. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Jiang, D. New insights into mycoviruses and exploration for the biological control of crop fungal diseases. Annu. Rev. Phytopathol. 2014, 52, 45–68. [Google Scholar] [CrossRef] [PubMed]
- Wickner, R.B. Double-stranded RNA replication in yeast: The killer system. Ann. Rev. Biochem. 1986, 55, 373–395. [Google Scholar] [CrossRef] [PubMed]
- Varga, J.; Kevei, F.; Vagvolgyi, C.; Vriesema, A.; Croft, J.H. Double-stranded RNA mycoviruses in section Nigri of the Aspergillus genus. Can. J. Microbiol. 1994, 40, 325–329. [Google Scholar] [CrossRef] [PubMed]
- Magliani, W.; Conti, S.; Gerloni, M.; Bertolotti, D.; Polonelli, L. Yeast killer systems. Clin. Microbiol. Rev. 1997, 10, 369–400. [Google Scholar] [PubMed]
- Schmitt, M.J.; Tipper, D.J. K28, a unique double-stranded RNA killer virus of Saccharomyces cerevisiae. Mol. Cell. Biol. 1990, 10, 4807–4815. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Cousino, N.; Gomez, P.; Esteban, R. L-A-lus, a new variant of the L-A totivirus found in wine yeasts with Klus killer toxin-encoding Mlus double-stranded RNA: Possible role of killer toxin-encoding satellite RNAs in the evolution of their helper viruses. Appl. Environ. Microbiol. 2013, 79, 4661–4674. [Google Scholar] [CrossRef] [PubMed]
- Bevan, E.A.; Herring, A.J.; Mitchell, D.J. Preliminary characterization of two species of dsRNA in yeast and their relationship to the “killer” character. Nature 1973, 245, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Starmer, W.T.; Ganter, P.F.; Aberdeen, V.; Lachance, M.A.; Phaff, H.J. The ecological role of killer yeasts in natural communities of yeasts. Can. J. Microbiol. 1987, 33, 783–796. [Google Scholar] [CrossRef] [PubMed]
- Rowley, P.A. The frenemies within: Viruses, retrotransposons, and plasmids that naturally infect Saccharomyces yeasts. Yeast 2017. [Google Scholar] [CrossRef] [PubMed]
- Palpacelli, V.; Ciani, M.; Rosini, G. Activity of different “killer” yeasts on strains of yeast species undesirable in the food industry. FEMS Microbiol. Lett. 1991, 68, 75–78. [Google Scholar] [CrossRef] [PubMed]
- Salek, A.; Schnettler, R.; Zimmermann, U. Stably inherited killer activity in industrial yeast strains obtained by electrotransformation. FEMS Microbiol. Lett. 1992, 75, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Javadekar, V.S.; SivaRaman, H.; Gokhale, D.V. Industrial yeast strain improvement: Construction of a highly flocculent yeast with a killer character by protoplast fusion. J. Ind. Microbiol. Biotechnol. 1995, 15, 94–102. [Google Scholar] [CrossRef]
- Buzzini, P.; Martini, A. Discrimination between Candida albicans and other pathogenic species of the genus Candida by their differential sensitivities to toxins of a panel of killer yeasts. J. Clin. Microbiol. 2001, 39, 3362–3364. [Google Scholar] [CrossRef] [PubMed]
- Vadkertiova, R.; Slavikova, E. Killer activity of yeasts isolated from natural environments against some medically important Candida species. Pol. J. Microbiol. 2007, 56, 39–43. [Google Scholar] [PubMed]
- Banjara, N.; Nickerson, K.W.; Suhr, M.J.; Hallen-Adams, H.E. Killer toxin from several food-derived Debaryomyces hansenii strains effective against pathogenic Candida yeasts. Int. J. Food Microbiol. 2016, 222, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Pagé, N.; Gérard-Vincent, M.; Ménard, P.; Beaulieu, M.; Azuma, M.; Dijkgraaf, G.J.P.; Li, H.; Marcoux, J.; Nguyen, T.; Dowse, T.; Sdicu, A.M.; Bussey, H. A Saccharomyces cerevisiae genome-wide mutant screen for altered sensitivity to K1 killer toxin. Genetics 2003, 163, 875–894. [Google Scholar] [CrossRef] [PubMed]
- Carroll, S.Y.; Stirling, P.C.; Stimpson, H.E.M.; Gießelmann, E.; Schmitt, M.J.; Drubin, D.G. A Yeast killer toxin screen provides insights into A/B toxin entry, trafficking, and killing mechanisms. Dev. Cell 2009, 17, 552–560. [Google Scholar] [CrossRef] [PubMed]
- Serviene, E.; Luksa, J.; Orentaite, I.; Lafontaine, D.L.J.; Urbonavicius, J. Screening the budding yeast genome reveals unique factors affecting K2 toxin susceptibility. PLoS ONE 2012, 7, e50779. [Google Scholar] [CrossRef] [PubMed]
- Pieczynska, M.D.; de Visser, J.A.G.M.; Korona, R. Incidence of symbiotic dsRNA “killer” viruses in wild and domesticated yeast. FEMS Yeast Res. 2013, 13, 856–859. [Google Scholar] [CrossRef] [PubMed]
- Pieczynska, M.D.; Korona, R.; De Visser, J.A.G.M. Experimental tests of host-virus coevolution in natural killer yeast strains. J. Evol. Biol. 2017, 30, 773–781. [Google Scholar] [CrossRef] [PubMed]
- Veses-Garcia, M.; Liu, X.; Rigden, D.J.; Kenny, J.G.; McCarthy, A.J.; Allison, H.E. Transcriptomic analysis of Shiga-toxigenic bacteriophage carriage reveals a profound regulatory effect on acid resistance in Escherichia coli. Appl. Environ. Microbiol. 2015, 81, 8118–8125. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Ding, H.; Zeng, Q. Transcriptomic response during phage infection of a marine cyanobacterium under phosphorus-limited conditions. Environ. Microbiol. 2016, 18, 450–460. [Google Scholar] [CrossRef] [PubMed]
- Hillung, J.; Garcia-Garcia, F.; Dopazo, J.; Cuevas, J.M.; Elena, S.F. The transcriptomics of an experimentally evolved plant-virus interaction. Sci. Rep. 2016, 6, 24901. [Google Scholar] [CrossRef] [PubMed]
- Castorena, K.M.; Stapleford, K.A.; Miller, D.J. Complementary transcriptomic, lipidomic, and targeted functional genetic analyses in cultured Drosophila cells highlight the role of glycerophospholipid metabolism in Flock House virus RNA replication. BMC Genom. 2010, 11, 183. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Rubio, L.; Evensen, O.; Krasnov, A.; Jorgensen, S.M.; Wadsworth, S.; Ruohonen, K.; Vecino, J.L.G.; Tocher, D.R. Effects of functional feeds on the lipid composition, transcriptomic responses and pathology in heart of Atlantic salmon (Salmo salar L.) before and after experimental challenge with Piscine Myocarditis Virus (PMCV). BMC Genom. 2014, 15, 462. [Google Scholar] [CrossRef] [PubMed]
- Yen, J.Y.; Garamszegi, S.; Geisbert, J.B.; Rubins, K.H.; Geisbert, T.W.; Honko, A.; Xia, Y.; Connor, J.H.; Hensley, L.E. Therapeutics of Ebola hemorrhagic fever: Whole-genome transcriptional analysis of successful disease mitigation. J. Infect. Dis. 2011, 204 (Suppl. 3), S1043–S1052. [Google Scholar] [CrossRef] [PubMed]
- Tsuge, M.; Oka, T.; Yamashita, N.; Saito, Y.; Fujii, Y.; Nagaoka, Y.; Yashiro, M.; Tsukahara, H.; Morishima, T. Gene expression analysis in children with complex seizures due to influenza A(H1N1)pdm09 or rotavirus gastroenteritis. J. Neurovirol. 2014, 20, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Allen, T.D.; Nuss, D.L. Specific and common alterations in host gene transcript accumulation following infection of the chestnut blight fungus by mild and severe hypoviruses. J. Virol. 2004, 78, 4145–4155. [Google Scholar] [CrossRef] [PubMed]
- Cho, W.K.; Yu, J.; Lee, K.-M.; Son, M.; Min, K.; Lee, Y.-W.; Kim, K.-H. Genome-wide expression profiling shows transcriptional reprogramming in Fusarium graminearum by Fusarium graminearum virus 1-DK21 infection. BMC Genom. 2012, 13, 173. [Google Scholar] [CrossRef] [PubMed]
- Eusebio-Cope, A.; Sun, L.; Tanaka, T.; Chiba, S.; Kasahara, S.; Suzuki, N. The chestnut blight fungus for studies on virus/host and virus/virus interactions: From a natural to a model host. Virology 2015, 477, 164–175. [Google Scholar] [CrossRef] [PubMed]
- Mcbride, R.C.; Boucher, N.; Park, D.S.; Turner, P.E.; Townsend, J.P. Yeast response to LA virus indicates coadapted global gene expression during mycoviral infection. FEMS Yeast Res. 2013, 13, 162–179. [Google Scholar] [CrossRef] [PubMed]
- Milewski, S.; Gabriel, I.; Olchowy, J. Enzymes of UDP-GlcNAc biosynthesis in yeast. Yeast 2006, 23, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Wickner, R.B.; Leibowitz, M.J. Chromosomal genes essential for replication of a double-stranded RNA plasmid of Saccharomyces cerevisiae: The killer character of yeast. J. Mol. Biol. 1976, 105, 427–443. [Google Scholar] [CrossRef]
- Toh-E, A.; Wickner, R.B. “Superkiller” mutations suppress chromosomal mutations affecting double-stranded RNA killer plasmid replication in saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 1980, 77, 527–530. [Google Scholar] [CrossRef] [PubMed]
- Icho, T.; Wickner, R.B. The MAK11 protein is essential for cell growth and replication of M double-stranded RNA and is apparently a membrane-associated protein. J. Biol. Chem. 1988, 263, 1467–1475. [Google Scholar] [PubMed]
- Coelho, P.S. R.; Bryan, A.C.; Kumar, A.; Shadel, G.S.; Snyder, M. A novel mitochondrial protein, Tar1p, is encoded on the antisense strand of the nuclear 25S rDNA. Genes Dev. 2002, 16, 2755–2760. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq.: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Ellahi, A.; Thurtle, D.M.; Rine, J. The chromatin and transcriptional landscape of native Saccharomyces cerevisiae telomeres and subtelomeric domains. Genetics 2015, 200, 505–521. [Google Scholar] [CrossRef] [PubMed]
- Bendjilali, N.; MacLeon, S.; Kalra, G.; Willis, S.D.; Hossian, A.K. M.N.; Avery, E.; Wojtowicz, O.; Hickman, M.J. Time-course analysis of gene expression during the Saccharomyces cerevisiae hypoxic response. G3 Genes Genomes Genet. 2017, 7, 221–231. [Google Scholar] [CrossRef] [PubMed]
- Ball, S.G.; Tirtiaux, C.; Wickner, R.B. Genetic control of L-A and L-(BC) dsRNA copy number in killer systems of Saccharomyces cerevisiae. Genetics 1984, 107, 199–217. [Google Scholar] [PubMed]
- Ohtake, Y.; Wickner, R.B. Yeast virus propagation depends critically on free 60S ribosomal subunit concentration. Mol. Cell. Biol. 1995, 15, 2772–2781. [Google Scholar] [CrossRef] [PubMed]
- Naumova, G.I.; Naumova, T.I. Comparative genetics of yeasts. XIII, Comparative study of Saccharomycetes-killers from different collections. Genetika 1973, 9, 140–145. (In Russian) [Google Scholar] [PubMed]
- Citavicius, D.; Inge-Vectomov, S.G. Saccharomyces cerevisiae multiple mutants: I. construction and general characterization. Genetika (Moscow) 1972, 1, 95–102. (In Russian) [Google Scholar]
- Aitmanaitė, L.; Konovalovas, A.; Servienė, E.; Serva, S. Healing yeast from L-A virus. unpublished work.
- Gulbiniene, G.; Kondratiene, L.; Jokantaite, T.; Serviene, E.; Melvydas, V.; Petkuniene, G. Occurrence of killer yeast strains in fruit and berry wine yeast populations. Food Technol. Biotechnol. 2004, 42, 159–163. [Google Scholar]
- Drinnenberg, I.A.; Fink, G.R.; Bartel, D.P. Compatibility with killer explains the rise of RNAi-deficient fungi. Science 2011, 333, 1592. [Google Scholar] [CrossRef] [PubMed]
- Boyle, E.I.; Weng, S.; Gollub, J.; Jin, H.; Botstein, D.; Cherry, J.M.; Sherlock, G. GO: TermFinder open source software for accessing Gene Ontology information and finding significantly enriched Gene Ontology terms associated with a list of genes. Bioinformatics 2004, 20, 3710–3715. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Franceschini, A.; Wyder, S.; Forslund, K.; Heller, D.; Huerta-Cepas, J.; Simonovic, M.; Roth, A.; Santos, A.; Tsafou, K.P.; et al. STRING v10: Protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015, 43, D447–D452. [Google Scholar] [CrossRef] [PubMed]
- Killcoyne, S.; Carter, G.W.; Smith, J.; Boyle, J. Cytoscape: A community-based framework for network modeling. Methods Mol. Biol. 2009, 563, 219–239. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef] [PubMed]
- Wong, M.L.; Medrano, J.F. Real-time PCR for mRNA quantitation. Biotechniques 2005, 39, 75–85. [Google Scholar] [CrossRef] [PubMed]






© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lukša, J.; Ravoitytė, B.; Konovalovas, A.; Aitmanaitė, L.; Butenko, A.; Yurchenko, V.; Serva, S.; Servienė, E. Different Metabolic Pathways Are Involved in Response of Saccharomyces cerevisiae to L-A and M Viruses. Toxins 2017, 9, 233. https://doi.org/10.3390/toxins9080233
Lukša J, Ravoitytė B, Konovalovas A, Aitmanaitė L, Butenko A, Yurchenko V, Serva S, Servienė E. Different Metabolic Pathways Are Involved in Response of Saccharomyces cerevisiae to L-A and M Viruses. Toxins. 2017; 9(8):233. https://doi.org/10.3390/toxins9080233
Chicago/Turabian StyleLukša, Juliana, Bazilė Ravoitytė, Aleksandras Konovalovas, Lina Aitmanaitė, Anzhelika Butenko, Vyacheslav Yurchenko, Saulius Serva, and Elena Servienė. 2017. "Different Metabolic Pathways Are Involved in Response of Saccharomyces cerevisiae to L-A and M Viruses" Toxins 9, no. 8: 233. https://doi.org/10.3390/toxins9080233
APA StyleLukša, J., Ravoitytė, B., Konovalovas, A., Aitmanaitė, L., Butenko, A., Yurchenko, V., Serva, S., & Servienė, E. (2017). Different Metabolic Pathways Are Involved in Response of Saccharomyces cerevisiae to L-A and M Viruses. Toxins, 9(8), 233. https://doi.org/10.3390/toxins9080233