Cytoskeleton as an Emerging Target of Anthrax Toxins

{kind=link}
{kind=link}
Abstract
:1. Introduction
2. General Overview on Bacillus anthracis Toxins and the Cytoskeleton
2.1. Bacillus anthracis Toxins
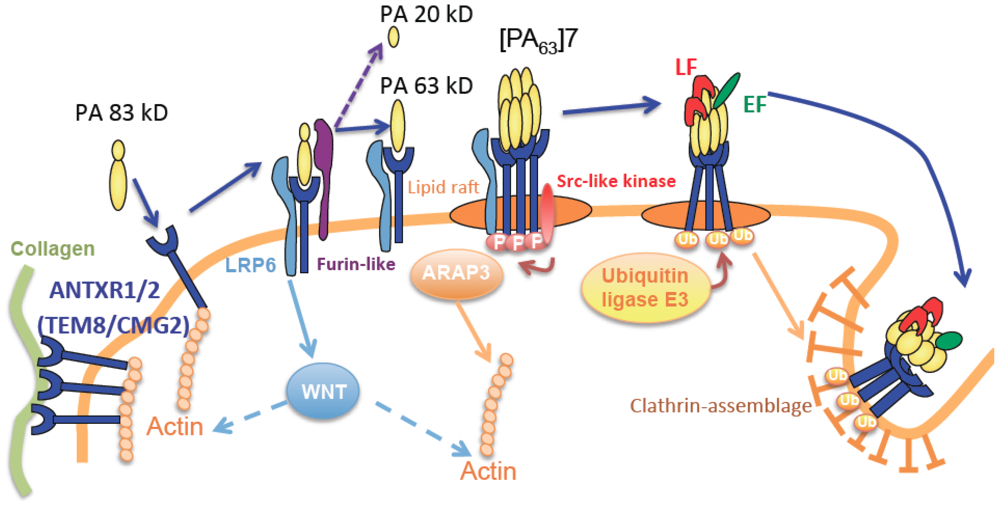
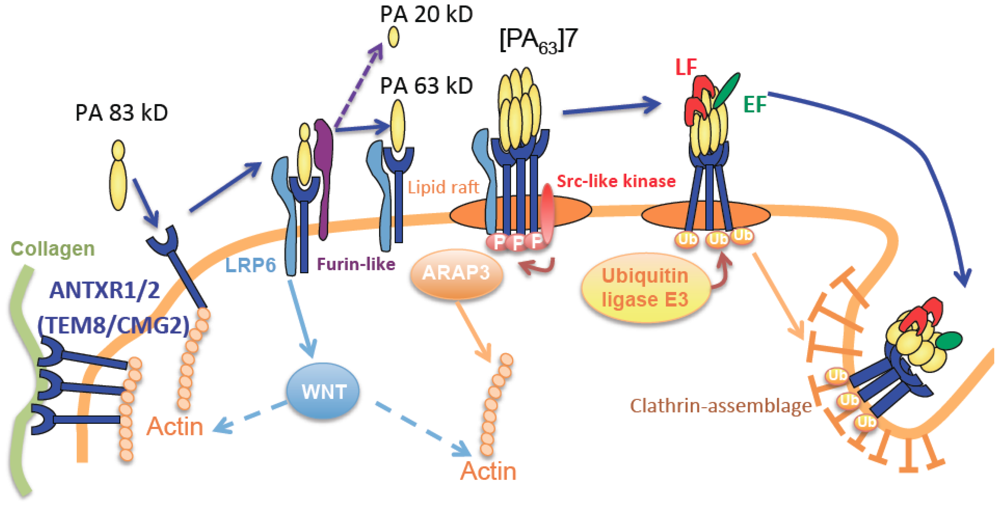
2.2. The Cytoskeleton
3. Association between Anthrax Receptor, Extra-Cellular Matrix and the Cytoskeleton
4. Anthrax Toxins and Disruption of the Cytoskeleton Network
4.1. LT Effects
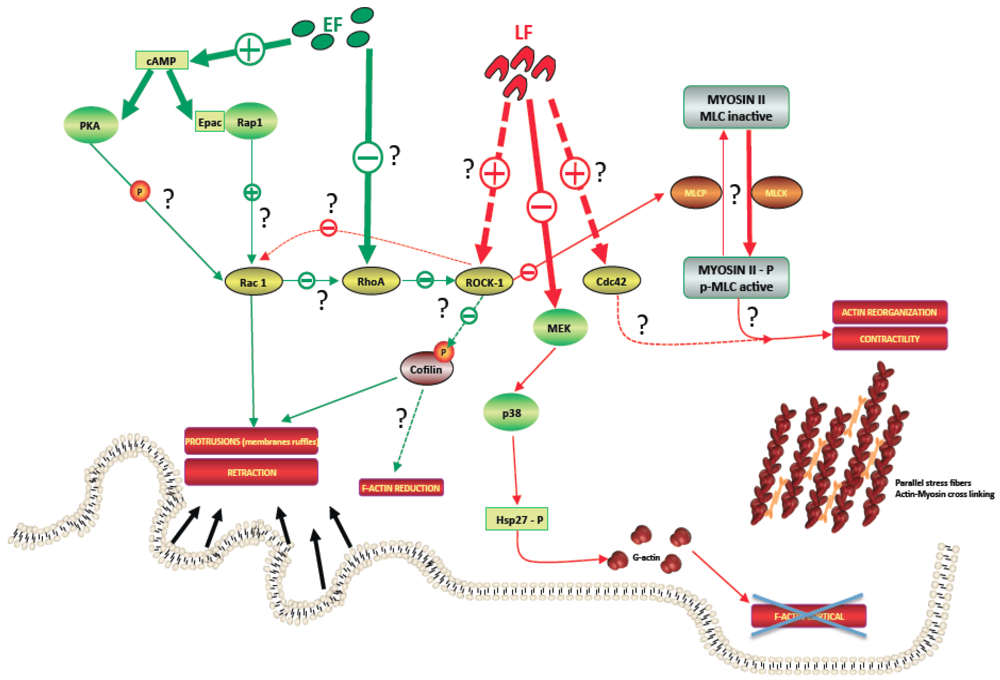
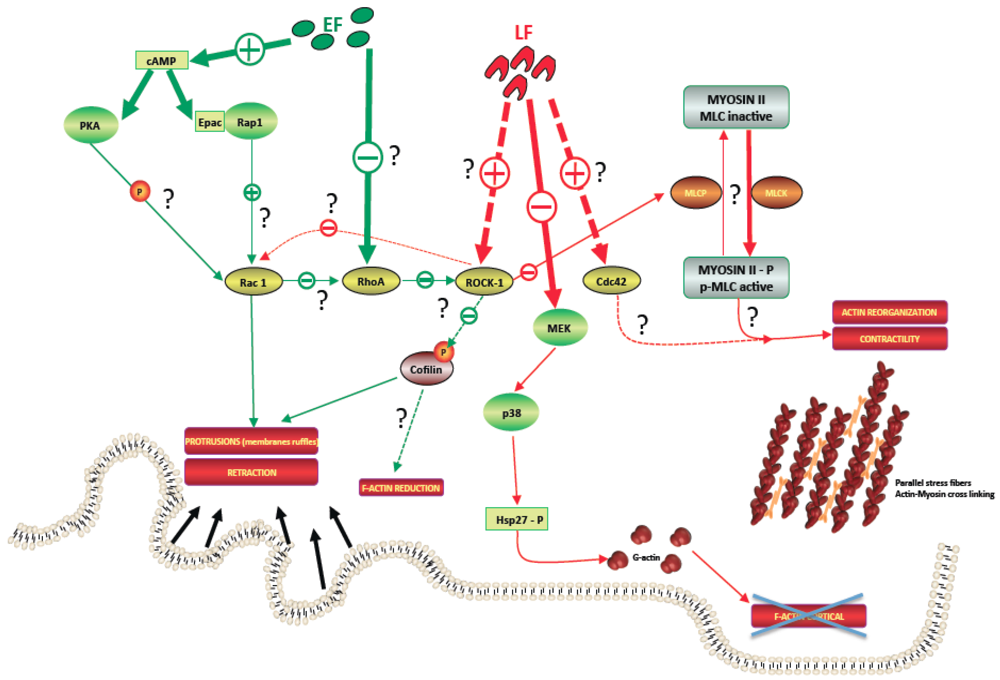
4.1.1. Actin Network
4.1.2. Microtubule Network
4.2. ET Effects
5. From the Cytoskeleton up to Cellular and Organ Disruption
5.1. B. anthracis Toxins, Cytoskeleton and Phagocytosis Disruption
5.2. Anthrax Toxins and Vascular Disruption
6. Conclusions-Perspectives
Acknowledgements
Conflict of Interest
References
- Mock, M.; Fouet, A. Anthrax. Annu. Rev. Microbiol. 2001, 55, 647–671. [Google Scholar] [Green Version]
- Inglesby, T.V.; O’Toole, T.; Henderson, D.A.; Bartlett, J.G.; Ascher, M.S.; Eitzen, E.; Friedlander, A.M.; Gerberding, J.; Hauer, J.; Hughes, J.; et al. Anthrax as a biological weapon, 2002: Updated recommendations for management. J. Am. Med. Assoc. 2002, 287, 2236–2252. [Google Scholar] [Green Version]
- Moayeri, M.; Leppla, S.H. The roles of anthrax toxin in pathogenesis. Curr. Opin. Microbiol. 2004, 7, 19–24. [Google Scholar] [Green Version]
- Moayeri, M.; Leppla, S.H. Cellular and systemic effects of anthrax lethal toxin and edema toxin. Mol. Aspects Med. 2009, 30, 439–455. [Google Scholar] [Green Version]
- Jernigan, D.B.; Raghunathan, P.L.; Bell, B.P.; Brechner, R.; Bresnitz, E.A.; Butler, J.C.; Cetron, M.; Cohen, M.; Doyle, T.; Fischer, M.; et al. Investigation of bioterrorism-related anthrax, United States, 2001: Epidemiologic findings. Emerg. Infect. Dis. 2002, 8, 1019–1028. [Google Scholar] [Green Version]
- Guarner, J.; Jernigan, J.A.; Shieh, W.J.; Tatti, K.; Flannagan, L.M.; Stephens, D.S.; Popovic, T.; Ashford, D.A.; Perkins, B.A.; Zaki, S.R. Pathology and pathogenesis of bioterrorism-related inhalational anthrax. Am. J. Pathol. 2003, 163, 701–709. [Google Scholar] [Green Version]
- Tournier, J.N.; Rossi Paccani, S.; Quesnel-Hellmann, A.; Baldari, C.T. Anthrax toxins: A weapon to systematically dismantle the host immune defenses. Mol. Aspects Med. 2009, 30, 456–466. [Google Scholar] [Green Version]
- Tournier, J.N.; Quesnel-Hellmann, A.; Cleret, A.; Vidal, D.R. Contribution of toxins to the pathogenesis of inhalational anthrax. Cell Microbiol. 2007, 9, 555–565. [Google Scholar] [Green Version]
- Van der Goot, G.; Young, J.A. Receptors of anthrax toxin and cell entry. Mol. Aspects Med. 2009, 30, 406–412. [Google Scholar] [Green Version]
- Yeager, L.A.; Chopra, A.K.; Peterson, J.W. Bacillus anthracis edema toxin suppresses human macrophage phagocytosis and cytoskeletal remodeling via the protein kinase A and exchange protein activated by cyclic AMP pathways. Infect. Immun. 2009, 77, 2530–2543. [Google Scholar] [Green Version]
- Lehmann, M.; Noack, D.; Wood, M.; Perego, M.; Knaus, U.G. Lung epithelial injury by B. anthracis lethal toxin is caused by MKK-dependent loss of cytoskeletal integrity. PLoS One 2009, 4. [Google Scholar] [CrossRef]
- Hong, J.; Doebele, R.C.; Lingen, M.W.; Quilliam, L.A.; Tang, W.J.; Rosner, M.R. Anthrax edema toxin inhibits endothelial cell chemotaxis via Epac and Rap1. J. Biol. Chem. 2007, 282, 19781–19787. [Google Scholar] [Green Version]
- During, R.L.; Gibson, B.G.; Li, W.; Bishai, E.A.; Sidhu, G.S.; Landry, J.; Southwick, F.S. Anthrax lethal toxin paralyzes actin-based motility by blocking Hsp27 phosphorylation. EMBO J. 2007, 26, 2240–2250. [Google Scholar] [Green Version]
- During, R.L.; Li, W.; Hao, B.; Koenig, J.M.; Stephens, D.S.; Quinn, C.P.; Southwick, F.S. Anthrax lethal toxin paralyzes neutrophil actin-based motility. J. Infect. Dis. 2005, 192, 837–845. [Google Scholar] [Green Version]
- Werner, E.; Kowalczyk, A.P.; Faundez, V. Anthrax toxin receptor 1/tumor endothelium marker 8 mediates cell spreading by coupling extracellular ligands to the actin cytoskeleton. J. Biol. Chem. 2006, 281, 23227–23236. [Google Scholar] [Green Version]
- Tang, W.J.; Guo, Q. The adenylyl cyclase activity of anthrax edema factor. Mol. Aspects Med. 2009, 30, 423–430. [Google Scholar] [Green Version]
- Tonello, F.; Montecucco, C. The anthrax lethal factor and its MAPK kinase-specific metalloprotease activity. Mol. Aspects Med. 2009, 30, 431–438. [Google Scholar] [Green Version]
- Young, J.A.; Collier, R.J. Anthrax toxin: Receptor binding, internalization, pore formation, and translocation. Annu. Rev. Biochem. 2007, 76, 243–265. [Google Scholar] [Green Version]
- Bradley, K.A.; Mogridge, J.; Mourez, M.; Collier, R.J.; Young, J.A. Identification of the cellular receptor for anthrax toxin. Nature 2001, 414, 225–229. [Google Scholar] [Green Version]
- Wei, W.; Lu, Q.; Chaudry, G.J.; Leppla, S.H.; Cohen, S.N. The LDL receptor-related protein LRP6 mediates internalization and lethality of anthrax toxin. Cell 2006, 124, 1141–1154. [Google Scholar] [Green Version]
- Young, J.J.; Bromberg-White, J.L.; Zylstra, C.; Church, J.T.; Boguslawski, E.; Resau, J.H.; Williams, B.O.; Duesbery, N.S. LRP5 and LRP6 are not required for protective antigen-mediated internalization or lethality of anthrax lethal toxin. PLoS Pathog 2007, 3. [Google Scholar] [CrossRef]
- Ryan, P.L.; Young, J.A. Evidence against a human cell-specific role for LRP6 in anthrax toxin entry. PLoS One 2008, 3. [Google Scholar] [CrossRef]
- Abrami, L.; Kunz, B.; Deuquet, J.; Bafico, A.; Davidson, G.; van der Goot, F.G. Functional interactions between anthrax toxin receptors and the WNT signalling protein LRP6. Cell Microbiol. 2008, 10, 2509–2519. [Google Scholar] [Green Version]
- Abrami, L.; Liu, S.; Cosson, P.; Leppla, S.H.; van der Goot, F.G. Anthrax toxin triggers endocytosis of its receptor via a lipid raft-mediated clathrin-dependent process. J. Cell Biol. 2003, 160, 321–328. [Google Scholar] [Green Version]
- Abrami, L.; Bischofberger, M.; Kunz, B.; Groux, R.; van der Goot, F.G. Endocytosis of the anthrax toxin is mediated by clathrin, actin and unconventional adaptors. PLoS Pathog. 2010, 6. [Google Scholar] [CrossRef]
- Dal Molin, F.; Tonello, F.; Ladant, D.; Zornetta, I.; Zamparo, I.; di Benedetto, G.; Zaccolo, M.; Montecucco, C. Cell entry and cAMP imaging of anthrax edema toxin. EMBO J. 2006, 25, 5405–5413. [Google Scholar] [Green Version]
- Liu, S.; Crown, D.; Miller-Randolph, S.; Moayeri, M.; Wang, H.; Hu, H.; Morley, T.; Leppla, S.H. Capillary morphogenesis protein-2 is the major receptor mediating lethality of anthrax toxin in vivo. Proc. Natl. Acad. Sci. USA 2009, 106, 12424–12429. [Google Scholar] [Green Version]
- Taylor, M.P.; Koyuncu, O.O.; Enquist, L.W. Subversion of the actin cytoskeleton during viral infection. Nat. Rev. Microbiol. 2011, 9, 427–439. [Google Scholar] [Green Version]
- Aktories, K.; Lang, A.E.; Schwan, C.; Mannherz, H.G. Actin as target for modification by bacterial protein toxins. FEBS J. 2011. [Google Scholar] [CrossRef]
- Le Clainche, C.; Carlier, M.F. Regulation of actin assembly associated with protrusion and adhesion in cell migration. Physiol. Rev. 2008, 88, 489–513. [Google Scholar] [Green Version]
- Ridley, A.J. Life at the leading edge. Cell 2011, 145, 1012–1022. [Google Scholar] [Green Version]
- Swanson, J.A. Shaping cups into phagosomes and macropinosomes. Nat. Rev. Mol. Cell Biol. 2008, 9, 639–649. [Google Scholar] [Green Version]
- Aktories, K. Bacterial protein toxins that modify host regulatory GTPases. Nat. Rev. Microbiol. 2011, 9, 487–498. [Google Scholar] [Green Version]
- Etienne-Manneville, S.; Hall, A. Rho GTPases in cell biology. Nature 2002, 420, 629–635. [Google Scholar] [Green Version]
- Spiering, D.; Hodgson, L. Dynamics of the Rho-family small GTPases in actin regulation and motility. Cell Adh. Migr. 2011, 5, 170–180. [Google Scholar] [Green Version]
- Bokoch, G.M. Regulation of innate immunity by Rho GTPases. Trends Cell Biol. 2005, 15, 163–171. [Google Scholar] [Green Version]
- Vicente-Manzanares, M.; Sanchez-Madrid, F. Role of the cytoskeleton during leukocyte responses. Nat. Rev. Immunol. 2004, 4, 110–122. [Google Scholar] [Green Version]
- Niedergang, F.; Chavrier, P. Regulation of phagocytosis by Rho GTPases. Curr. Top. Microbiol. Immunol. 2005, 291, 43–60. [Google Scholar] [Green Version]
- Luders, J.; Stearns, T. Microtubule-organizing centres: A re-evaluation. Nat. Rev. Mol. Cell Biol. 2007, 8, 161–167. [Google Scholar] [Green Version]
- Leopold, P.L.; Pfister, K.K. Viral strategies for intracellular trafficking: Motors and microtubules. Traffic 2006, 7, 516–523. [Google Scholar] [Green Version]
- Eng, E.W.; Bettio, A.; Ibrahim, J.; Harrison, R.E. MTOC reorientation occurs during FcgammaR-mediated phagocytosis in macrophages. Mol. Biol. Cell 2007, 18, 2389–2399. [Google Scholar] [Green Version]
- Cryan, L.M.; Rogers, M.S. Targeting the anthrax receptors, TEM-8 and CMG-2, for anti-angiogenic therapy. Front. Biosci. 2011, 16, 1574–1588. [Google Scholar] [CrossRef]
- Dowling, O.; Difeo, A.; Ramirez, M.C.; Tukel, T.; Narla, G.; Bonafe, L.; Kayserili, H.; Yuksel-Apak, M.; Paller, A.S.; Norton, K.; et al. Mutations in capillary morphogenesis gene-2 result in the allelic disorders juvenile hyaline fibromatosis and infantile systemic hyalinosis. Am. J. Hum. Genet. 2003, 73, 957–966. [Google Scholar] [CrossRef]
- Hanks, S.; Adams, S.; Douglas, J.; Arbour, L.; Atherton, D.J.; Balci, S.; Bode, H.; Campbell, M.E.; Feingold, M.; Keser, G.; et al. Mutations in the gene encoding capillary morphogenesis protein 2 cause juvenile hyaline fibromatosis and infantile systemic hyalinosis. Am. J. Hum. Genet. 2003, 73, 791–800. [Google Scholar] [CrossRef]
- Jinnin, M.; Medici, D.; Park, L.; Limaye, N.; Liu, Y.; Boscolo, E.; Bischoff, J.; Vikkula, M.; Boye, E.; Olsen, B.R. Suppressed NFAT-dependent VEGFR1 expression and constitutive VEGFR2 signaling in infantile hemangioma. Nat. Med. 2008, 14, 1236–1246. [Google Scholar]
- Abrami, L.; Kunz, B.; van der Goot, F.G. Anthrax toxin triggers the activation of src-like kinases to mediate its own uptake. Proc. Natl. Acad. Sci. USA 2010, 107, 1420–1424. [Google Scholar]
- Lu, Q.; Wei, W.; Kowalski, P.E.; Chang, A.C.; Cohen, S.N. EST-based genome-wide gene inactivation identifies ARAP3 as a host protein affecting cellular susceptibility to anthrax toxin. Proc. Natl. Acad. Sci. USA 2004, 101, 17246–17251. [Google Scholar]
- Bell, S.E.; Mavila, A.; Salazar, R.; Bayless, K.J.; Kanagala, S.; Maxwell, S.A.; Davis, G.E. Differential gene expression during capillary morphogenesis in 3D collagen matrices: Regulated expression of genes involved in basement membrane matrix assembly, cell cycle progression, cellular differentiation and G-protein signaling. J. Cell Sci. 2001, 114, 2755–2773. [Google Scholar]
- Nanda, A.; Carson-Walter, E.B.; Seaman, S.; Barber, T.D.; Stampfl, J.; Singh, S.; Vogelstein, B.; Kinzler, K.W.; St Croix, B. TEM8 interacts with the cleaved C5 domain of collagen alpha 3(VI). Cancer Res. 2004, 64, 817–820. [Google Scholar]
- Hotchkiss, K.A.; Basile, C.M.; Spring, S.C.; Bonuccelli, G.; Lisanti, M.P.; Terman, B.I. TEM8 expression stimulates endothelial cell adhesion and migration by regulating cell-matrix interactions on collagen. Exp. Cell Res. 2005, 305, 133–144. [Google Scholar]
- Garlick, K.M.; Mogridge, J. Direct interaction between anthrax toxin receptor 1 and the actin cytoskeleton. Biochemistry 2009, 48, 10577–10581. [Google Scholar]
- Go, M.Y.; Chow, E.M.; Mogridge, J. The cytoplasmic domain of anthrax toxin receptor 1 affects binding of the protective antigen. Infect. Immun. 2009, 77, 52–59. [Google Scholar]
- Rolando, M.; Stefani, C.; Flatau, G.; Auberger, P.; Mettouchi, A.; Mhlanga, M.; Rapp, U.; Galmiche, A.; Lemichez, E. Transcriptome dysregulation by anthrax lethal toxin plays a key role in induction of human endothelial cell cytotoxicity. Cell Microbiol. 2010, 12, 891–905. [Google Scholar]
- Warfel, J.M.; Steele, A.D.; D’Agnillo, F. Anthrax lethal toxin induces endothelial barrier dysfunction. Am. J. Pathol. 2005, 166, 1871–1881. [Google Scholar]
- Rolando, M.; Munro, P.; Stefani, C.; Auberger, P.; Flatau, G.; Lemichez, E. Injection of Staphylococcus aureus EDIN by the Bacillus anthracis protective antigen machinery induces vascular permeability. Infect. Immun. 2009, 77, 3596–3601. [Google Scholar]
- Warfel, J.M.; D’Agnillo, F. Anthrax lethal toxin-mediated disruption of endothelial VE-cadherin is attenuated by inhibition of the Rho-associated kinase pathway. Toxins 2011, 3, 1278–1293. [Google Scholar]
- Szarowicz, S.E.; During, R.L.; Li, W.; Quinn, C.P.; Tang, W.J.; Southwick, F.S. Bacillus anthracis edema toxin impairs neutrophil actin-based motility. Infect. Immun. 2009, 77, 2455–2464. [Google Scholar]
- Sapra, R.; Gaucher, S.P.; Lachmann, J.S.; Buffleben, G.M.; Chirica, G.S.; Comer, J.E.; Peterson, J.W.; Chopra, A.K.; Singh, A.K. Proteomic analyses of murine macrophages treated with Bacillus anthracis lethal toxin. Microb. Pathog. 2006, 41, 157–167. [Google Scholar]
- Chandra, H.; Gupta, P.K.; Sharma, K.; Mattoo, A.R.; Garg, S.K.; Gade, W.N.; Sirdeshmukh, R.; Maithal, K.; Singh, Y. Proteome analysis of mouse macrophages treated with anthrax lethal toxin. Biochim. Biophys. Acta 2005, 1747, 151–159. [Google Scholar]
- Nour, A.M.; Yeung, Y.G.; Santambrogio, L.; Boyden, E.D.; Stanley, E.R.; Brojatsch, J. Anthrax lethal toxin triggers the formation of a membrane-associated inflammasome complex in murine macrophages. Infect. Immun. 2009, 77, 1262–1271. [Google Scholar]
- Vitale, G.; Pellizzari, R.; Recchi, C.; Napolitani, G.; Mock, M.; Montecucco, C. Anthrax lethal factor cleaves the N-terminus of MAPKKs and induces tyrosine/threonine phosphorylation of MAPKs in cultured macrophages. Biochem. Biophys. Res. Commun. 1998, 248, 706–711. [Google Scholar]
- Tucker, A.E.; Salles, I.I.; Voth, D.E.; Ortiz-Leduc, W.; Wang, H.; Dozmorov, I.; Centola, M.; Ballard, J.D. Decreased glycogen synthase kinase 3-beta levels and related physiological changes in Bacillus anthracis lethal toxin-treated macrophages. Cell Microbiol. 2003, 5, 523–532. [Google Scholar]
- Hong, J.; Beeler, J.; Zhukovskaya, N.L.; He, W.; Tang, W.J.; Rosner, M.R. Anthrax edema factor potency depends on mode of cell entry. Biochem. Biophys. Res. Commun. 2005, 335, 850–857. [Google Scholar]
- Kim, C.; Wilcox-Adelman, S.; Sano, Y.; Tang, W.J.; Collier, R.J.; Park, J.M. Antiinflammatory cAMP signaling and cell migration genes co-opted by the anthrax bacillus. Proc. Natl. Acad. Sci. USA 2008, 105, 6150–6155. [Google Scholar]
- Gnade, B.T.; Moen, S.T.; Chopra, A.K.; Peterson, J.W.; Yeager, L.A. Emergence of anthrax edema toxin as a master manipulator of macrophage and B cell functions. Toxins 2010, 2, 1881–1897. [Google Scholar]
- Yahr, T.L.; Vallis, A.J.; Hancock, M.K.; Barbieri, J.T.; Frank, D.W. ExoY, an adenylate cyclase secreted by the Pseudomonas aeruginosa type III system. Proc. Natl. Acad. Sci. USA 1998, 95, 13899–13904. [Google Scholar]
- Hewlett, E.L.; Urban, M.A.; Manclark, C.R.; Wolff, J. Extracytoplasmic adenylate cyclase of Bordetella pertussis. Proc. Natl. Acad. Sci. USA 1976, 73, 1926–1930. [Google Scholar]
- Ohnishi, H.; Miyake, M.; Kamitani, S.; Horiguchi, Y. The morphological changes in cultured cells caused by Bordetella pertussis adenylate cyclase toxin. FEMS Microbiol. Lett. 2008, 279, 174–179. [Google Scholar]
- Carbonetti, N.H. Pertussis toxin and adenylate cyclase toxin: Key virulence factors of Bordetella pertussis and cell biology tools. Future Microbiol. 2010, 5, 455–469. [Google Scholar]
- Vojtova, J.; Kofronova, O.; Sebo, P.; Benada, O. Bordetella adenylate cyclase toxin induces a cascade of morphological changes of sheep erythrocytes and localizes into clusters in erythrocyte membranes. Microsc. Res. Tech. 2006, 69, 119–129. [Google Scholar]
- Kamanova, J.; Kofronova, O.; Masin, J.; Genth, H.; Vojtova, J.; Linhartova, I.; Benada, O.; Just, I.; Sebo, P. Adenylate cyclase toxin subverts phagocyte function by RhoA inhibition and unproductive ruffling. J. Immunol. 2008, 181, 5587–5597. [Google Scholar]
- Weingart, C.L.; Weiss, A.A. Bordetella pertussis virulence factors affect phagocytosis by human neutrophils. Infect. Immun. 2000, 68, 1735–1739. [Google Scholar]
- Weingart, C.L.; Mobberley-Schuman, P.S.; Hewlett, E.L.; Gray, M.C.; Weiss, A.A. Neutralizing antibodies to adenylate cyclase toxin promote phagocytosis of Bordetella pertussis by human neutrophils. Infect. Immun. 2000, 68, 7152–7155. [Google Scholar]
- Prasain, N.; Alexeyev, M.; Balczon, R.; Stevens, T. Soluble adenylyl cyclase-dependent microtubule disassembly reveals a novel mechanism of endothelial cell retraction. Am. J. Physiol. Lung Cell Mol. Physiol. 2009, 297, L73–L83. [Google Scholar]
- Sayner, S.L. Emerging themes of cAMP regulation of the pulmonary endothelial barrier. Am. J. Physiol. Lung Cell Mol. Physiol. 2011, 300, L667–L678. [Google Scholar]
- Hritonenko, V.; Mun, J.J.; Tam, C.; Simon, N.C.; Barbieri, J.T.; Evans, D.J.; Fleiszig, S.M. Adenylate cyclase activity of Pseudomonas aeruginosa ExoY can mediate bleb-niche formation in epithelial cells and contributes to virulence. Microb. Pathog. 2011, 51, 305–312. [Google Scholar]
- Buck, M.; Chojkier, M. C/EBPbeta phosphorylation rescues macrophage dysfunction and apoptosis induced by anthrax lethal toxin. Am. J. Physiol. Cell Physiol. 2007, 293, C1788–C1796. [Google Scholar]
- Ribot, W.J.; Panchal, R.G.; Brittingham, K.C.; Ruthel, G.; Kenny, T.A.; Lane, D.; Curry, B.; Hoover, T.A.; Friedlander, A.M.; Bavari, S. Anthrax lethal toxin impairs innate immune functions of alveolar macrophages and facilitates Bacillus anthracis survival. Infect. Immun. 2006, 74, 5029–5034. [Google Scholar]
- Kau, J.H.; Sun, D.S.; Huang, H.S.; Lien, T.S.; Huang, H.H.; Lin, H.C.; Chang, H.H. Sublethal doses of anthrax lethal toxin on the suppression of macrophage phagocytosis. PLoS One 2010, 5. [Google Scholar] [CrossRef]
- O’Brien, J.; Friedlander, A.; Dreier, T.; Ezzell, J.; Leppla, S. Effects of anthrax toxin components on human neutrophils. Infect. Immun. 1985, 47, 306–310. [Google Scholar]
- Stanley, J.L.; Smith, H. Purification of factor I and recognition of a third factor of the anthrax toxin. J. Gen. Microbiol. 1961, 26, 49–63. [Google Scholar]
- Sirisanthana, T.; Brown, A.E. Anthrax of the gastrointestinal tract. Emerg. Infect. Dis. 2002, 8, 649–651. [Google Scholar]
- Firoved, A.M.; Miller, G.F.; Moayeri, M.; Kakkar, R.; Shen, Y.; Wiggins, J.F.; McNally, E.M.; Tang, W.J.; Leppla, S.H. Bacillus anthracis edema toxin causes extensive tissue lesions and rapid lethality in mice. Am. J. Pathol. 2005, 167, 1309–1320. [Google Scholar]
- Guichard, A.; Nizet, V.; Bier, E. New insights into the biological effects of anthrax toxins: Linking cellular to organismal responses. Microbes Infect. 2012, 14, 97–118. [Google Scholar]
- Ebrahimi, C.M.; Sheen, T.R.; Renken, C.W.; Gottlieb, R.A.; Doran, K.S. Contribution of lethal toxin and edema toxin to the pathogenesis of anthrax meningitis. Infect. Immun. 2011, 79, 2510–2518. [Google Scholar]
- Van Sorge, N.M.; Ebrahimi, C.M.; McGillivray, S.M.; Quach, D.; Sabet, M.; Guiney, D.G.; Doran, K.S. Anthrax toxins inhibit neutrophil signaling pathways in brain endothelium and contribute to the pathogenesis of meningitis. PLoS One 2008, 3. [Google Scholar] [CrossRef]
- Warfel, J.M.; Steele, A.D.; D’Agnillo, F. Anthrax lethal toxin induces endothelial barrier dysfunction. Am. J. Pathol. 2005, 166, 1871–1881. [Google Scholar]
- Bolcome, R.E., III; Sullivan, S.E.; Zeller, R.; Barker, A.P.; Collier, R.J.; Chan, J. Anthrax lethal toxin induces cell death-independent permeability in zebrafish vasculature. Proc. Natl. Acad. Sci. USA 2008, 105, 2439–2444. [Google Scholar]
- Guichard, A.; McGillivray, S.M.; Cruz-Moreno, B.; van Sorge, N.M.; Nizet, V.; Bier, E. Anthrax toxins cooperatively inhibit endocytic recycling by the Rab11/Sec15 exocyst. Nature 2010, 467, 854–858. [Google Scholar]
- Guichard, A.; Park, J.M.; Cruz-Moreno, B.; Karin, M.; Bier, E. Anthrax lethal factor and edema factor act on conserved targets in Drosophila. Proc. Natl. Acad. Sci. USA 2006, 103, 3244–3249. [Google Scholar]
- Bier, E.; Guichard, A. Deconstructing host-pathogen interactions in Drosophila. Dis. Model. Mech. 2012, 5, 48–61. [Google Scholar]
- Lemichez, E.; Lecuit, M.; Nassif, X.; Bourdoulous, S. Breaking the wall: Targeting of the endothelium by pathogenic bacteria. Nat. Rev. Microbiol. 2010, 8, 93–104. [Google Scholar]
- Maddugoda, M.P.; Stefani, C.; Gonzalez-Rodriguez, D.; Saarikangas, J.; Torrino, S.; Janel, S.; Munro, P.; Doye, A.; Prodon, F.; Aurrand-Lions, M.; et al. cAMP signaling by anthrax edema toxin induces transendothelial cell tunnels, which are resealed by MIM via Arp2/3-driven actin polymerization. Cell Host Microbe 2011, 10, 464–474. [Google Scholar] [CrossRef]
- Tournier, J.N.; Quesnel-Hellmann, A.; Mathieu, J.; Montecucco, C.; Tang, W.J.; Mock, M.; Vidal, D.R.; Goossens, P.L. Anthrax edema toxin cooperates with lethal toxin to impair cytokine secretion during infection of dendritic cells. J. Immunol. 2005, 174, 4934–4941. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Trescos, Y.; Tournier, J.-N. Cytoskeleton as an Emerging Target of Anthrax Toxins. Toxins 2012, 4, 83-97. https://doi.org/10.3390/toxins4020083
Trescos Y, Tournier J-N. Cytoskeleton as an Emerging Target of Anthrax Toxins. Toxins. 2012; 4(2):83-97. https://doi.org/10.3390/toxins4020083
Chicago/Turabian StyleTrescos, Yannick, and Jean-Nicolas Tournier. 2012. "Cytoskeleton as an Emerging Target of Anthrax Toxins" Toxins 4, no. 2: 83-97. https://doi.org/10.3390/toxins4020083
APA StyleTrescos, Y., & Tournier, J.-N. (2012). Cytoskeleton as an Emerging Target of Anthrax Toxins. Toxins, 4(2), 83-97. https://doi.org/10.3390/toxins4020083