Binding of Hanatoxin to the Voltage Sensor of Kv2.1

Abstract
:1. Introduction

2. Results and Discussion
2.1. Binding to the VSR
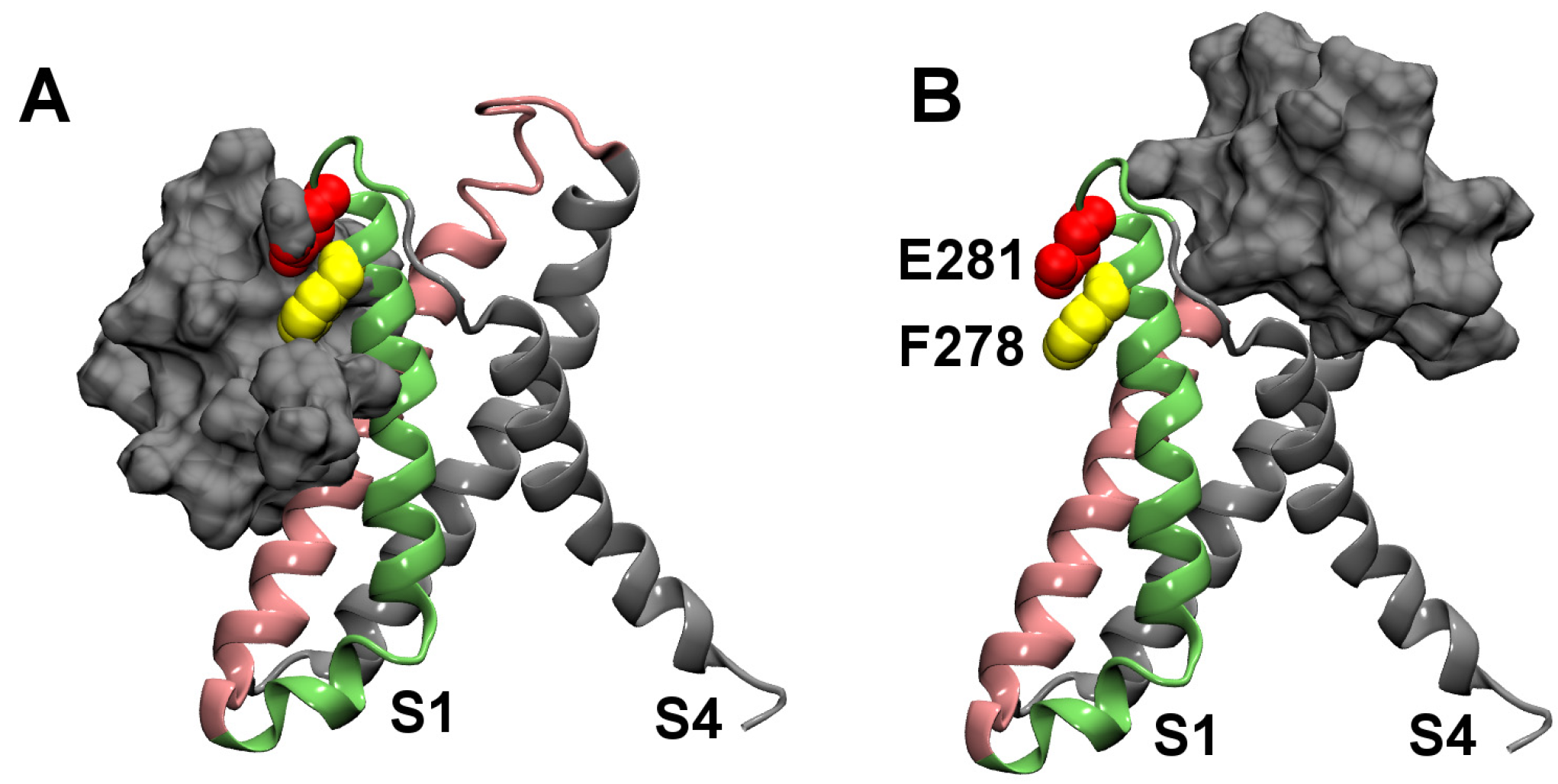

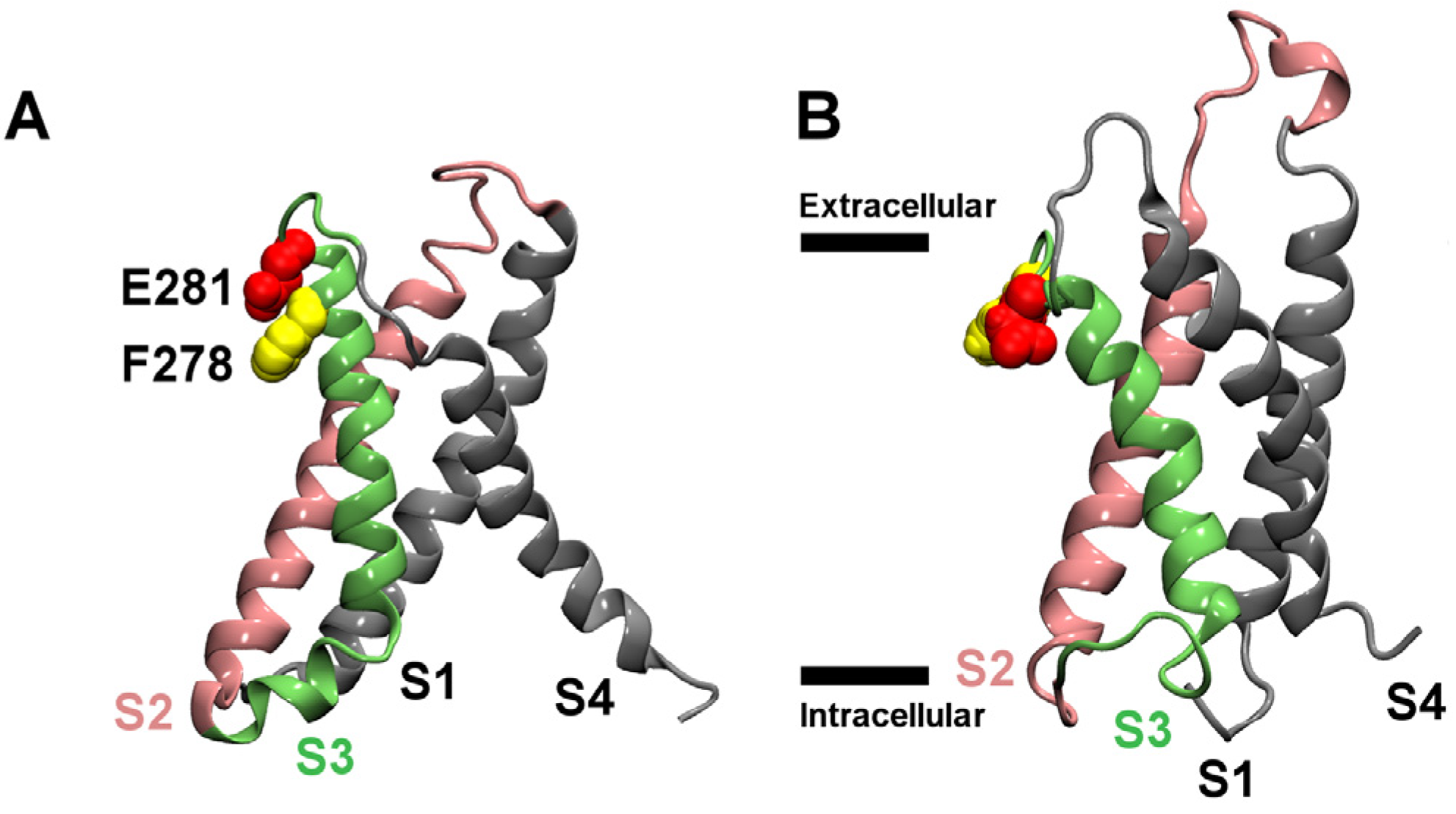
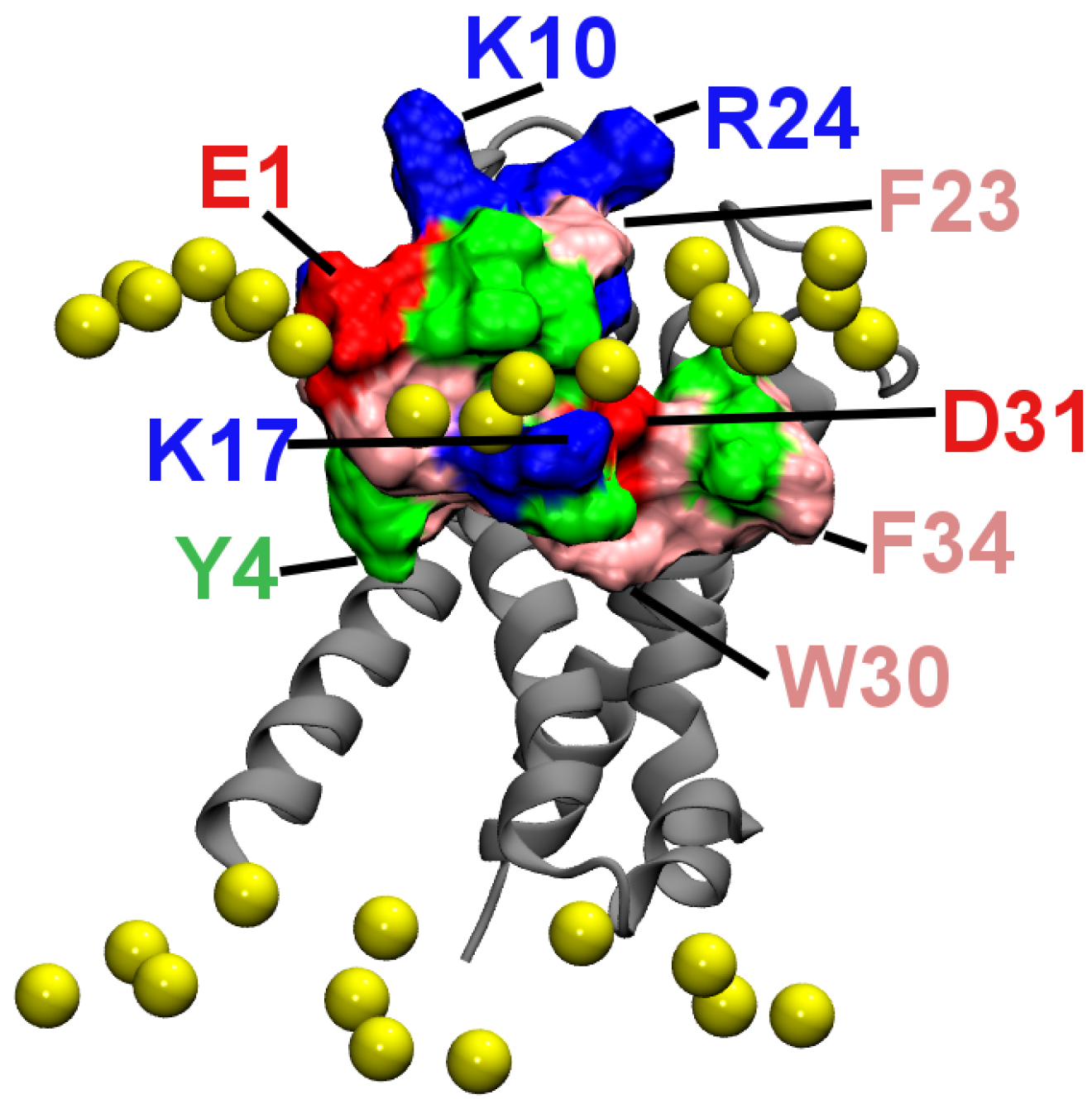
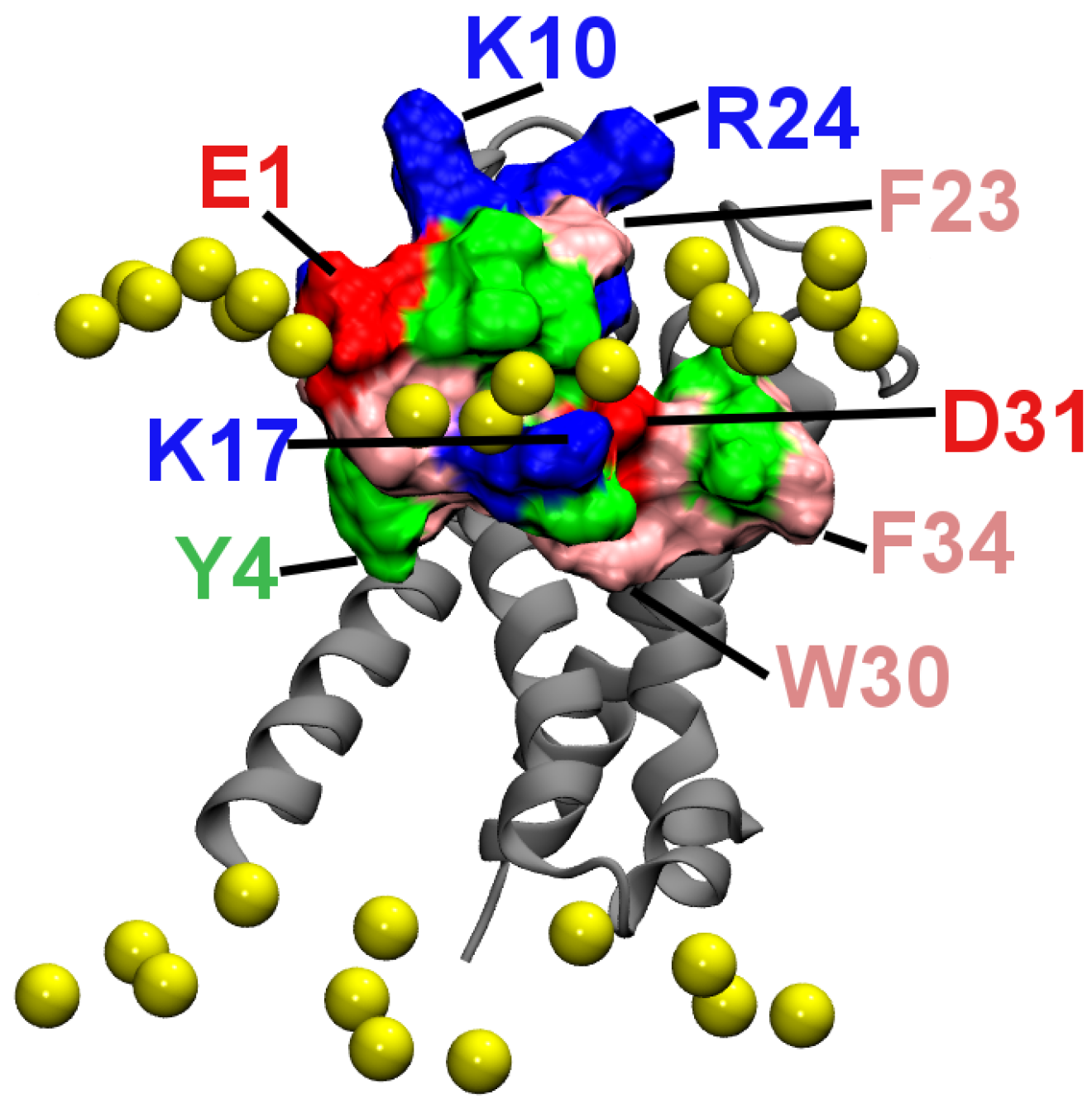
2.2. Binding to the VSO

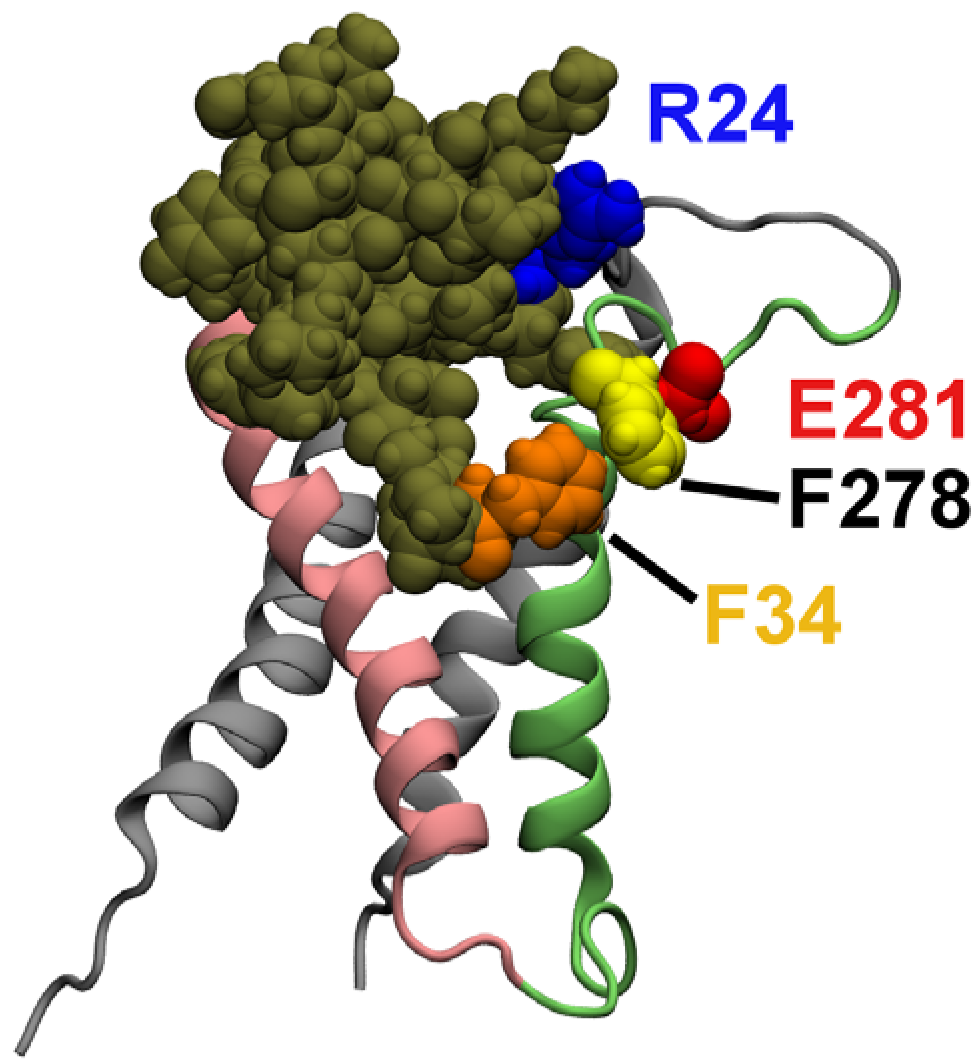
2.3. Computational Mutagenesis
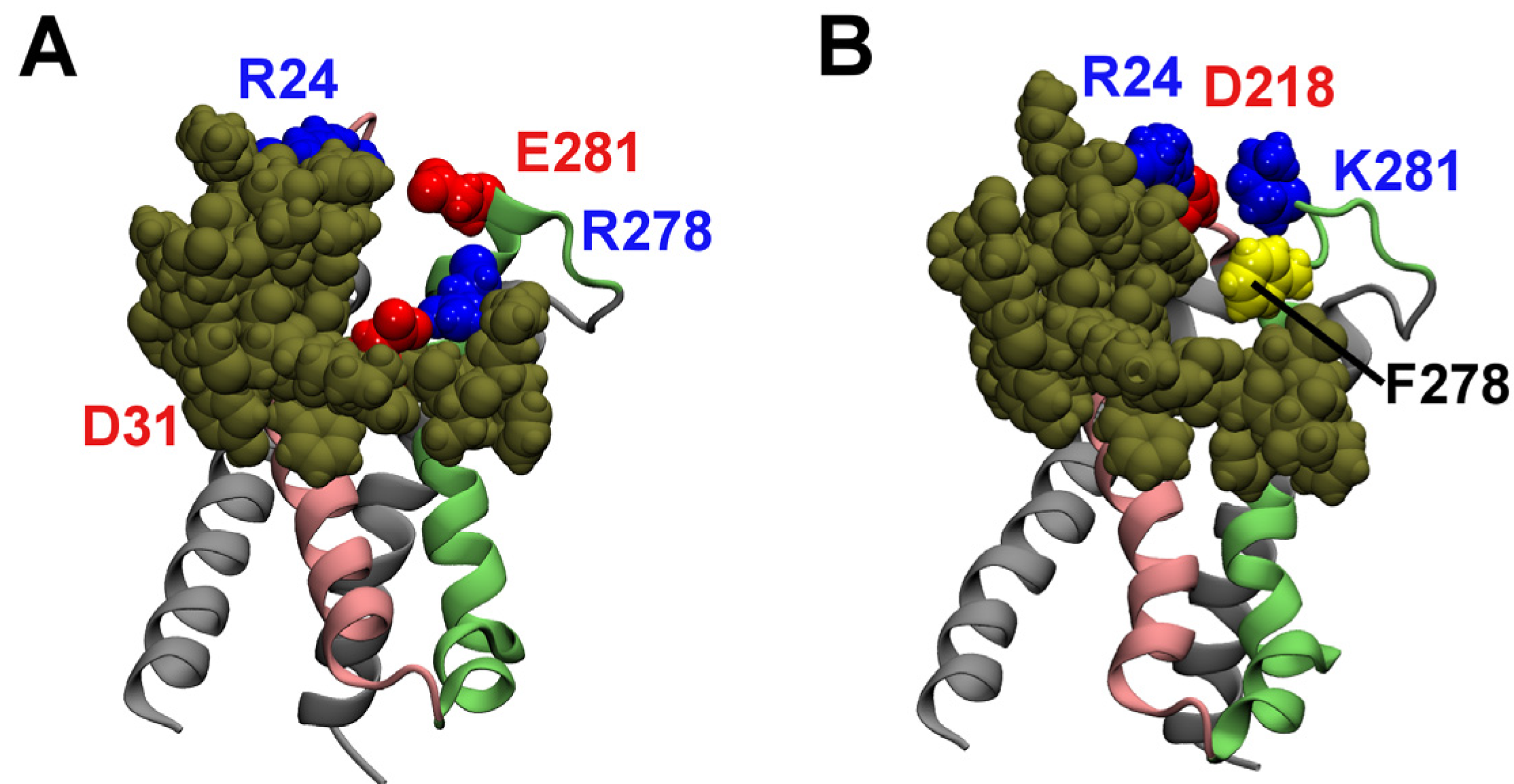

{kind=link}
{kind=link}
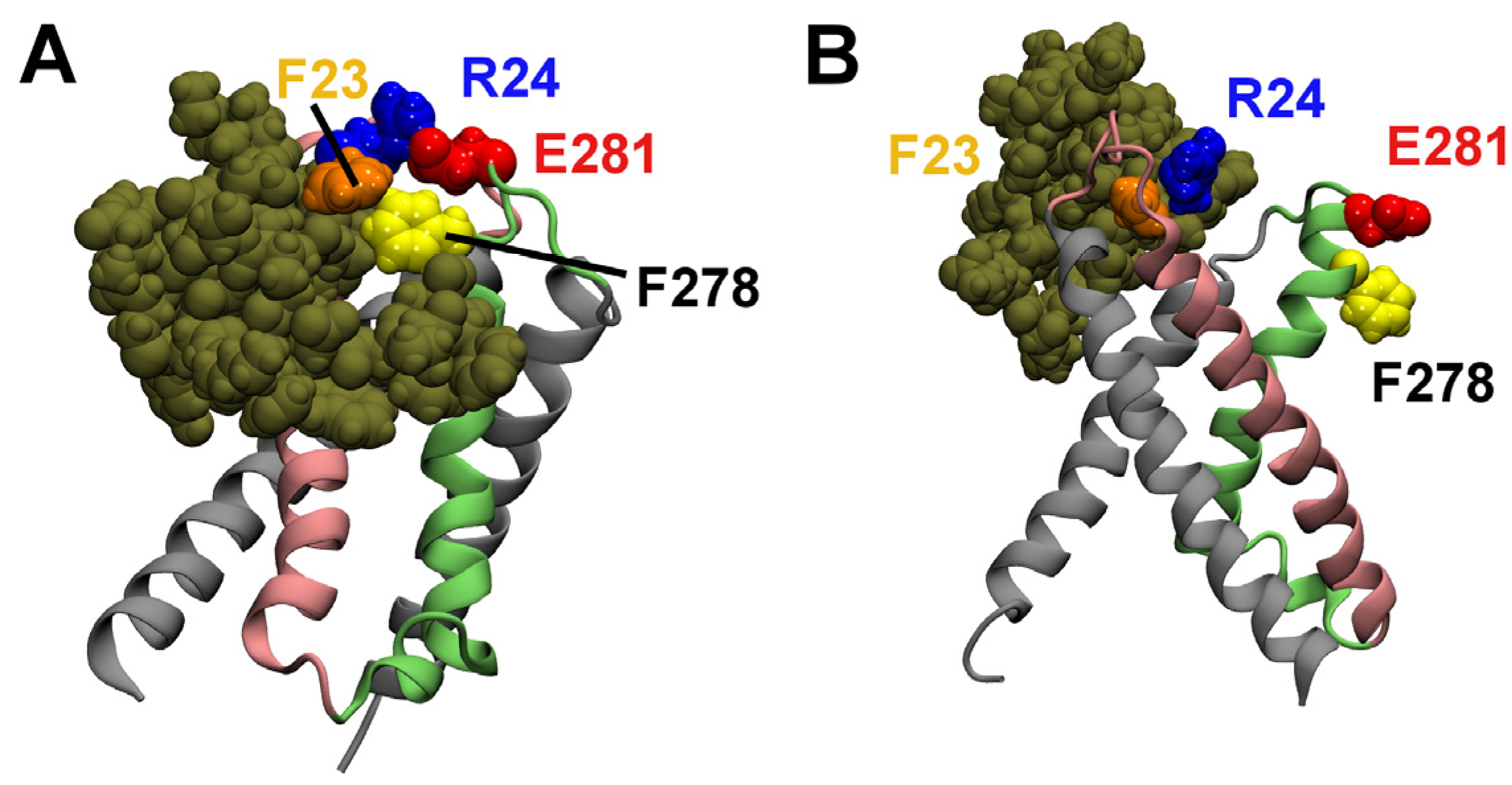{kind=link}
{kind=link}
{kind=link}
{kind=link}
| VS | SASA (Å2) | ΔGbind (kT) |
|---|---|---|
| WT | 790 ± 20 | −16.1 |
| F278R | 690 ± 27 | −9.7 |
| E281K | 751 ± 53 | −11.0 |
2.4. Membrane Partition
3. Methods
3.1. Initial Structures
3.2. Molecular Docking
3.3. Molecular Dynamics Simulations
4. Conclusions
Acknowledgments
Conflict of Interest
References
- Lee, S.Y.; MacKinnon, R. A membrane-access mechanism of ion channel inhibition by voltage sensor toxins from spider venom. Nature 2004, 430, 232–235. [Google Scholar]
- Jung, H.J.; Lee, J.Y.; Kim, S.H.; Eu, Y.J.; Shin, S.Y.; Milescu, M.; Swartz, K.J.; Kim, J.I. Solution structure and lipid membrane partitioning of VSTx1, an inhibitor of the KvAP potassium channel. Biochemistry 2005, 44, 6015–6023. [Google Scholar]
- Milescu, M.; Vobecky, J.; Roh, S.H.; Kim, S.H.; Jung, H.J.; Kim, J.I.; Swartz, K.J. Tarantula toxins interact with voltage sensors within lipid membranes. J. Gen. Physiol. 2007, 130, 497–511. [Google Scholar] [CrossRef]
- Milescu, M.; Bosmans, F.; Lee, S.; Alabi, A.A.; Kim, J.I.; Swartz, K.J. Interactions between lipids and voltage sensor paddles detected with tarantula toxins. Nat. Struct. Mol. Biol. 2009, 16, 1080–1085. [Google Scholar]
- Swartz, K.J. Tarantula toxins interacting with voltage sensors in potassium channels. Toxicon 2007, 49, 213–230. [Google Scholar] [CrossRef]
- Phillips, L.R.; Milescu, M.; Li-Smerin, Y.Y.; Mindell, J.A.; Kim, J.I.; Swartz, K.J. Voltage-sensor activation with a tarantula toxin as cargo. Nature 2005, 436, 857–860. [Google Scholar]
- Swartz, K.J.; MacKinnon, R. An inhibitor of the Kv2.1 potassium channel isolated from the venom of a Chilean tarantula. Neuron 1995, 15, 941–949. [Google Scholar] [CrossRef]
- Marvin, L.; De, E.; Cosette, P.; Gagnon, J.; Molle, G.; Lange, C. Isolation, amino acid sequence and functional assays of SGTx1. The first toxin purified from the venom of the spider scodra griseipes. Eur. J. Biochem. 1999, 265, 572–579. [Google Scholar] [CrossRef]
- Ruta, V.; Jiang, Y.; Lee, A.; Chen, J.; MacKinnon, R. Functional analysis of an archaebacterial voltage-dependent K+ channel. Nature 2003, 422, 180–185. [Google Scholar]
- Sanguinetti, M.C.; Johnson, J.H.; Hammerland, L.G.; Kelbaugh, P.R.; Volkmann, R.A.; Saccomano, N.A.; Mueller, A.L. Heteropodatoxins: Peptides isolated from spider venom that block Kv4.2 potassium channels. Mol. Pharmacol. 1997, 51, 491–498. [Google Scholar]
- Diochot, S.; Drici, M.D.; Moinier, D.; Fink, M.; Lazdunski, M. Effects of phrixotoxins on the Kv4 family of potassium channels and implications for the role of Ito1 in cardiac electrogenesis. Br. J. Pharmacol. 1999, 126, 251–263. [Google Scholar] [CrossRef]
- Alabi, A.A.; Bahamonde, M.I.; Jung, H.J.; Kim, J.I.; Swartz, K.J. Portability of paddle motif function and pharmacology in voltage sensors. Nature 2007, 450, 370–376. [Google Scholar]
- Zarayskiy, V.V.; Balasubramanian, G.; Bondarenko, V.E.; Morales, M.J. Heteropoda toxin 2 is a gating modifier toxin specific for voltage-gated K+ channels of the Kv4 family. Toxicon 2005, 45, 431–442. [Google Scholar] [CrossRef]
- Norton, R.S.; Pallaghy, P.K. The cystine knot structure of ion channel toxins and related polypeptides. Toxicon 1998, 36, 1573–1583. [Google Scholar] [CrossRef]
- Takahashi, H.; Kim, J.I.; Min, H.J.; Sato, K.; Swartz, K.J.; Shimada, I. Solution structure of hanatoxin1, a gating modifier of voltage-dependent K+ channels: Common surface features of gating modifier toxins. J. Mol. Biol. 2000, 297, 771–780. [Google Scholar] [CrossRef]
- Lee, C.W.; Kim, S.; Roh, S.H.; Endoh, H.; Kodera, Y.; Maeda, T.; Kohno, T.; Wang, J.M.; Swartz, K.J.; Kim, J.I. Solution structure and functional characterization of SGTx1, a modifier of Kv2.1 channel gating. Biochemistry 2004, 43, 890–897. [Google Scholar]
- Wang, J.M.; Roh, S.H.; Kim, S.; Lee, C.W.; Kim, J.I.; Swartz, K.J. Molecular surface of tarantula toxins interacting with voltage sensors in KV channels. J. Gen. Physiol. 2004, 123, 455–467. [Google Scholar] [CrossRef]
- Nishizawa, M.; Nishizawa, K. Interaction between K+ channel gate modifier hanatoxin and lipid bilayer membranes analyzed by molecular dynamics simulation. Eur. Biophys. J. 2006, 35, 373–381. [Google Scholar] [CrossRef]
- Wee, C.L.; Bemporad, D.; Sands, Z.A.; Gavaghan, D.; Sansom, M.S.P. SGTx1, a Kv channel gating-modifier toxin, binds to the interfacial region of lipid bilayers. Biophys. J. 2007, 92, L07–L09. [Google Scholar] [CrossRef]
- Bemporad, D.; Sands, Z.A.; Wee, C.L.; Grottesi, A.; Sansom, M.S. P. Vstx1, a modifier of Kv channel gating, localizes to the interfacial region of lipid bilayers. Biochemistry 2006, 45, 11844–11855. [Google Scholar] [CrossRef]
- Wee, C.L.; Gavaghan, D.; Sansom, M.S.P. Interactions between a voltage sensor and a toxin via multiscale simulations. Biophys. J. 2010, 98, 1558–1565. [Google Scholar] [CrossRef]
- Swartz, K.J.; MacKinnon, R. Hanatoxin modifies the gating of a voltage-dependent K+ channel through multiple binding sites. Neuron 1997, 18, 665–673. [Google Scholar] [CrossRef]
- Swartz, K.J.; MacKinnon, R. Mapping the receptor site for hanatoxin, a gating modifier of voltage-dependent K+ channels. Neuron 1997, 18, 675–682. [Google Scholar] [CrossRef]
- Li-Smerin, Y.; Swartz, K.J. Localization and molecular determinants of the hanatoxin receptors on the voltage-sensing domains of a K+ channel. J. Gen. Physiol. 2000, 115, 673–684. [Google Scholar] [CrossRef]
- Li-Smerin, Y.; Swartz, K.J. Helical structure of the COOH terminus of S3 and its contribution to the gating modifier toxin receptor in voltage-gated ion channels. J. Gen. Physiol. 2001, 117, 205–217. [Google Scholar] [CrossRef]
- Ruta, V.; MacKinnon, R. Localization of the voltage-sensor toxin receptor on KvAP. Biochemistry 2004, 43, 10071–10079. [Google Scholar] [CrossRef]
- Chakrapani, S.; Cuello, L.G.; Cortes, D.M.; Perozo, E. Structural dynamics of an isolated voltage-sensor domain in a lipid bilayer. Structure 2008, 16, 398–409. [Google Scholar] [CrossRef]
- Kumar, S.; Nussinov, R. Close-range electrostatic interactions in proteins. ChemBioChem 2002, 3, 604–617. [Google Scholar] [CrossRef]
- Kollman, P.A.; Massova, I.; Reyes, C.; Kuhn, B.; Huo, S.; Chong, L.; Lee, M.; Lee, T.; Duan, Y.; Wang, W.; et al. Calculating structures and free energies of complex molecules: Combining molecular mechanics and continuum models. Acc. Chem. Res. 2000, 33, 889–897. [Google Scholar] [CrossRef]
- Wang, J.M.; Hou, T.J.; Xu, X.J. Recent advances in free energy calculations with a combination of molecular mechanics and continuum models. Curr. Comput. Aided Drug Des. 2006, 2, 287–306. [Google Scholar] [CrossRef]
- Cohen, L.; Karbat, I.; Gilles, N.; Ilan, N.; Benveniste, M.; Gordon, D.; Gurevitz, M. Common features in the functional surface of scorpion β-toxins and elements that confer specificity for insect and mammalian voltage-gated sodium channels. J. Biol. Chem. 2005, 280, 5045–5053. [Google Scholar]
- Posokhov, Y.O.; Gottlieb, P.A.; Morales, M.J.; Sachs, F.; Ladokhin, A.S. Is lipid bilayer binding a common property of inhibitor cysteine knot ion-channel blockers? Biophys. J. 2007, 93, L20–L22. [Google Scholar] [CrossRef]
- Chen, R.; Chung, S.H. Conserved functional surface of anti-mammalian scorpion β-toxins. J. Phys. Chem. B 2012, 116, 4796–4800. [Google Scholar] [CrossRef]
- Chen, R.; Chung, S.H. Binding modes and functional surface of anti-mammalian scorpion α-toxins to sodium channels. Biochemistry 2012, 51, 7775–7782. [Google Scholar] [CrossRef]
- Cestèle, S.; Qu, Y.; Rogers, J.C.; Rochat, H.; Scheuer, T.; Catterall, W.A. Voltage sensor-trapping: Enhanced activation of sodium channels by β-scorpion toxin bound to the S3-S4 loop in domain II. Neuron 1998, 21, 919–931. [Google Scholar] [CrossRef]
- Jensen, M.Ø.; Jogini, V.; Borhani, D.W.; Leffler, A.E.; Dror, R.O.; Shaw, D.E. Mechanism of voltage gating in potassium channels. Science 2012, 336, 229–233. [Google Scholar] [CrossRef]
- Amaral, C.; Carnevale, V.; Klein, M.L.; Treptow, W. Exploring conformational states of the bacterial voltage-gated sodium channel NavAb via molecular dynamics simulations. Proc. Natl. Acad. Sci. USA 2012. [Google Scholar] [CrossRef]
- Guex, N.; Peitsch, M.C. SWISS-MODEL and the Swiss-PdbViewer: An environment for comparative protein modeling. Electrophoresis 1997, 18, 2714–2723. [Google Scholar] [CrossRef]
- Schwede, T.; Kopp, J.; Guex, N.; Peitsch, M.C. SWISS-MODEL: An automated protein homology-modeling server. Nucleic Acids Res. 2003, 31, 3381–3385. [Google Scholar] [CrossRef]
- Arnold, K.; Bordoli, L.; Kopp, J.; Schwede, T. The SWISS-MODEL Workspace: A web-based environment for protein structure homology modeling. Bioinformatics 2006, 22, 195–201. [Google Scholar] [CrossRef]
- Pathak, M.M.; Yarov-Yarovoy, V.; Agarwal, G.; Roux, B.; Barth, P.; Kohout, S.; Tombola, F.; Isacoff, E.Y. Closing in on the resting state of the Shaker K+ channel. Neuron 2007, 56, 124–140. [Google Scholar] [CrossRef]
- Long, S.B.; Campbell, E.B.; Mackinnon, R. Crystal structure of a mammalian voltage-dependent Shaker family K+ channel. Science 2005, 309, 897–903. [Google Scholar] [CrossRef]
- Chen, X.; Wang, Q.; Ni, F.; Ma, J. Structure of the full-length Shaker potassium channel Kv1.2 by normal-mode-based X-ray crystallographic refinement. Proc. Natl. Acad. Sci. USA 2010, 107, 11352–11357. [Google Scholar] [CrossRef]
- Hillisch, A.; Pineda, L.F.; Hilgenfeld, R. Utility of homology models in the drug discovery process. Drug Discov. Today 2004, 9, 659–669. [Google Scholar] [CrossRef]
- Mintseris, J.; Pierce, B.; Wiehe, K.; Anderson, R.; Chen, R.; Weng, Z. Integrating statistical pair potentials into protein complex prediction. Proteins 2007, 69, 511–520. [Google Scholar] [CrossRef]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kalé, L.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef]
- MacKerell, A.D.; Bashford, D.; Bellott, M.; Dunbrack, R.L.; Evanseck, J.D.; Field, M.J.; Fischer, S.; Gao, J.; Guo, H.; Ha, S.; et al. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B 1998, 102, 3586–3616. [Google Scholar]
- MacKerell, A.D.; Feig, M.; Brooks, C.L. Extending the treatment of backbone energetics in protein force fields: Limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulations. J. Comput. Chem. 2004, 25, 1400–1415. [Google Scholar] [CrossRef]
- Klauda, J.B.; Venable, R.M.; Freites, J.A.; O’Connor, J.W.; Tobias, D.J.; Mondragon-Ramirez, C.; Vorobyov, I.; MacKerell, A.D., Jr.; Pastor, R.W. Update of the CHARMM all-atom additive force field for lipids: Validation on six lipid types. J. Phys. Chem. B 2010, 114, 7830–7843. [Google Scholar]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1982, 79, 926–935. [Google Scholar]
- Ryckaert, J.P.; Ciccotti, G.; Berendsen, H.J.C. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef]
- Miyamoto, S.; Kollman, P.A. SETTLE: An analytical version of the SHAKE and RATTLE algorithm for rigid water models. J. Comput. Chem. 1992, 13, 952–962. [Google Scholar] [CrossRef]
- Martyna, G.J.; Tobias, D.J.; Klein, M.L. Constant pressure molecular dynamics algorithms. J. Chem. Phys. 1994, 101, 4177–4189. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Chen, R.; Robinson, A.; Chung, S.-H. Binding of Hanatoxin to the Voltage Sensor of Kv2.1. Toxins 2012, 4, 1552-1564. https://doi.org/10.3390/toxins4121552
Chen R, Robinson A, Chung S-H. Binding of Hanatoxin to the Voltage Sensor of Kv2.1. Toxins. 2012; 4(12):1552-1564. https://doi.org/10.3390/toxins4121552
Chicago/Turabian StyleChen, Rong, Anna Robinson, and Shin-Ho Chung. 2012. "Binding of Hanatoxin to the Voltage Sensor of Kv2.1" Toxins 4, no. 12: 1552-1564. https://doi.org/10.3390/toxins4121552
APA StyleChen, R., Robinson, A., & Chung, S.-H. (2012). Binding of Hanatoxin to the Voltage Sensor of Kv2.1. Toxins, 4(12), 1552-1564. https://doi.org/10.3390/toxins4121552