The Enterotoxicity of Clostridium difficile Toxins

Abstract
:1. Introduction
2. C. difficile Infection in Humans
3. Animal Models of CDI
4. Mechanism of Action and Functional Domains of TcdA and TcdB
4.1. Structure of TcdA and TcdB
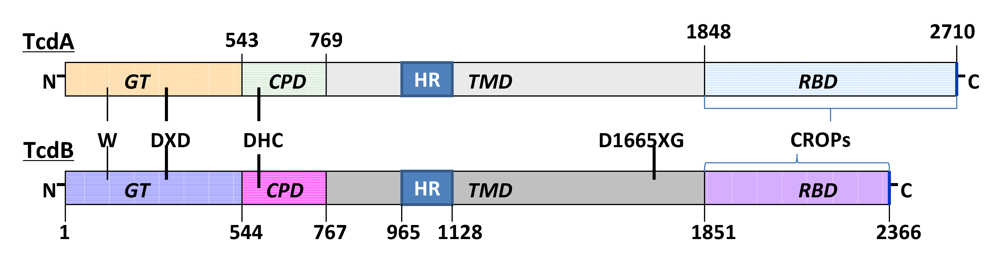
4.2. The C-terminal receptor binding domain (RBD) mediates receptor binding
4.3. The central translocation domain (TMD), autocatalytic cysteine proteinase domain (CPD) and uptake of TcdA and TcdB
4.4. N-terminal glucosylatransferase domain (GT)
4.5. Small GTPase proteins as targets of TcdA and TcdB
4.6. C. difficile binary toxin (CDT)
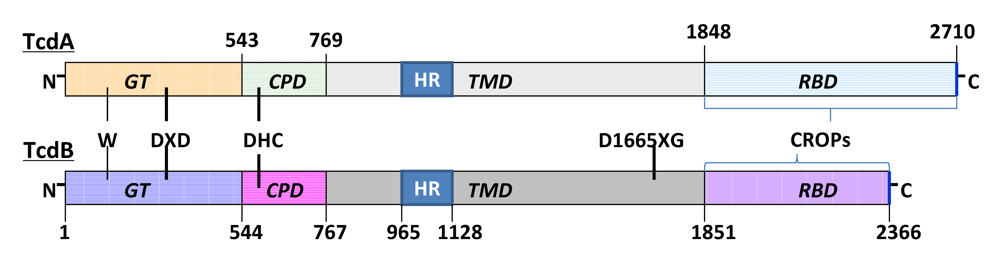
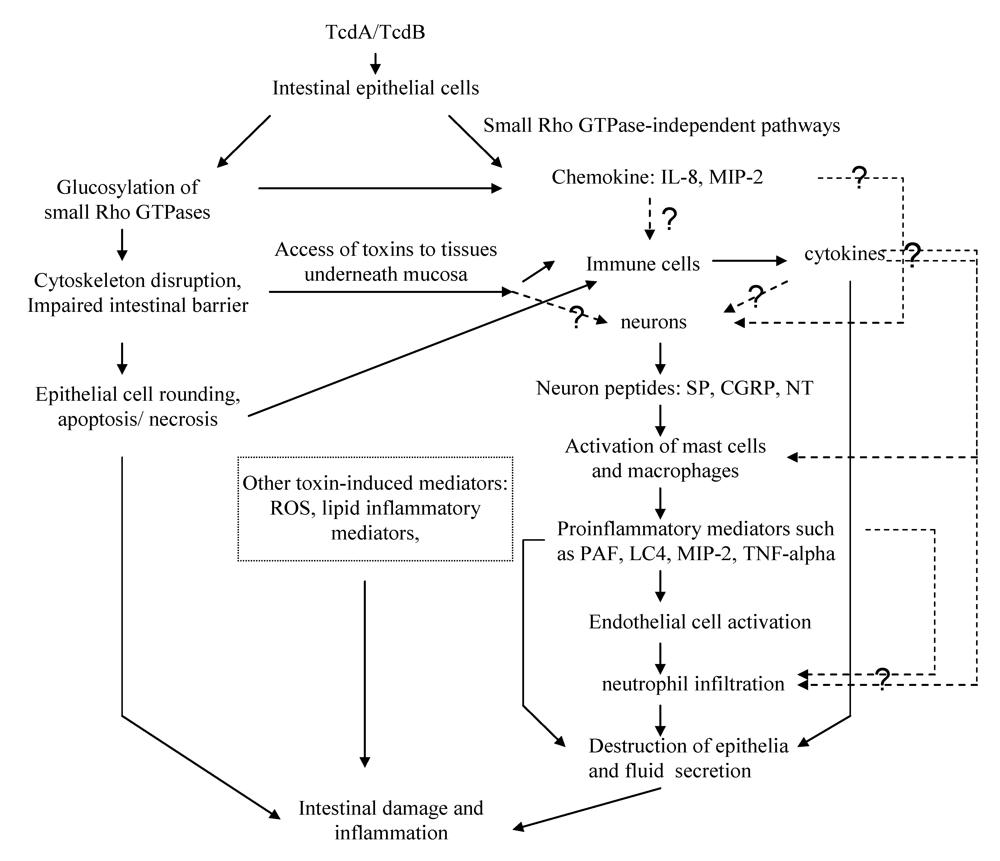
5. The Mechanisms Underlying the Enterotoxicity of TcdA and TcdB

{kind=link}
{kind=link}
| Proinflammatory mediators | Sources (cells or tissue) | Known or proposed functions | References |
|---|---|---|---|
| IL-8 | Intestinal epithelia, macrophages, peripheral blood monocytes | Neutrophil recruitment | [21,23,132] |
| GRO-α (growth-related oncogene) | Intestinal epithelia | Neutrophil recruitment | [21,133] |
| MIP-1 (macrophage inflammatory protein-1) | Intestinal epithelia, macrophages | Neutrophil recruitment | [133,134] |
| MIP-2 (macrophage inflammatory protein-2) | Intestinal epithelia, macrophages | Neutrophil recruitment | [133,134] |
| ENA-78 (epithelial neutrophil-activating peptide-78) | Intestinal epithelia | Neutrophil recruitment | [21,133] |
| MCP-1 (monocytes-chemotactic protein-1) | Intestinal epithelia | Neutrophil recruitment | [21] |
| ICAM-1 (intercellular adhesion molecule-1) | Endothelial cells, neutrophils, epithelia | Neutrophil adhesion to endothelial cells | [135,136,137] |
| IL-1 | Macrophages, dendritic cells, | Neutrophil recruitment, enhancing IL-8 production, | [135,136,138,139] |
| IL-6 | Monocytes, dendritic cells | not specified | [24] |
| TNF-α | Macrophages, monocytes | Neutrophil recruitment | [24,138,139] |
| IFN-γ | Neutrophils | Enhancing chemokine and ICAM-1 expression expression | [134] |
| LB4 (Leukotriene B4) | Macrophages, mast cells | Neutrophil recruitment, activation of transient receptor potential vailloid (TRPV1) and SP release | [138,140,141] |
| LC4 (Leukotriene C4) | Mast cells | Stimulation of fluid secretion by intestinal epithelia | [140] |
| RMCP II (rat mast cell protease II) | Mast cells | Indicator of mast cell activation | [140] |
| SP (substance P) | Intestinal neurons | Activation of mast cells and macrophages | [142,143] |
| CGRP (calcitonin gene-related peptide) | Intestinal neurons | Activation of mast cells and macrophages | [142,143] |
| NT (neurotensin) | Intestinal neurons | Activation of mast cells | [144] |
| VIP (vasoactive intestinal polypeptide) | Colonic submucosal neurons | Activated partially via an IL-1β-dependent pathway ( its role in the intestine is to increase motility | [145] |
| PAR2 (protease activated receptor 2) | Enterocytes, neurons, endothelial cells, neutrophils | Intestinal inflammation | [146] |
| Inflammasome | Macrophages | IL-1β production | [147] |
| Melanin-concentrating hormone (MCH) | Intestinal tissue | Upregulation of IL-8 transcription | [148] |
| Reactive oxygen species (ROS) | Neutrophils | Direct damage of proteins and lipids, induction of IL-8 and ICAM-1 | [149,150,151,152] |
| Cyclooxygenase-2 (COX-2) | Human colonocyte, human intestinal xenograft | Formation of PGE2 | [150,153] |
| Prostaglandin E2 (PGE2) | Human colonocyte, human intestinal xenograft | Intestinal inflammation | [150] |
| Phospholipase A2 | Rabbit ileal tissue, human T-84 cells | Synthesis of inflammatory lipid mediators | [154] |
| Platelet-activating factor (PAF) | Rabbit ileal tissue | Stimulation of fluid secretion by intestinal epithelial cells | [155] |
| Na+/H+ exchanger (NHE) | Intestinal epithelial cells | Involvement in Na+ absorption and fluid homeostasis | [156] |
| Angiotensin II (ANGII) | Rabbit ileal tissues | Regulation of intestinal secretion and absorption | [157] |
| Epidermal growth factor receptor (EGFR) | Human colonic epithelial cells | Activation of IL-8 | [158] |
5.1. Disruption of the tight junctions of epithelial barriers
5.2. Apoptosis and necrosis of epithelial cells and other cell types
5.3. The role of chemokines released by epithelial cells
5.4. Immune cells and proinflammatory cytokines/mediators
5.5. Role of neuronal cells
5.6. Role of other toxin-induced mediators in CDI
5.6.1. Reactive oxygen species (ROS)
5.6.2. Lipid inflammatory mediators
5.6.3. Na+/H+ exchanger (NHE)
5.6.4. Angiotensin II
5.6.5. Epidermal growth factor receptor (EGFR)
6. Concluding Remarks
References
- Cloud, J.; Kelly, C.P. Update on Clostridium difficile associated disease. Curr. Opin. Gastroenterol. 2007, 23, 4–9. [Google Scholar]
- Elliott, B.; Chang, B.J.; Golledge, C.L.; Riley, T.V. Clostridium difficile-associated diarrhoea. Intern. Med. J. 2007, 37, 561–568. [Google Scholar]
- Kelly, C.P.; Pothoulakis, C.; LaMont, J.T. Clostridium difficile colitis. N. Engl. J. Med. 1994, 330, 257–262. [Google Scholar]
- Voth, D.E.; Ballard, J.D. Clostridium difficile toxins: mechanism of action and role in disease. Clin. Microbiol. Rev. 2005, 18, 24–263. [Google Scholar]
- Carter, G.P.; Lyras, D.; Allen, D.L.; Mackin, K.E.; Howarth, P.M.; O'Connor, J.R.; Rood, J.I. Binary toxin production in Clostridium difficile is regulated by CdtR, a LytTR family response regulator. J. Bacteriol. 2007, 189, 7290–7301. [Google Scholar]
- McMaster-Baxter, N.L.; Musher, D.M. Clostridium difficile: recent epidemiologic findings and advances in therapy. Pharmacotherapy 2007, 27, 1029–1039. [Google Scholar]
- Blossom, D.B.; McDonald, L.C. The challenges posed by reemerging Clostridium difficile infection. Clin. Infect. Dis. 2007, 45, 222–227. [Google Scholar]
- Stare, B.G.; Delmee, M.; Rupnik, M. Variant forms of the binary toxin CDT locus and tcdC gene in Clostridium difficile strains. J. Med. Microbiol. 2007, 56, 329–335. [Google Scholar]
- Geric, B.; Carman, R.J.; Rupnik, M.; Genheimer, C.W.; Sambol, S.P.; Lyerly, D.M.; Gerding, D.N.; Johnson, S. Binary toxin-producing, large clostridial toxin-negative Clostridium difficile strains are enterotoxic but do not cause disease in hamsters. J. Infect. Dis. 2006, 193, 1143–1150. [Google Scholar]
- Schwan, C.; Stecher, B.; Tzivelekidis, T.; van Ham, M.; Rohde, M.; Hardt, W.D.; Wehland, J.; Aktories, K. Clostridium difficile toxin CDT induces formation of microtubule-based protrusions and increases adherence of bacteria. PLoS Pathog. 2009, 5, e1000626. [Google Scholar]
- Borriello, S.P.; Davies, H.A.; Kamiya, S.; Reed, P.J.; Seddon, S. Virulence factors of Clostridium difficile. Rev. Infect. Dis. 1990, 12, S185–S191, (Suppl. 2).. [Google Scholar] [CrossRef] [PubMed]
- Seddon, S.V.; Hemingway, I.; Borriello, S.P. Hydrolytic enzyme production by Clostridium difficile and its relationship to toxin production and virulence in the hamster model. J. Med. Microbiol. 1990, 31, 169–174. [Google Scholar]
- Borriello, S.P. Pathogenesis of Clostridium difficile infection. J. Antimicrob. Chemother. 1998, 41, 13–19, (Suppl. C).. [Google Scholar] [CrossRef]
- Calabi, E.; Calabi, F.; Phillips, A.D.; Fairweather, N.F. Binding of Clostridium difficile surface layer proteins to gastrointestinal tissues. Infect. Immun. 2002, 70, 5770–5778. [Google Scholar]
- O'Brien, J.B.; McCabe, M.S.; Athie-Morales, V.; McDonald, G.S.; Ni Eidhin, D.B.; Kelleher, D.P. Passive immunisation of hamsters against Clostridium difficile infection using antibodies to surface layer proteins. FEMS Microbiol. Lett. 2005, 246, 199–205. [Google Scholar]
- Halabi-Cabezon, I.; Huelsenbeck, J.; May, M.; Ladwein, M.; Rottner, K.; Just, I.; Genth, H. Prevention of the cytopathic effect induced by Clostridium difficile Toxin B by active Rac1. FEBS Lett. 2008, 582, 3751–3756. [Google Scholar]
- Gerhard, R.; Nottrott, S.; Schoentaube, J.; Tatge, H.; Olling, A.; Just, I. Glucosylation of Rho GTPases by Clostridium difficile toxin A triggers apoptosis in intestinal epithelial cells. J. Med. Microbiol. 2008, 57, 765–770. [Google Scholar]
- Brito, G.A.; Fujji, J.; Carneiro-Filho, B.A.; Lima, A.A.; Obrig, T.; Guerrant, R.L. Mechanism of Clostridium difficile toxin A-induced apoptosis in T84 cells. J. Infect. Dis. 2002, 186, 1438–1447. [Google Scholar]
- Qa'Dan, M.; Ramsey, M.; Daniel, J.; Spyres, L.M.; Safiejko-Mroczka, B.; Ortiz-Leduc, W.; Ballard, J.D. Clostridium difficile toxin B activates dual caspase-dependent and caspase-independent apoptosis in intoxicated cells. Cell. Microbiol. 2002, 4, 425–434. [Google Scholar]
- Savidge, T.C.; Pan, W.H.; Newman, P.; O'Brien, M.; Anton, P.M.; Pothoulakis, C. Clostridium difficile toxin B is an inflammatory enterotoxin in human intestine. Gastroenterology 2003, 125, 413–420. [Google Scholar]
- Kim, J.M.; Kim, J.S.; Jun, H.C.; Oh, Y.K.; Song, I.S.; Kim, C.Y. Differential expression and polarized secretion of CXC and CC chemokines by human intestinal epithelial cancer cell lines in response to Clostridium difficile toxin A. Microbiol. Immunol. 2002, 46, 333–342. [Google Scholar]
- Ng, E.K.; Panesar, N.; Longo, W.E.; Shapiro, M.J.; Kaminski, D.L.; Tolman, K.C.; Mazuski, J.E. Human intestinal epithelial and smooth muscle cells are potent producers of IL-6. Mediators Inflamm. 2003, 12, 3–8. [Google Scholar]
- Linevsky, J.K.; Pothoulakis, C.; Keates, S.; Warny, M.; Keates, A.C.; Lamont, J.T.; Kelly, C.P. IL-8 release and neutrophil activation by Clostridium difficile toxin-exposed human monocytes. Am. J. Physiol. 1997, 273, G1333–G1340. [Google Scholar]
- Flegel, W.A.; Muller, F.; Daubener, W.; Fischer, H.G.; Hadding, U.; Northoff, H. Cytokine response by human monocytes to Clostridium difficile toxin A and toxin B. Infect. Immun. 1991, 59, 3659–3666. [Google Scholar]
- Kelly, C.P.; Becker, S.; Linevsky, J.K.; Joshi, M.A.; O'Keane, J.C.; Dickey, B.F.; LaMont, J.T.; Pothoulakis, C. Neutrophil recruitment in Clostridium difficile toxin A enteritis in the rabbit. J. Clin. Invest. 1994, 93, 1257–1265. [Google Scholar]
- Pothoulakis, C.; Lamont, J.T. Microbes and microbial toxins: paradigms for microbial-mucosal interactions II. The integrated response of the intestine to Clostridium difficile toxins. Am. J. Physiol. Gastrointest. Liver Physiol. 2001, 280, G178–G183. [Google Scholar]
- Lyerly, D.M.; Lockwood, D.E.; Richardson, S.H.; Wilkins, T.D. Biological activities of toxins A and B of Clostridium difficile. Infect. Immun. 1982, 35, 1147–1150. [Google Scholar]
- Lyerly, D.M.; Saum, K.E.; MacDonald, D.K.; Wilkins, T.D. Effects of Clostridium difficile toxins given intragastrically to animals. Infect. Immun. 1985, 47, 349–352. [Google Scholar]
- Riegler, M.; Sedivy, R.; Pothoulakis, C.; Hamilton, G.; Zacherl, J.; Bischof, G.; Cosentini, E.; Feil, W.; Schiessel, R.; LaMont, J.T.; et al. Clostridium difficile toxin B is more potent than toxin A in damaging human colonic epithelium in vitro. J. Clin. Invest. 1995, 95, 2004–2011. [Google Scholar]
- Lyras, D.; O'Connor, J.R.; Howarth, P.M.; Sambol, S.P.; Carter, G.P.; Phumoonna, T.; Poon, R.; Adams, V.; Vedantam, G.; Johnson, S.; Gerding, D.N.; Rood, J.I. Toxin B is essential for virulence of Clostridium difficile. Nature 2009, 458, 1176–1179. [Google Scholar]
- Shin, B.M.; Kuak, E.Y.; Yoo, S.J.; Shin, W.C.; Yoo, H.M. Emerging toxin A-B+ variant strain of Clostridium difficile responsible for pseudomembranous colitis at a tertiary care hospital in Korea. Diagn. Microbiol. Infect. Dis. 2008, 60, 333–337. [Google Scholar]
- Hamm, E.E.; Voth, D.E.; Ballard, J.D. Identification of Clostridium difficile toxin B cardiotoxicity using a zebrafish embryo model of intoxication. Proc. Natl. Acad. Sci. USA 2006, 103, 14176–14181. [Google Scholar]
- Pavliakova, D.; Moncrief, J.S.; Lyerly, D.M.; Schiffman, G.; Bryla, D.A.; Robbins, J.B.; Schneerson, R. Clostridium difficile recombinant toxin A repeating units as a carrier protein for conjugate vaccines: studies of pneumococcal type 14, Escherichia coli K1, and Shigella flexneri type 2a polysaccharides in mice. Infect. Immun. 2000, 68, 2161–2166. [Google Scholar]
- He, X.; Wang, J.; Steele, J.; Sun, X.; Nie, W.; Tzipori, S.; Feng, H. An ultrasensitive rapid immunocytotoxicity assay for detecting Clostridium difficile toxins. J. Microbiol. Methods 2009, 78, 97–100. [Google Scholar]
- Steele, J.; Feng, H.; Parry, N.; Tzipori, S. Piglet models of acute or chronic Clostridium difficile illness. J. Infect. Dis. 2010, 201, 428–434. [Google Scholar]
- Roberts, K.; Smith, C.F.; Snelling, A.M.; Kerr, K.G.; Banfield, K.R.; Sleigh, P.A.; Beggs, C.B. Aerial dissemination of Clostridium difficile spores. BMC Infect. Dis. 2008, 8, 7. [Google Scholar]
- Dubberke, E.R.; Reske, K.A.; Noble-Wang, J.; Thompson, A.; Killgore, G.; Mayfield, J.; Camins, B.; Woeltje, K.; McDonald, J.R.; McDonald, L.C.; Fraser, V.J. Prevalence of Clostridium difficile environmental contamination and strain variability in multiple health care facilities. Am. J. Infect. Control 2007, 35, 315–318. [Google Scholar]
- Bartlett, J.G. Narrative review: the new epidemic of Clostridium difficile-associated enteric disease. Ann. Intern. Med. 2006, 145, 758–764. [Google Scholar]
- Johnson, S.; Kent, S.A.; O'Leary, K.J.; Merrigan, M.M.; Sambol, S.P.; Peterson, L.R.; Gerding, D.N. Fatal pseudomembranous colitis associated with a variant Clostridium difficile strain not detected by toxin A immunoassay. Ann. Intern. Med. 2001, 135, 434–438. [Google Scholar]
- Jacob, S.S.; Sebastian, J.C.; Hiorns, D.; Jacob, S.; Mukerjee, P.K. Clostridium difficile and acute respiratory distress syndrome. Heart Lung 2004, 33, 265–268. [Google Scholar]
- Dobson, G.; Hickey, C.; Trinder, J. Clostridium difficile colitis causing toxic megacolon, severe sepsis and multiple organ dysfunction syndrome. Intensive Care Med. 2003, 29, 1030. [Google Scholar]
- Cunney, R.J.; Magee, C.; McNamara, E.; Smyth, E.G.; Walshe, J. Clostridium difficile colitis associated with chronic renal failure. Nephrol. Dial. Transplant. 1998, 13, 2842–2846. [Google Scholar]
- Sakurai, T.; Hajiro, K.; Takakuwa, H.; Nishi, A.; Aihara, M.; Chiba, T. Liver abscess caused by Clostridium difficile. Scand. J. Infect. Dis. 2001, 33, 69–70. [Google Scholar]
- Zar, F.; Bakkanagari, S.; Moorthi, K.M.L.S.T.; Davis, M. A Comparison of Vancomycin and Metronidazole for the Treatment of Clostridium difficile—Associated Diarrhea, Stratified by Disease Severity. Clin. Infect. Dis. 2007, 45, 302–307. [Google Scholar]
- Barbut, F.; Richard, A.; Hamadi, K.; Chomette, V.; Burghoffer, B.; Petit, J.C. Epidemiology of recurrences or reinfections of Clostridium difficile-associated diarrhea. J. Clin. Microbiol. 2000, 38, 2386–2388. [Google Scholar]
- Tonna, I.; Welsby, P.D. Pathogenesis and treatment of Clostridium difficile infection. Postgrad. Med. J. 2005, 81, 367–369. [Google Scholar]
- McVay, C.S.; Rolfe, R.D. In vitro and in vivo activities of nitazoxanide against Clostridium difficile. Antimicrob. Agents Chemother. 2000, 44, 2254–2258. [Google Scholar]
- Anton, P.M.; O'Brien, M.; Kokkotou, E.; Eisenstein, B.; Michaelis, A.; Rothstein, D.; Paraschos, S.; Kelly, C.P.; Pothoulakis, C. Rifalazil treats and prevents relapse of Clostridium difficile-associated diarrhea in hamsters. Antimicrob. Agents Chemother. 2004, 48, 3975–3979. [Google Scholar]
- Hinkson, P.L.; Dinardo, C.; DeCiero, D.; Klinger, J.D.; Barker, R.H., Jr. Tolevamer, an anionic polymer, neutralizes toxins produced by the BI/027 strains of Clostridium difficile. Antimicrob. Agents Chemother. 2008, 52, 2190–2195. [Google Scholar]
- Taylor, C.P.; Tummala, S.; Molrine, D.; Davidson, L.; Farrell, R.J.; Lembo, A.; Hibberd, P.L.; Lowy, I.; Kelly, C.P. Open-label, dose escalation phase I study in healthy volunteers to evaluate the safety and pharmacokinetics of a human monoclonal antibody to Clostridium difficile toxin A. Vaccine 2008, 26, 3404–3409. [Google Scholar]
- Kotloff, K.L.; Wasserman, S.S.; Losonsky, G.A.; Thomas, W., Jr.; Nichols, R.; Edelman, R.; Bridwell, M.; Monath, T.P. Safety and immunogenicity of increasing doses of a Clostridium difficile toxoid vaccine administered to healthy adults. Infect. Immun. 2001, 69, 988–995. [Google Scholar]
- Sougioultzis, S.; Kyne, L.; Drudy, D.; Keates, S.; Maroo, S.; Pothoulakis, C.; Giannasca, P.J.; Lee, C.K.; Warny, M.; Monath, T.P.; Kelly, C.P. Clostridium difficile toxoid vaccine in recurrent C. difficile-associated diarrhea. Gastroenterology 2005, 128, 764–770. [Google Scholar] [CrossRef] [PubMed]
- Archibald, L.K.; Banerjee, S.N.; Jarvis, W.R. Secular trends in hospital-acquired Clostridium difficile disease in the United States, 1987–2001. J. Infect. Dis. 2004, 189, 1585–1589. [Google Scholar]
- Loo, V.G.; Poirier, L.; Miller, M.A.; Oughton, M.; Libman, M.D.; Michaud, S.; Bourgault, A.M.; Nguyen, T.; Frenette, C.; Kelly, M.; Vibien, A.; Brassard, P.; Fenn, S.; Dewar, K.; Hudson, T.J.; Horn, R.; Rene, P.; Monczak, Y.; Dascal, A. A predominantly clonal multi-institutional outbreak of Clostridium difficile-associated diarrhea with high morbidity and mortality. N. Engl. J. Med. 2005, 353, 2442–2449. [Google Scholar]
- McDonald, L.C.; Killgore, G.E.; Thompson, A.; Owens, R.C., Jr.; Kazakova, S.V.; Sambol, S.P.; Johnson, S.; Gerding, D.N. An epidemic, toxin gene-variant strain of Clostridium difficile. N. Engl. J. Med. 2005, 353, 2433–2441. [Google Scholar]
- Larson, H.E.; Barclay, F.E.; Honour, P.; Hill, I.D. Epidemiology of Clostridium difficile in infants. J. Infect. Dis. 1982, 146, 727–733. [Google Scholar]
- Al-Jumaili, I.J.; Shibley, M.; Lishman, A.H.; Record, C.O. Incidence and origin of Clostridium difficile in neonates. J. Clin. Microbiol. 1984, 19, 77–78. [Google Scholar]
- Boenning, D.A.; Fleisher, G.R.; Campos, J.M.; Hulkower, C.W.; Quinlan, R.W. Clostridium difficile in a pediatric outpatient population. Pediatr. Infect. Dis. 1982, 1, 336–338. [Google Scholar]
- Bolton, R.P.; Tait, S.K.; Dear, P.R.; Losowsky, M.S. Asymptomatic neonatal colonisation by Clostridium difficile. Arch. Dis. Child. 1984, 59, 466–472. [Google Scholar]
- Collignon, A.; Cotte-Laffitte, J.; Quero, A.M.; Torlotin, J.C. Clostridium difficile and its cytotoxin in the stools of young hospitalized children. Influence of antibiotic treatment. Pathol. Biol. (Paris) 1986, 34, 977–982. [Google Scholar] [PubMed]
- Libby, J.M.; Donta, S.T.; Wilkins, T.D. Clostridium difficile toxin A in infants. J. Infect. Dis. 1983, 148, 606. [Google Scholar]
- Chang, T.W.; Sullivan, N.M.; Wilkins, T.D. Insusceptibility of fetal intestinal mucosa and fetal cells to Clostridium difficile toxins. Acta Pharmacol. Sin. 1986, 7, 448–453. [Google Scholar]
- Giesemann, T.; Guttenberg, G.; Aktories, K. Human alpha-defensins inhibit Clostridium difficile toxin B. Gastroenterology 2008, 134, 2049–2058. [Google Scholar]
- Abrams, G.D.; Allo, M.; Rifkin, G.D.; Fekety, R.; Silva, J., Jr. Mucosal damage mediated by clostridial toxin in experimental clindamycin-associated colitis. Gut 1980, 21, 493–499. [Google Scholar]
- Czuprynski, C.J.; Johnson, W.J.; Balish, E.; Wilkins, T. Pseudomembranous colitis in Clostridium difficile-monoassociated rats. Infect. Immun. 1983, 39, 1368–1376. [Google Scholar]
- Fekety, R.; Silva, J.; Toshniwal, R.; Allo, M.; Armstrong, J.; Browne, R.; Ebright, J.; Rifkin, G. Antibiotic-associated colitis: effects of antibiotics on Clostridium difficile and the disease in hamsters. Rev. Infect. Dis. 1979, 1, 386–397. [Google Scholar]
- Knoop, F.C. Clindamycin-associated enterocolitis in guinea pigs: evidence for a bacterial toxin. Infect. Immun. 1979, 23, 31–33. [Google Scholar]
- Chang, T.W.; Bartlett, J.G.; Gorbach, S.L.; Onderdonk, A.B. Clindamycin-induced enterocolitis in hamsters as a model of pseudomembranous colitis in patients. Infect. Immun. 1978, 20, 526–529. [Google Scholar]
- Corthier, G.; Dubos, F.; Raibaud, P. Modulation of cytotoxin production by Clostridium difficile in the intestinal tracts of gnotobiotic mice inoculated with various human intestinal bacteria. Appl. Environ. Microbiol. 1985, 49, 250–252. [Google Scholar]
- Lusk, R.H.; Fekety, R.; Silva, J.; Browne, R.A.; Ringler, D.H.; Abrams, G.D. Clindamycin-induced enterocolitis in hamsters. J. Infect. Dis. 1978, 137, 464–475. [Google Scholar]
- Price, A.B.; Larson, H.E.; Crow, J. Morphology of experimental antibiotic-associated enterocolitis in the hamster: a model for human pseudomembranous colitis and antibiotic-associated diarrhoea. Gut 1979, 20, 467–475. [Google Scholar]
- Rehg, J.E.; Pakes, S.P. Implication of Clostridium difficile and Clostridium perfringens iota toxins in experimental lincomycin-associated colitis of rabbits. Lab. Anim. Sci. 1982, 32, 253–257. [Google Scholar]
- Sugiyama, T.; Mukai, M.; Yamashita, R.; Sunakawa, K. Experimental models of Clostridium difficile enterocolitis in gnotobiotic mice. Prog. Clin. Biol. Res. 1985, 181, 203–206. [Google Scholar]
- Chen, X.; Katchar, K.; Goldsmith, J.D.; Nanthakumar, N.; Cheknis, A.; Gerding, D.N.; Kelly, C.P. A mouse model of Clostridium difficile-associated disease. Gastroenterology 2008, 135, 1984–1992. [Google Scholar]
- Keel, M.K.; Songer, J.G. The comparative pathology of Clostridium difficile-associated disease. Vet. Pathol. 2006, 43, 225–240. [Google Scholar]
- Rolfe, R.D.; Iaconis, J.P. Intestinal colonization of infant hamsters with Clostridium difficile. Infect. Immun. 1983, 42, 480–486. [Google Scholar]
- Lyerly, D.M.; Krivan, H.C.; Wilkins, T.D. Clostridium difficile: its disease and toxins. Clin. Microbiol. Rev. 1988, 1, 1–18. [Google Scholar]
- Mitchell, T.J.; Ketley, J.M.; Haslam, S.C.; Stephen, J.; Burdon, D.W.; Candy, D.C.; Daniel, R. Effect of toxin A and B of Clostridium difficile on rabbit ileum and colon. Gut 1986, 27, 78–85. [Google Scholar]
- Lima, A.A.; Lyerly, D.M.; Wilkins, T.D.; Innes, D.J.; Guerrant, R.L. Effects of Clostridium difficile toxins A and B in rabbit small and large intestine in vivo and on cultured cells in vitro. Infect. Immun. 1988, 56, 582–588. [Google Scholar]
- Bongaerts, G.P.; Lyerly, D.M. Role of toxins A and B in the pathogenesis of Clostridium difficile disease. Infect. Immun. 1994, 17, 1–12. [Google Scholar]
- Ketley, J.M.; Mitchell, T.J.; Candy, D.C.; Burdon, D.W.; Stephen, J. The effects of Clostridium difficile crude toxins and toxin A on ileal and colonic loops in immune and non-immune rabbits. J. Med. Microbiol. 1987, 24, 41–52. [Google Scholar]
- Kim, P.H.; Iaconis, J.P.; Rolfe, R.D. Immunization of adult hamsters against Clostridium difficile-associated ileocecitis and transfer of protection to infant hamsters. Infect. Immun. 1987, 55, 2984–2992. [Google Scholar]
- Corthier, G.; Muller, M.C.; Wilkins, T.D.; Lyerly, D.; L'Haridon, R. Protection against experimental pseudomembranous colitis in gnotobiotic mice by use of monoclonal antibodies against Clostridium difficile toxin A. Infect. Immun. 1991, 59, 1192–1195. [Google Scholar]
- Leav, B.A.; Blair, B.; Leney, M.; Knauber, M.; Reilly, C.; Lowy, I.; Gerding, D.N.; Kelly, C.P.; Katchar, K.; Baxter, R.; Ambrosino, D.; Molrine, D. Serum anti-toxin B antibody correlates with protection from recurrent Clostridium difficile infection (CDI). Vaccine 2010, 28, 965–969. [Google Scholar]
- Kyne, L.; Warny, M.; Qamar, A.; Kelly, C.P. Asymptomatic carriage of Clostridium difficile and serum levels of IgG antibody against toxin A. N. Engl. J. Med. 2000, 342, 390–397. [Google Scholar]
- Bacon, A.E., III; Fekety, R. Immunoglobulin G directed against toxins A and B of Clostridium difficile in the general population and patients with antibiotic-associated diarrhea. Diagn. Microbiol. Infect. Dis. 1994, 18, 205–209. [Google Scholar] [CrossRef] [PubMed]
- Libby, J.M.; Jortner, B.S.; Wilkins, T.D. Effects of the two toxins of Clostridium difficile in antibiotic-associated cecitis in hamsters. Infect. Immun. 1982, 36, 822–829. [Google Scholar]
- Lonnroth, I.; Lange, S. Toxin A of Clostridium difficile: production, purification and effect in mouse intestine. Acta Pathol. Microbiol. Immunol. Scand. B 1983, 91, 395–400. [Google Scholar]
- von Eichel-Streiber, C.; Boquet, P.; Sauerborn, M.; Thelestam, M. Large clostridial cytotoxins—a family of glycosyltransferases modifying small GTP-binding proteins. Trends Microbiol. 1996, 4, 375–382. [Google Scholar]
- Drudy, D.; Fanning, S.; Kyne, L. Toxin A-negative, toxin B-positive Clostridium difficile. Int. J. Infect. Dis. 2007, 11, 5–10. [Google Scholar]
- Lawley, T.D.; Clare, S.; Walker, A.W.; Goulding, D.; Stabler, R.A.; Croucher, N.; Mastroeni, P.; Scott, P.; Raisen, C.; Mottram, L.; Fairweather, N.F.; Wren, B.W.; Parkhill, J.; Dougan, G. Antibiotic treatment of Clostridium difficile carrier mice triggers a supershedder state, spore-mediated transmission, and severe disease in immunocompromised hosts. Infect. Immun. 2009, 77, 3661–3669. [Google Scholar]
- Lyerly, D.M.; Barroso, L.A.; Wilkins, T.D.; Depitre, C.; Corthier, G. Characterization of a toxin A-negative, toxin B-positive strain of Clostridium difficile. Infect. Immun. 1992, 60, 4633–4639. [Google Scholar]
- Borriello, S.P.; Wren, B.W.; Hyde, S.; Seddon, S.V.; Sibbons, P.; Krishna, M.M.; Tabaqchali, S.; Manek, S.; Price, A.B. Molecular, immunological, and biological characterization of a toxin A-negative, toxin B-positive strain of Clostridium difficile. Infect. Immun. 1992, 60, 4192–4199. [Google Scholar]
- Kink, J.A.; Williams, J.A. Antibodies to recombinant Clostridium difficile toxins A and B are an effective treatment and prevent relapse of C. difficile-associated disease in a hamster model of infection. Infect. Immun. 1998, 66, 2018–2025. [Google Scholar] [PubMed]
- Babcock, G.J.; Broering, T.J.; Hernandez, H.J.; Mandell, R.B.; Donahue, K.; Boatright, N.; Stack, A.M.; Lowy, I.; Graziano, R.; Molrine, D.; Ambrosino, D.M.; Thomas, W.D., Jr. Human monoclonal antibodies directed against toxins A and B prevent Clostridium difficile-induced mortality in hamsters. Infect. Immun. 2006, 74, 6339–6347. [Google Scholar]
- Eglow, R.; Pothoulakis, C.; Itzkowitz, S.; Israel, E.J.; O'Keane, C.J.; Gong, D.; Gao, N.; Xu, Y.L.; Walker, W.A.; LaMont, J.T. Diminished Clostridium difficile toxin A sensitivity in newborn rabbit ileum is associated with decreased toxin A receptor. J. Clin. Invest. 1992, 90, 822–829. [Google Scholar]
- Rolfe, R.D. Binding kinetics of Clostridium difficile toxins A and B to intestinal brush border membranes from infant and adult hamsters. Infect. Immun. 1991, 59, 1223–1230. [Google Scholar]
- Taylor, N.S.; Thorne, G.M.; Bartlett, J.G. Comparison of two toxins produced by Clostridium difficile. Infect. Immun. 1981, 34, 1036–1043. [Google Scholar]
- Sutton, P.A.; Li, S.; Webb, J.; Solomon, K.; Brazier, J.; Mahida, Y.R. Essential role of toxin A in C. difficile 027 and reference strain supernatant-mediated disruption of Caco-2 intestinal epithelial barrier function. Clin. Exp. Immunol. 2008, 153, 439–447. [Google Scholar] [PubMed]
- Just, I.; Gerhard, R. Large clostridial cytotoxins. Rev. Physiol. Biochem. Pharmacol. 2004, 152, 23–47. [Google Scholar]
- Jank, T.; Aktories, K. Structure and mode of action of clostridial glucosylating toxins: the ABCD model. Trends Microbiol. 2008, 16, 222–229. [Google Scholar]
- Ho, J.G.; Greco, A.; Rupnik, M.; Ng, K.K. Crystal structure of receptor-binding C-terminal repeats from Clostridium difficile toxin A. Proc. Natl. Acad. Sci. USA 2005, 102, 18373–18378. [Google Scholar]
- Greco, A.; Ho, J.G.; Lin, S.J.; Palcic, M.M.; Rupnik, M.; Ng, K.K. Carbohydrate recognition by Clostridium difficile toxin A. Nat. Struct. Mol. Biol. 2006, 13, 460–461. [Google Scholar]
- Reinert, D.J.; Jank, T.; Aktories, K.; Schulz, G.E. Structural basis for the function of Clostridium difficile toxin B. J. Mol. Biol. 2005, 351, 973–981. [Google Scholar]
- Pruitt, R.N.; Chagot, B.; Cover, M.; Chazin, W.J.; Spiller, B.; Lacy, D.B. Structure-function analysis of inositol hexakisphosphate-induced autoprocessing in Clostridium difficile toxin A. J. Biol. Chem. 2009, 284, 21934–21940. [Google Scholar]
- Albesa-Jove, D.; Bertrand, T.; Carpenter, E.P.; Swain, G.V.; Lim, J.; Zhang, J.; Haire, L.F.; Vasisht, N.; Braun, V.; Lange, A.; von Eichel-Streiber, C.; Svergun, D.I.; Fairweather, N.F.; Brown, K.A. Four distinct structural domains in Clostridium difficile toxin B visualized using SAXS. J. Mol. Biol. 2010, 396, 1260–1270. [Google Scholar]
- Krivan, H.C.; Clark, G.F.; Smith, D.F.; Wilkins, T.D. Cell surface binding site for Clostridium difficile enterotoxin: evidence for a glycoconjugate containing the sequence Gal alpha 1-3Gal beta 1-4GlcNAc. Infect. Immun. 1986, 53, 573–581. [Google Scholar]
- Tucker, K.D.; Wilkins, T.D. Toxin A of Clostridium difficile binds to the human carbohydrate antigens I, X, and Y. Infect. Immun. 1991, 59, 73–78. [Google Scholar]
- Rolfe, R.D.; Song, W. Purification of a functional receptor for Clostridium difficile toxin A from intestinal brush border membranes of infant hamsters. Clin. Infect. Dis. 1993, 16, S219–S227, (Suppl. 4).. [Google Scholar] [PubMed]
- Teneberg, S.; Lonnroth, I.; Torres Lopez, J.F.; Galili, U.; Halvarsson, M.O.; Angstrom, J.; Karlsson, K.A. Molecular mimicry in the recognition of glycosphingolipids by Gal alpha 3 Gal beta 4 GlcNAc beta-binding Clostridium difficile toxin A, human natural anti alpha-galactosyl IgG and the monoclonal antibody Gal-13: characterization of a binding-active human glycosphingolipid, non-identical with the animal receptor. Glycobiology 1996, 6, 599–609. [Google Scholar] [CrossRef] [PubMed]
- Koike, T.; Kuzuya, M.; Asai, T.; Kanda, S.; Cheng, X.W.; Watanabe, K.; Banno, Y.; Nozawa, Y.; Iguchi, A. Activation of MMP-2 by Clostridium difficile toxin B in bovine smooth muscle cells. Biochem. Biophys. Res. Commun. 2000, 277, 43–46. [Google Scholar]
- Jank, T.; Giesemann, T.; Aktories, K. Rho-glucosylating Clostridium difficile toxins A and B: new insights into structure and function. Glycobiology 2007, 17, 15R–22R. [Google Scholar]
- Na, X.; Kim, H.; Moyer, M.P.; Pothoulakis, C.; LaMont, J.T. gp96 is a human colonocyte plasma membrane binding protein for Clostridium difficile toxin A. Infect. Immun. 2008, 76, 2862–2871. [Google Scholar]
- Stubbe, H.; Berdoz, J.; Kraehenbuhl, J.P.; Corthesy, B. Polymeric IgA is superior to monomeric IgA and IgG carrying the same variable domain in preventing Clostridium difficile toxin A damaging of T84 monolayers. J. Immunol. 2000, 164, 1952–1960. [Google Scholar]
- Reineke, J.; Tenzer, S.; Rupnik, M.; Koschinski, A.; Hasselmayer, O.; Schrattenholz, A.; Schild, H.; von Eichel-Streiber, C. Autocatalytic cleavage of Clostridium difficile toxin B. Nature 2007, 446, 415–419. [Google Scholar]
- Karlsson, K.A. Microbial recognition of target-cell glycoconjugates. Curr. Opin. Struct. Biol. 1995, 5, 622–635. [Google Scholar]
- Florin, I.; Thelestam, M. Internalization of Clostridium difficile cytotoxin into cultured human lung fibroblasts. Biochim. Biophys. Acta 1983, 763, 383–392. [Google Scholar]
- Florin, I.; Thelestam, M. Lysosomal involvement in cellular intoxication with Clostridium difficile toxin B. Microb. Pathog. 1986, 1, 373–385. [Google Scholar]
- Henriques, B.; Florin, I.; Thelestam, M. Cellular internalisation of Clostridium difficile toxin A. Microb. Pathog. 1987, 2, 455–463. [Google Scholar]
- Giesemann, T.; Jank, T.; Gerhard, R.; Maier, E.; Just, I.; Benz, R.; Aktories, K. Cholesterol-dependent pore formation of Clostridium difficile toxin A. J. Biol. Chem. 2006, 281, 10808–10815. [Google Scholar]
- Qa'Dan, M.; Spyres, L.M.; Ballard, J.D. pH-induced conformational changes in Clostridium difficile toxin B. Infect. Immun. 2000, 68, 2470–2474. [Google Scholar]
- Jank, T.; Giesemann, T.; Aktories, K. Clostridium difficile glucosyltransferase toxin B-essential amino acids for substrate binding. J. Biol. Chem. 2007, 282, 35222–35231. [Google Scholar]
- Just, I.; Selzer, J.; Wilm, M.; von Eichel-Streiber, C.; Mann, M.; Aktories, K. Glucosylation of Rho proteins by Clostridium difficile toxin B. Nature 1995, 375, 500–503. [Google Scholar]
- Sehr, P.; Joseph, G.; Genth, H.; Just, I.; Pick, E.; Aktories, K. Glucosylation and ADP ribosylation of rho proteins: effects on nucleotide binding, GTPase activity, and effector coupling. Biochemistry 1998, 37, 5296–5304. [Google Scholar]
- Chaves-Olarte, E.; Freer, E.; Parra, A.; Guzman-Verri, C.; Moreno, E.; Thelestam, M. R-Ras glucosylation and transient RhoA activation determine the cytopathic effect produced by toxin B variants from toxin A-negative strains of Clostridium difficile. J. Biol. Chem. 2003, 278, 7956–7963. [Google Scholar]
- Soehn, F.; Wagenknecht-Wiesner, A.; Leukel, P.; Kohl, M.; Weidmann, M.; von Eichel-Streiber, C.; Braun, V. Genetic rearrangements in the pathogenicity locus of Clostridium difficile strain 8864—implications for transcription, expression and enzymatic activity of toxins A and B. Mol. Gen. Genet. 1998, 258, 222–232. [Google Scholar]
- Mehlig, M.; Moos, M.; Braun, V.; Kalt, B.; Mahony, D.E.; von Eichel-Streiber, C. Variant toxin B and a functional toxin A produced by Clostridium difficile C34. FEMS Microbiol. Lett. 2001, 198, 171–176. [Google Scholar]
- Rupnik, M. Heterogeneity of large clostridial toxins: importance of Clostridium difficile toxinotypes. FEMS Microbiol. Rev. 2008, 32, 541–555. [Google Scholar]
- Goncalves, C.; Decre, D.; Barbut, F.; Burghoffer, B.; Petit, J.C. Prevalence and characterization of a binary toxin (actin-specific ADP-ribosyltransferase) from Clostridium difficile. J. Clin. Microbiol. 2004, 42, 1933–1939. [Google Scholar]
- Geric, B.; Johnson, S.; Gerding, D.N.; Grabnar, M.; Rupnik, M. Frequency of binary toxin genes among Clostridium difficile strains that do not produce large clostridial toxins. J. Clin. Microbiol. 2003, 41, 5227–5232. [Google Scholar]
- Sundriyal, A.; Roberts, A.K.; Shone, C.C.; Acharya, K.R. Structural basis for substrate recognition in the enzymatic component of ADP-ribosyltransferase toxin CDTa from Clostridium difficile. J. Biol. Chem. 2009, 284, 28713–28719. [Google Scholar]
- Mahida, Y.R.; Makh, S.; Hyde, S.; Gray, T.; Borriello, S.P. Effect of Clostridium difficile toxin A on human intestinal epithelial cells: induction of interleukin 8 production and apoptosis after cell detachment. Gut 1996, 38, 337–347. [Google Scholar]
- Castagliuolo, I.; Keates, A.C.; Wang, C.C.; Pasha, A.; Valenick, L.; Kelly, C.P.; Nikulasson, S.T.; LaMont, J.T.; Pothoulakis, C. Clostridium difficile toxin A stimulates macrophage-inflammatory protein-2 production in rat intestinal epithelial cells. J. Immunol. 1998, 160, 6039–6045. [Google Scholar]
- Ishida, Y.; Maegawa, T.; Kondo, T.; Kimura, A.; Iwakura, Y.; Nakamura, S.; Mukaida, N. Essential involvement of IFN-gamma in Clostridium difficile toxin A-induced enteritis. J. Immunol. 2004, 172, 3018–3025. [Google Scholar]
- Tixier, E.; Lalanne, F.; Just, I.; Galmiche, J.P.; Neunlist, M. Human mucosa/submucosa interactions during intestinal inflammation: involvement of the enteric nervous system in interleukin-8 secretion. Cell. Microbiol. 2005, 7, 1798–1810. [Google Scholar]
- Kelly, C.P.; Keates, S.; Siegenberg, D.; Linevsky, J.K.; Pothoulakis, C.; Brady, H.R. IL-8 secretion and neutrophil activation by HT-29 colonic epithelial cells. Am. J. Physiol. 1994, 267, G991–G997. [Google Scholar]
- Canny, G.; Drudy, D.; Macmathuna, P.; O'Farrelly, C.; Baird, A.W. Toxigenic C. difficile induced inflammatory marker expression by human intestinal epithelial cells is asymmetrical. Life Sci. 2006, 78, 920–925. [Google Scholar] [PubMed]
- Souza, M.H.; Melo-Filho, A.A.; Rocha, M.F.; Lyerly, D.M.; Cunha, F.Q.; Lima, A.A.; Ribeiro, R.A. The involvement of macrophage-derived tumour necrosis factor and lipoxygenase products on the neutrophil recruitment induced by Clostridium difficile toxin B. Immunology 1997, 91, 281–288. [Google Scholar]
- Rocha, M.F.; Maia, M.E.; Bezerra, L.R.; Lyerly, D.M.; Guerrant, R.L.; Ribeiro, R.A.; Lima, A.A. Clostridium difficile toxin A induces the release of neutrophil chemotactic factors from rat peritoneal macrophages: role of interleukin-1beta, tumor necrosis factor alpha, and leukotrienes. Infect. Immun. 1997, 65, 2740–2746. [Google Scholar]
- Pothoulakis, C.; Castagliuolo, I.; LaMont, J.T. Nerves and Intestinal Mast Cells Modulate Responses to Enterotoxins. News Physiol. Sci. 1998, 13, 58–63. [Google Scholar]
- McVey, D.C.; Vigna, S.R. The role of leukotriene B4 in Clostridium difficile toxin A-induced ileitis in rats. Gastroenterology 2005, 128, 1306–1316. [Google Scholar]
- Castagliuolo, I.; Keates, A.C.; Qiu, B.; Kelly, C.P.; Nikulasson, S.; Leeman, S.E.; Pothoulakis, C. Increased substance P responses in dorsal root ganglia and intestinal macrophages during Clostridium difficile toxin A enteritis in rats. Proc. Natl. Acad. Sci. USA 1997, 94, 4788–4793. [Google Scholar]
- Keates, A.C.; Castagliuolo, I.; Qiu, B.; Nikulasson, S.; Sengupta, A.; Pothoulakis, C. CGRP upregulation in dorsal root ganglia and ileal mucosa during Clostridium difficile toxin A-induced enteritis. J. Biol. Chem. 1998, 274, G196–G202. [Google Scholar]
- Castagliuolo, I.; Wang, C.C.; Valenick, L.; Pasha, A.; Nikulasson, S.; Carraway, R.E.; Pothoulakis, C. Neurotensin is a proinflammatory neuropeptide in colonic inflammation. J. Clin. Invest. 1999, 103, 843–849. [Google Scholar]
- Neunlist, M.; Barouk, J.; Michel, K.; Just, I.; Oreshkova, T.; Schemann, M.; Galmiche, J.P. Toxin B of Clostridium difficile activates human VIP submucosal neurons, in part via an IL-1beta-dependent pathway. Am. J. Physiol. Gastrointest. Liver Physiol. 2003, 285, G1049–G1055. [Google Scholar]
- Cottrell, G.S.; Amadesi, S.; Pikios, S.; Camerer, E.; Willardsen, J.A.; Murphy, B.R.; Caughey, G.H.; Wolters, P.J.; Coughlin, S.R.; Peterson, A.; Knecht, W.; Pothoulakis, C.; Bunnett, N.W.; Grady, E.F. Protease-activated receptor 2, dipeptidyl peptidase I, and proteases mediate Clostridium difficile toxin A enteritis. Gastroenterology 2007, 132, 2422–2437. [Google Scholar]
- Ng, J.; Hirota, S.A.; Gross, O.; Li, Y.; Ulke-Lemee, A.; Potentier, M.S.; Schenck, L.P.; Vilaysane, A.; Seamone, M.E.; Feng, H.; Armstrong, G.D.; Tschopp, J.; Macdonald, J.A.; Muruve, D.A.; Beck, P.L. Clostridium difficile toxin-induced inflammation and intestinal injury are mediated by the inflammasome. Gastroenterology 2010. [Google Scholar]
- Kokkotou, E.; Espinoza, D.O.; Torres, D.; Karagiannides, I.; Kosteletos, S.; Savidge, T.; O'Brien, M.; Pothoulakis, C. Melanin-concentrating hormone (MCH) modulates C difficile toxin A-mediated enteritis in mice. Gut 2009, 58, 34–40. [Google Scholar]
- Qiu, B.; Pothoulakis, C.; Castagliuolo, I.; Nikulasson, S.; LaMont, J.T. Participation of reactive oxygen metabolites in Clostridium difficile toxin A-induced enteritis in rats. Am. J. Physiol. 1999, 276, G485–G490. [Google Scholar]
- Kim, H.; Rhee, S.H.; Kokkotou, E.; Na, X.; Savidge, T.; Moyer, M.P.; Pothoulakis, C.; LaMont, J.T. Clostridium difficile toxin A regulates inducible cyclooxygenase-2 and prostaglandin E2 synthesis in colonocytes via reactive oxygen species and activation of p38 MAPK. J. Biol. Chem. 2005, 280, 21237–21245. [Google Scholar]
- He, D.; Sougioultzis, S.; Hagen, S.; Liu, J.; Keates, S.; Keates, A.C.; Pothoulakis, C.; Lamont, J.T. Clostridium difficile toxin A triggers human colonocyte IL-8 release via mitochondrial oxygen radical generation. Gastroenterology 2002, 122, 1048–1057. [Google Scholar]
- Roebuck, K.A. Oxidant stress regulation of IL-8 and ICAM-1 gene expression: differential activation and binding of the transcription factors AP-1 and NF-kappaB (Review). Int. J. Mol. Med. 1999, 4, 223–230. [Google Scholar]
- Alcantara, C.; Stenson, W.F.; Steiner, T.S.; Guerrant, R.L. Role of inducible cyclooxygenase and prostaglandins in Clostridium difficile toxin A-induced secretion and inflammation in an animal model. J. Infect. Dis. 2001, 184, 648–652. [Google Scholar]
- Lima, A.A.; Nascimento, N.R.; Fang, G.D.; Yotseff, P.; Toyama, M.H.; Guerrant, R.L.; Fonteles, M.C. Role of phospholipase A(2) and tyrosine kinase in Clostridium difficile toxin A-induced disruption of epithelial integrity, histologic inflammatory damage and intestinal secretion. J. Appl. Toxicol. 2008, 28, 849–857. [Google Scholar]
- Fonteles, M.; Fang, G.; Thielman, N.M.; Yotseff, P.S.; Guerrant, R.L. Role of platelet activating factor in the inflammatory and secretory effects of Clostridium difficile toxin A. J. Lipid Mediat. Cell Signal. 1995, 11, 133–143. [Google Scholar]
- Hayashi, H.; Szaszi, K.; Coady-Osberg, N.; Furuya, W.; Bretscher, A.P.; Orlowski, J.; Grinstein, S. Inhibition and redistribution of NHE3, the apical Na+/H+ exchanger, by Clostridium difficile toxin B. J. Gen. Physiol. 2004, 123, 491–504. [Google Scholar]
- Alcantara, C.S.; Jin, X.H.; Brito, G.A.; Carneiro-Filho, B.A.; Barrett, L.J.; Carey, R.M.; Guerrant, R.L. Angiotensin II subtype 1 receptor blockade inhibits Clostridium difficile toxin A-induced intestinal secretion in a rabbit model. J. Infect. Dis. 2005, 191, 2090–2096. [Google Scholar]
- Na, X.; Zhao, D.; Koon, H.W.; Kim, H.; Husmark, J.; Moyer, M.P.; Pothoulakis, C.; LaMont, J.T. Clostridium difficile toxin B activates the EGF receptor and the ERK/MAP kinase pathway in human colonocytes. Gastroenterology 2005, 128, 1002–1011. [Google Scholar]
- Feltis, B.A.; Wiesner, S.M.; Kim, A.S.; Erlandsen, S.L.; Lyerly, D.L.; Wilkins, T.D.; Wells, C.L. Clostridium difficile toxins A and B can alter epithelial permeability and promote bacterial paracellular migration through HT-29 enterocytes. Shock 2000, 14, 629–634. [Google Scholar]
- Aktories, K.; Just, I. Monoglucosylation of low-molecular-mass GTP-binding Rho proteins by clostridial cytotoxins. Trends Cell Biol. 1995, 5, 441–443. [Google Scholar]
- Hecht, G.; Pothoulakis, C.; LaMont, J.T.; Madara, J.L. Clostridium difficile toxin A perturbs cytoskeletal structure and tight junction permeability of cultured human intestinal epithelial monolayers. J. Clin. Invest. 1988, 82, 1516–1524. [Google Scholar]
- Hecht, G.; Koutsouris, A.; Pothoulakis, C.; LaMont, J.T.; Madara, J.L. Clostridium difficile toxin B disrupts the barrier function of T84 monolayers. Gastroenterology 1992, 102, 416–423. [Google Scholar]
- Johal, S.S.; Solomon, K.; Dodson, S.; Borriello, S.P.; Mahida, Y.R. Differential effects of varying concentrations of Clostridium difficile toxin A on epithelial barrier function and expression of cytokines. J. Infect. Dis. 2004, 189, 2110–2119. [Google Scholar]
- Moore, R.; Pothoulakis, C.; LaMont, J.T.; Carlson, S.; Madara, J.L. C. difficile toxin A increases intestinal permeability and induces Cl- secretion. Am. J. Physiol. 1990, 259, G165–G172. [Google Scholar]
- Triadafilopoulos, G.; Pothoulakis, C.; O'Brien, M.J.; LaMont, J.T. Differential effects of Clostridium difficile toxins A and B on rabbit ileum. Gastroenterology 1987, 93, 273–279. [Google Scholar]
- Triadafilopoulos, G.; Pothoulakis, C.; Weiss, R.; Giampaolo, C.; Lamont, J.T. Comparative study of Clostridium difficile toxin A and cholera toxin in rabbit ileum. Gastroenterology 1989, 97, 1186–1192. [Google Scholar]
- Chen, M.L.; Pothoulakis, C.; LaMont, J.T. Protein kinase C signaling regulates ZO-1 translocation and increased paracellular flux of T84 colonocytes exposed to Clostridium difficile toxin A. J. Biol. Chem. 2002, 277, 4247–4254. [Google Scholar]
- Teichert, M.; Tatge, H.; Schoentaube, J.; Just, I.; Gerhard, R. Application of mutated Clostridium difficile toxin A for determination of glucosyltransferase-dependent effects. Infect. Immun. 2006, 74, 6006–6010. [Google Scholar]
- Kim, H.; Rhee, S.H.; Pothoulakis, C.; LaMont, J.T. Clostridium difficile toxin A binds colonocyte Src causing dephosphorylation of focal adhesion kinase and paxillin. Exp. Cell Res. 2009, 315, 3336–3344. [Google Scholar]
- Fiorentini, C.; Fabbri, A.; Falzano, L.; Fattorossi, A.; Matarrese, P.; Rivabene, R.; Donelli, G. Clostridium difficile toxin B induces apoptosis in intestinal cultured cells. Infect. Immun. 1998, 66, 2660–2665. [Google Scholar]
- Solomon, K.; Webb, J.; Ali, N.; Robins, R.A.; Mahida, Y.R. Monocytes are highly sensitive to Clostridium difficile toxin A-induced apoptotic and nonapoptotic cell death. Infect. Immun. 2005, 73, 1625–1634. [Google Scholar]
- Mahida, Y.R.; Galvin, A.; Makh, S.; Hyde, S.; Sanfilippo, L.; Borriello, S.P.; Sewell, H.F. Effect of Clostridium difficile toxin A on human colonic lamina propria cells: early loss of macrophages followed by T-cell apoptosis. Infect. Immun. 1998, 66, 5462–5469. [Google Scholar]
- Hippenstiel, S.; Schmeck, B.; N'Guessan, P.D.; Seybold, J.; Krull, M.; Preissner, K.; Eichel-Streiber, C.V.; Suttorp, N. Rho protein inactivation induced apoptosis of cultured human endothelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2002, 283, L830–L838. [Google Scholar]
- Huelsenbeck, J.; Dreger, S.; Gerhard, R.; Barth, H.; Just, I.; Genth, H. Difference in the cytotoxic effects of toxin B from Clostridium difficile strain VPI 10463 and toxin B from variant Clostridium difficile strain 1470. Infect. Immun. 2007, 75, 801–809. [Google Scholar]
- Matarrese, P.; Falzano, L.; Fabbri, A.; Gambardella, L.; Frank, C.; Geny, B.; Popoff, M.R.; Malorni, W.; Fiorentini, C. Clostridium difficile toxin B causes apoptosis in epithelial cells by thrilling mitochondria. Involvement of ATP-sensitive mitochondrial potassium channels. J. Biol. Chem. 2007, 282, 9029–9041. [Google Scholar] [PubMed]
- Pfeifer, G.; Schirmer, J.; Leemhuis, J.; Busch, C.; Meyer, D.K.; Aktories, K.; Barth, H. Cellular uptake of Clostridium difficile toxin B. Translocation of the N-terminal catalytic domain into the cytosol of eukaryotic cells. J. Biol. Chem. 2003, 278, 44535–44541. [Google Scholar] [PubMed]
- Warny, M.; Keates, A.C.; Keates, S.; Castagliuolo, I.; Zacks, J.K.; Aboudola, S.; Qamar, A.; Pothoulakis, C.; LaMont, J.T.; Kelly, C.P. p38 MAP kinase activation by Clostridium difficile toxin A mediates monocyte necrosis, IL-8 production, and enteritis. J. Clin. Invest. 2000, 105, 1147–1156. [Google Scholar]
- Warny, M.; Kelly, C.P. Monocytic cell necrosis is mediated by potassium depletion and caspase-like proteases. Am. J. Physiol. 1999, 276, C717–C724. [Google Scholar]
- Kim, H.; Kokkotou, E.; Na, X.; Rhee, S.H.; Moyer, M.P.; Pothoulakis, C.; Lamont, J.T. Clostridium difficile toxin A-induced colonocyte apoptosis involves p53-dependent p21(WAF1/CIP1) induction via p38 mitogen-activated protein kinase. Gastroenterology 2005, 129, 1875–1888. [Google Scholar]
- Chae, S.; Eckmann, L.; Miyamoto, Y.; Pothoulakis, C.; Karin, M.; Kagnoff, M.F. Epithelial cell I kappa B-kinase beta has an important protective role in Clostridium difficile toxin A-induced mucosal injury. J. Immunol. 2006, 177, 1214–1220. [Google Scholar]
- Hirota, S.A.; Fines, K.; Ng, J.; Traboulsi, D.; Lee, J.; Ihara, E.; Li, Y.; Willmore, W.G.; Chung, D.; Scully, M.M.; Louie, T.; Medlicott, S.; Lejeune, M.; Chadee, K.; Armstrong, G.; Colgan, S.P.; Muruve, D.A.; Macdonald, J.A.; Beck, P.L. Hypoxia-inducible factor signaling provides protection in Clostridium difficile-induced intestinal Injury. Gastroenterology 2010, 139, 259–269, e3.. [Google Scholar] [PubMed]
- Johnson, S.; Gerding, D.N. Clostridium difficile-associated diarrhea. Clin. Infect. Dis. 1998, 26, 1027–1034. [Google Scholar]
- Kelly, C.P.; LaMont, J.T. Clostridium difficile infection. Annu. Rev. Med. 1998, 49, 375–390. [Google Scholar]
- Burakoff, R.; Zhao, L.; Celifarco, A.J.; Rose, K.L.; Donovan, V.; Pothoulakis, C.; Percy, W.H. Effects of purified Clostridium difficile toxin A on rabbit distal colon. Gastroenterology 1995, 109, 348–354. [Google Scholar]
- Springer, T.A. Traffic signals for lymphocyte recirculation and leukocyte emigration: the multistep paradigm. Cell 1994, 76, 301–314. [Google Scholar]
- Miller, M.D.; Krangel, M.S. Biology and biochemistry of the chemokines: a family of chemotactic and inflammatory cytokines. Crit. Rev. Immunol. 1992, 12, 17–46. [Google Scholar]
- Hoch, R.C.; Schraufstatter, I.U.; Cochrane, C.G. In vivo, in vitro, and molecular aspects of interleukin-8 and the interleukin-8 receptors. J. Lab. Clin. Med. 1996, 128, 134–145. [Google Scholar]
- Matsukawa, A.; Yoshimura, T.; Maeda, T.; Ohkawara, S.; Takagi, K.; Yoshinaga, M. Neutrophil accumulation and activation by homologous IL-8 in rabbits. IL-8 induces destruction of cartilage and production of IL-1 and IL-1 receptor antagonist in vivo. J. Immunol. 1995, 154, 5418–5425. [Google Scholar] [PubMed]
- Sun, X.; He, X.; Tzipori, S.; Gerhard, R.; Feng, H. Essential role of the glucosyltransferase activity in Clostridium difficile toxin-induced secretion of TNF-alpha by macrophages. Microb. Pathog. 2009, 46, 298–305. [Google Scholar]
- Yeh, C.Y.; Lin, C.N.; Chang, C.F.; Lin, C.H.; Lien, H.T.; Chen, J.Y.; Chia, J.S. C-terminal repeats of Clostridium difficile toxin A induce production of chemokine and adhesion molecules in endothelial cells and promote migration of leukocytes. Infect. Immun. 2008, 76, 1170–1178. [Google Scholar]
- Kim, J.M.; Lee, J.Y.; Yoon, Y.M.; Oh, Y.K.; Youn, J.; Kim, Y.J. NF-kappa B activation pathway is essential for the chemokine expression in intestinal epithelial cells stimulated with Clostridium difficile toxin A. Scand. J. Immunol. 2006, 63, 453–460. [Google Scholar]
- Jefferson, K.K.; Smith, M.F., Jr.; Bobak, D.A. Roles of intracellular calcium and NF-kappa B in the Clostridium difficile toxin A-induced up-regulation and secretion of IL-8 from human monocytes. J. Immunol. 1999, 163, 5183–5191. [Google Scholar]
- Wershil, B.K.; Castagliuolo, I.; Pothoulakis, C. Direct evidence of mast cell involvement in Clostridium difficile toxin A-induced enteritis in mice. Gastroenterology 1998, 114, 956–964. [Google Scholar]
- Torimoto, K.; Sato, N.; Okubo, M.; Yagihashi, A.; Wada, Y.; Hara, I.; Hayasaka, H.; Kikuchi, K. Development of multiple necrotizing enteritis induced by a tumor necrosis factor-like cytokine from lipopolysaccharide-stimulated peritoneal macrophages in rats. Am. J. Pathol. 1990, 137, 1103–1111. [Google Scholar]
- Pothoulakis, C.; Sullivan, R.; Melnick, D.A.; Triadafilopoulos, G.; Gadenne, A.S.; Meshulam, T.; LaMont, J.T. Clostridium difficile toxin A stimulates intracellular calcium release and chemotactic response in human granulocytes. Am. J. Pathol. 1988, 81, 1741–1745. [Google Scholar]
- Prepens, U.; Just, I.; von Eichel-Streiber, C.; Aktories, K. Inhibition of Fc epsilon-RI-mediated activation of rat basophilic leukemia cells by Clostridium difficile toxin B (monoglucosyltransferase). J. Biol. Chem. 1996, 271, 7324–7329. [Google Scholar]
- Wex, C.B.; Koch, G.; Aktories, K. Effects of Clostridium difficile toxin B on activation of rat peritoneal mast cells. Naunyn Schmiedebergs Arch. Pharmacol. 1997, 355, 328–334. [Google Scholar]
- Meyer, G.K.; Neetz, A.; Brandes, G.; Tsikas, D.; Butterfield, J.H.; Just, I.; Gerhard, R. Clostridium difficile toxins A and B directly stimulate human mast cells. Infect. Immun. 2007, 75, 3868–3876. [Google Scholar]
- Steinman, R.M. The dendritic cell system and its role in immunogenicity. Annu. Rev. Immunol. 1991, 9, 271–296. [Google Scholar]
- Sozzani, S.; Allavena, P.; Vecchi, A.; Mantovani, A. Chemokines and dendritic cell traffic. J. Clin. Immunol. 2000, 20, 151–160. [Google Scholar]
- Niess, J.H.; Brand, S.; Gu, X.; Landsman, L.; Jung, S.; McCormick, B.A.; Vyas, J.M.; Boes, M.; Ploegh, H.L.; Fox, J.G.; Littman, D.R.; Reinecker, H.C. CX3CR1-mediated dendritic cell access to the intestinal lumen and bacterial clearance. Science 2005, 307, 254–258. [Google Scholar]
- Lee, J.Y.; Kim, H.; Cha, M.Y.; Park, H.G.; Kim, Y.J.; Kim, I.Y.; Kim, J.M. Clostridium difficile toxin A promotes dendritic cell maturation and chemokine CXCL2 expression through p38, IKK, and the NF-kappaB signaling pathway. J. Mol. Med. 2009, 87, 169–180. [Google Scholar]
- Pothoulakis, C.; LaMont, J.T. Clostridium difficile colitis and diarrhea. Gastroenterol. Clin. North Am. 1993, 22, 623–637. [Google Scholar]
- Melo Filho, A.A.; Souza, M.H.; Lyerly, D.M.; Cunha, F.Q.; Lima, A.A.; Ribeiro, R.A. Role of tumor necrosis factor and nitric oxide in the cytotoxic effects of Clostridium difficile toxin A and toxin B on macrophages. Toxicon 1997, 35, 743–752. [Google Scholar]
- Branka, J.E.; Vallette, G.; Jarry, A.; Bou-Hanna, C.; Lemarre, P.; Van, P.N.; Laboisse, C.L. Early functional effects of Clostridium difficile toxin A on human colonocytes. Gastroenterology 1997, 112, 1887–1894. [Google Scholar]
- Castagliuolo, I.; LaMont, J.T.; Letourneau, R.; Kelly, C.; O'Keane, J.C.; Jaffer, A.; Theoharides, T.C.; Pothoulakis, C. Neuronal involvement in the intestinal effects of Clostridium difficile toxin A and Vibrio cholerae enterotoxin in rat ileum. Gastroenterology 1994, 107, 657–665. [Google Scholar]
- Castagliuolo, I.; Riegler, M.; Pasha, A.; Nikulasson, S.; Lu, B.; Gerard, C.; Gerard, N.P.; Pothoulakis, C. Neurokinin-1 (NK-1) receptor is required in Clostridium difficile- induced enteritis. J. Clin. Invest. 1998, 101, 1547–1550. [Google Scholar]
- Anton, P.M.; Gay, J.; Mykoniatis, A.; Pan, A.; O'Brien, M.; Brown, D.; Karalis, K.; Pothoulakis, C. Corticotropin-releasing hormone (CRH) requirement in Clostridium difficile toxin A-mediated intestinal inflammation. Proc. Natl. Acad. Sci. USA 2004, 101, 8503–8508. [Google Scholar]
- Kokkotou, E.; Torres, D.; Moss, A.C.; O'Brien, M.; Grigoriadis, D.E.; Karalis, K.; Pothoulakis, C. Corticotropin-releasing hormone receptor 2-deficient mice have reduced intestinal inflammatory responses. J. Immunol. 2006, 177, 3355–3361. [Google Scholar]
- Wlk, M.; Wang, C.C.; Venihaki, M.; Liu, J.; Zhao, D.; Anton, P.M.; Mykoniatis, A.; Pan, A.; Zacks, J.; Karalis, K.; Pothoulakis, C. Corticotropin-releasing hormone antagonists possess anti-inflammatory effects in the mouse ileum. Gastroenterology 2002, 123, 505–515. [Google Scholar]
- Qiu, B.; Pothoulakis, C.; Castagliuolo, I.; Nikulasson, Z.; LaMont, J.T. Nitric oxide inhibits rat intestinal secretion by Clostridium difficile toxin A but not Vibrio cholerae enterotoxin. Gastroenterology 1996, 111, 409–418. [Google Scholar]
- Lima, A.A.; Nascimento, N.R.; Fang, G.D.; Yotseff, P.; Toyama, M.H.; Guerrant, R.L.; Fonteles, M.C. Role of phospholipase A2 and tyrosine kinase in Clostridium difficile toxin A-induced disruption of epithelial integrity, histologic inflammatory damage and intestinal secretion. J. Appl. Toxicol. 2008, 28, 849–857. [Google Scholar]
- Orlowski, J.; Grinstein, S. Na+/H+ exchangers of mammalian cells. J. Biol. Chem. 1997, 272, 22373–22376. [Google Scholar]
- Biemesderfer, D.; Pizzonia, J.; Abu-Alfa, A.; Exner, M.; Reilly, R.; Igarashi, P.; Aronson, P.S. NHE3: a Na+/H+ exchanger isoform of renal brush border. J. Biol. Chem. 1993, 265, F736–F742. [Google Scholar]
© 2010 by the authors; licensee MDPI, Basel, Switzerland This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Sun, X.; Savidge, T.; Feng, H. The Enterotoxicity of Clostridium difficile Toxins. Toxins 2010, 2, 1848-1880. https://doi.org/10.3390/toxins2071848
Sun X, Savidge T, Feng H. The Enterotoxicity of Clostridium difficile Toxins. Toxins. 2010; 2(7):1848-1880. https://doi.org/10.3390/toxins2071848
Chicago/Turabian StyleSun, Xingmin, Tor Savidge, and Hanping Feng. 2010. "The Enterotoxicity of Clostridium difficile Toxins" Toxins 2, no. 7: 1848-1880. https://doi.org/10.3390/toxins2071848
APA StyleSun, X., Savidge, T., & Feng, H. (2010). The Enterotoxicity of Clostridium difficile Toxins. Toxins, 2(7), 1848-1880. https://doi.org/10.3390/toxins2071848