Detection of Extremely Low Level Ciguatoxins through Monitoring of Lithium Adduct Ions by Liquid Chromatography-Triple Quadrupole Tandem Mass Spectrometry



Abstract
1. Introduction
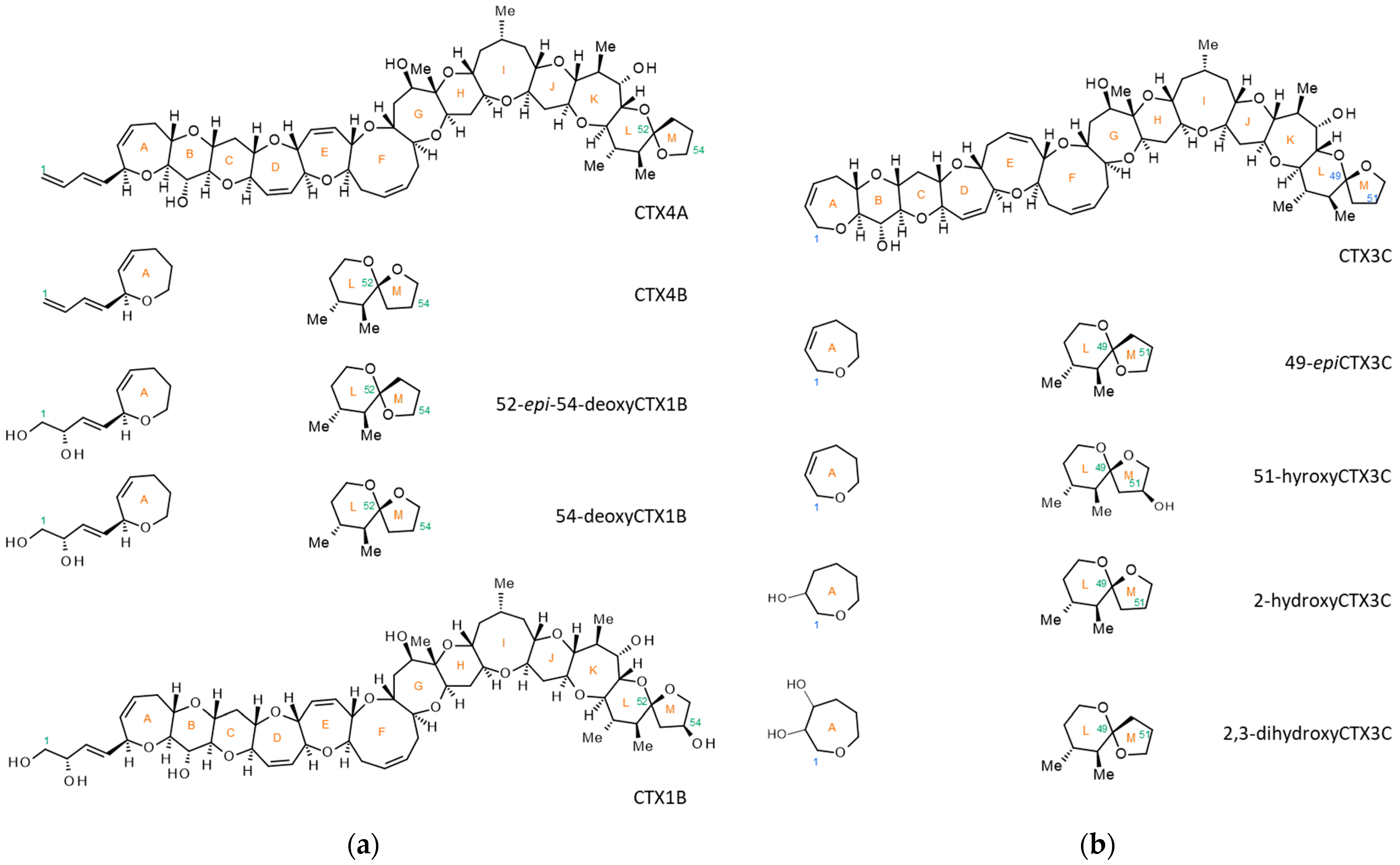
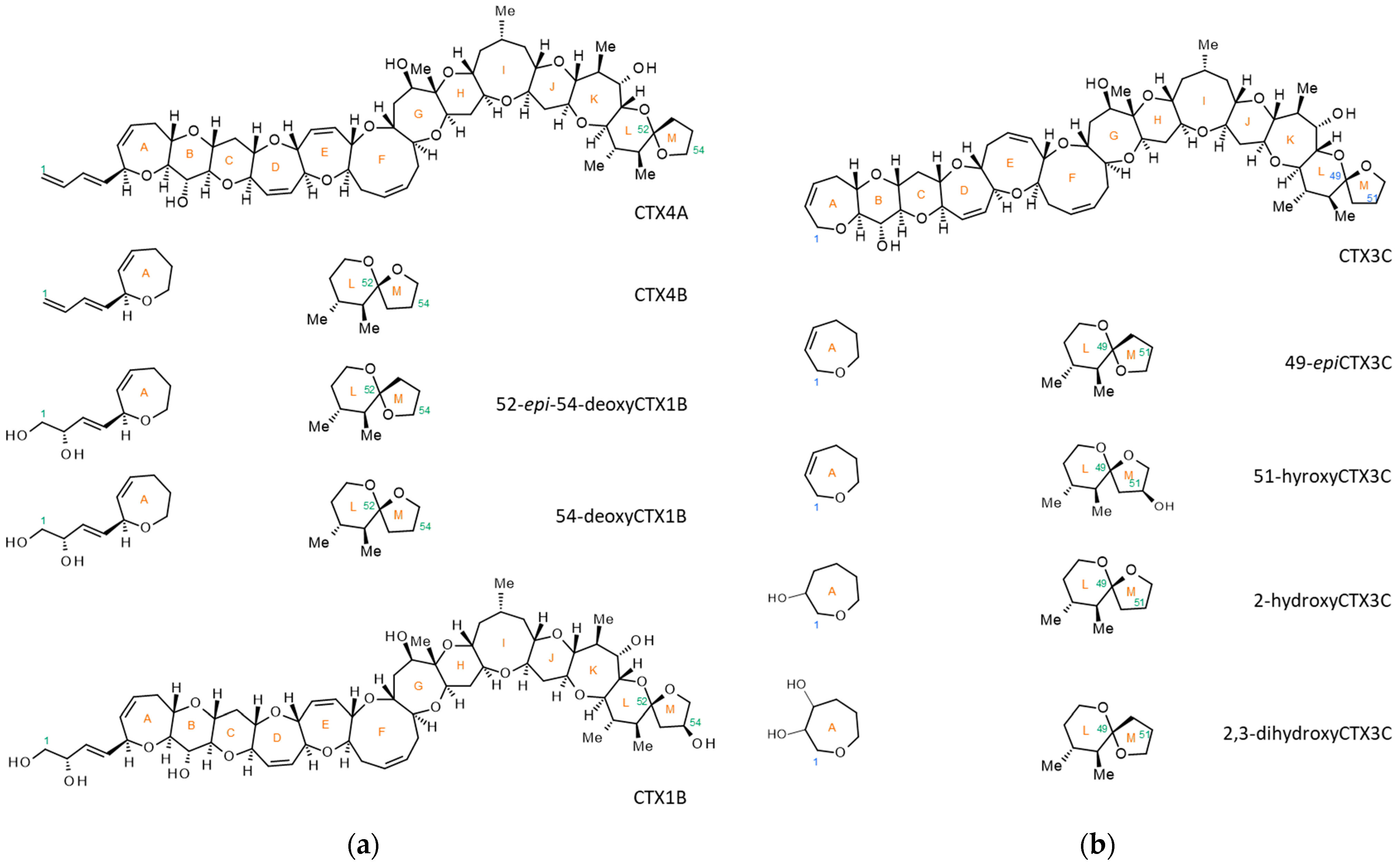
2. Results and Discussion
2.1. Preliminary Experiments with Previously Reported Methods
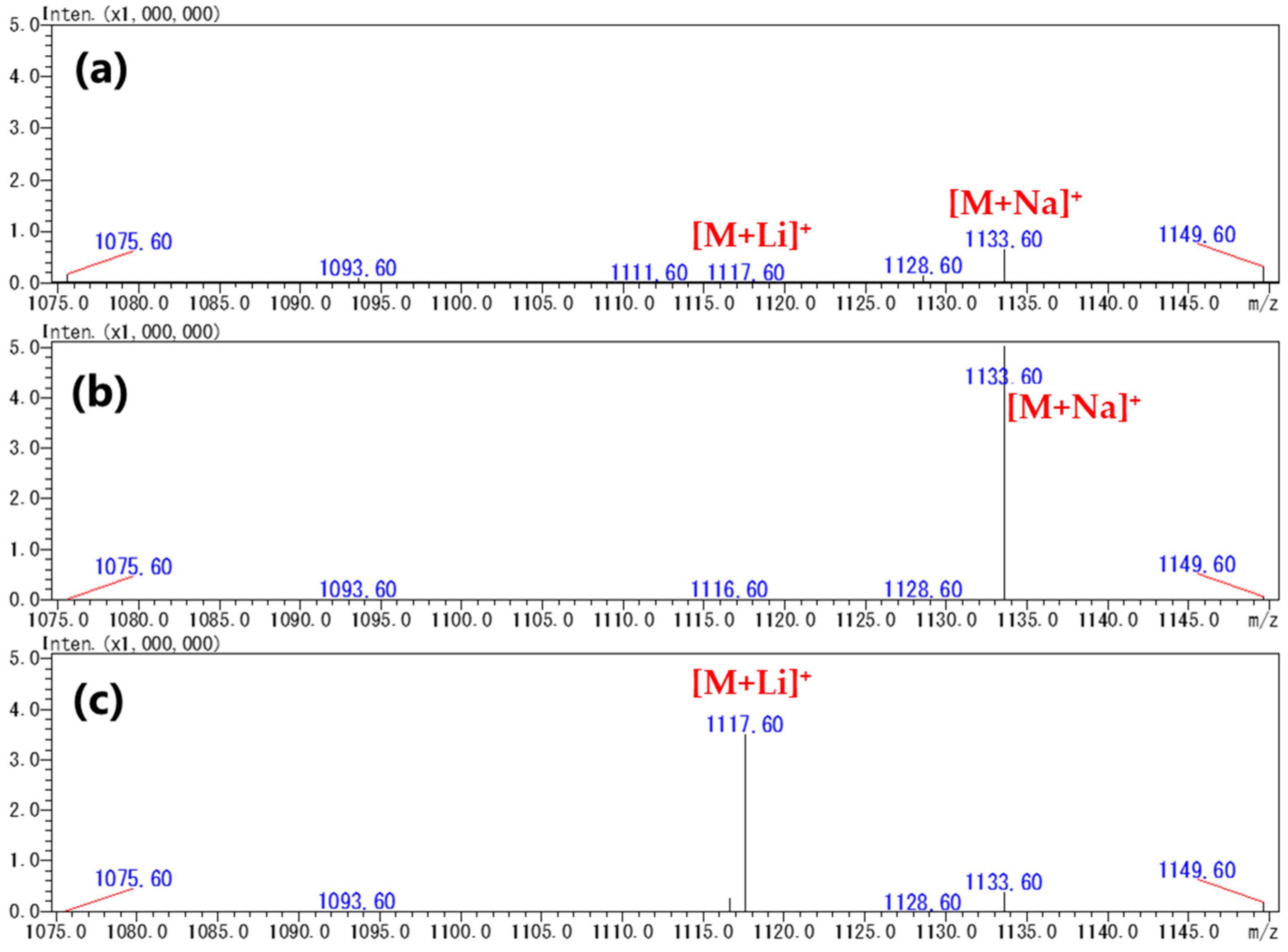
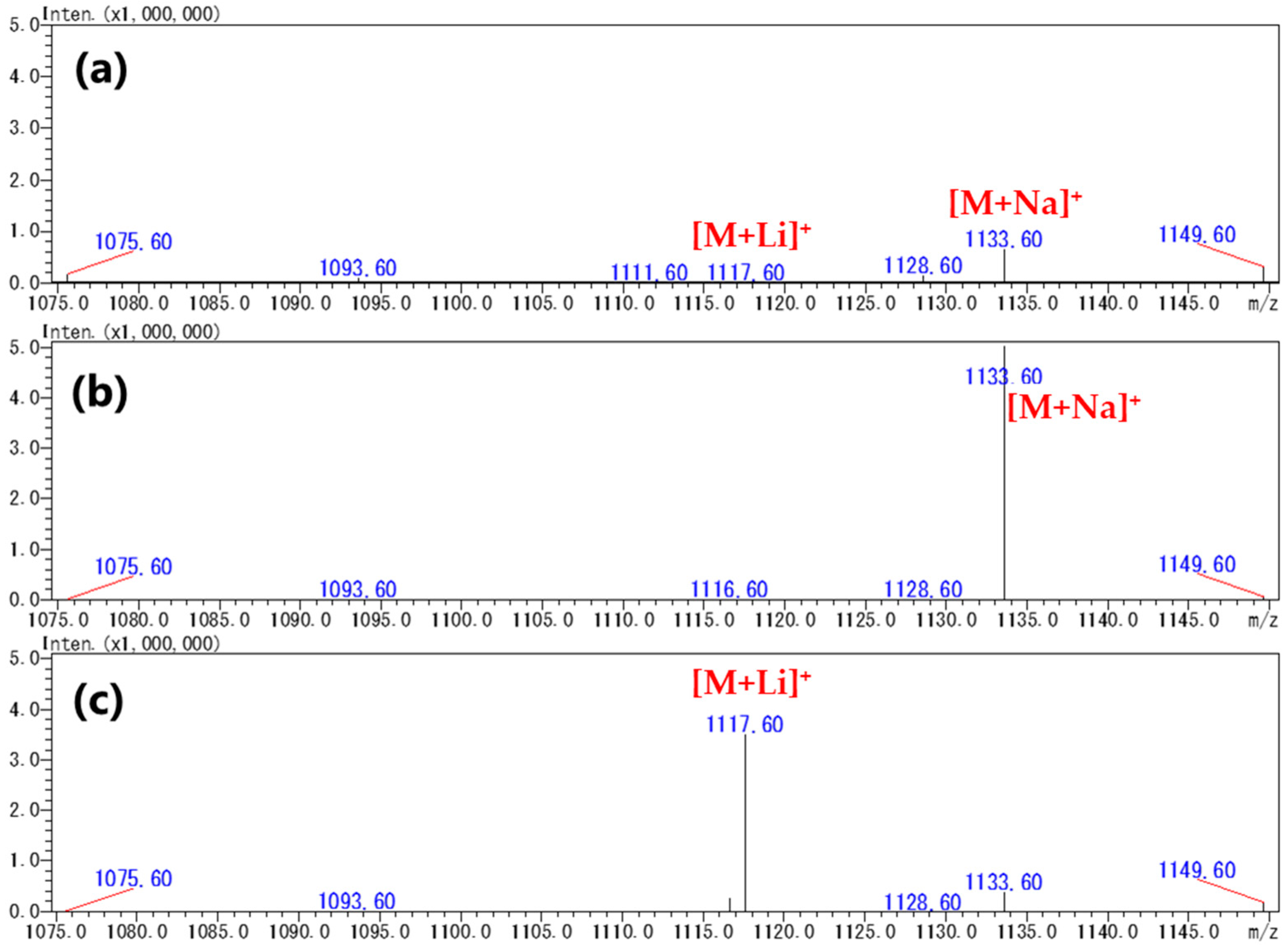
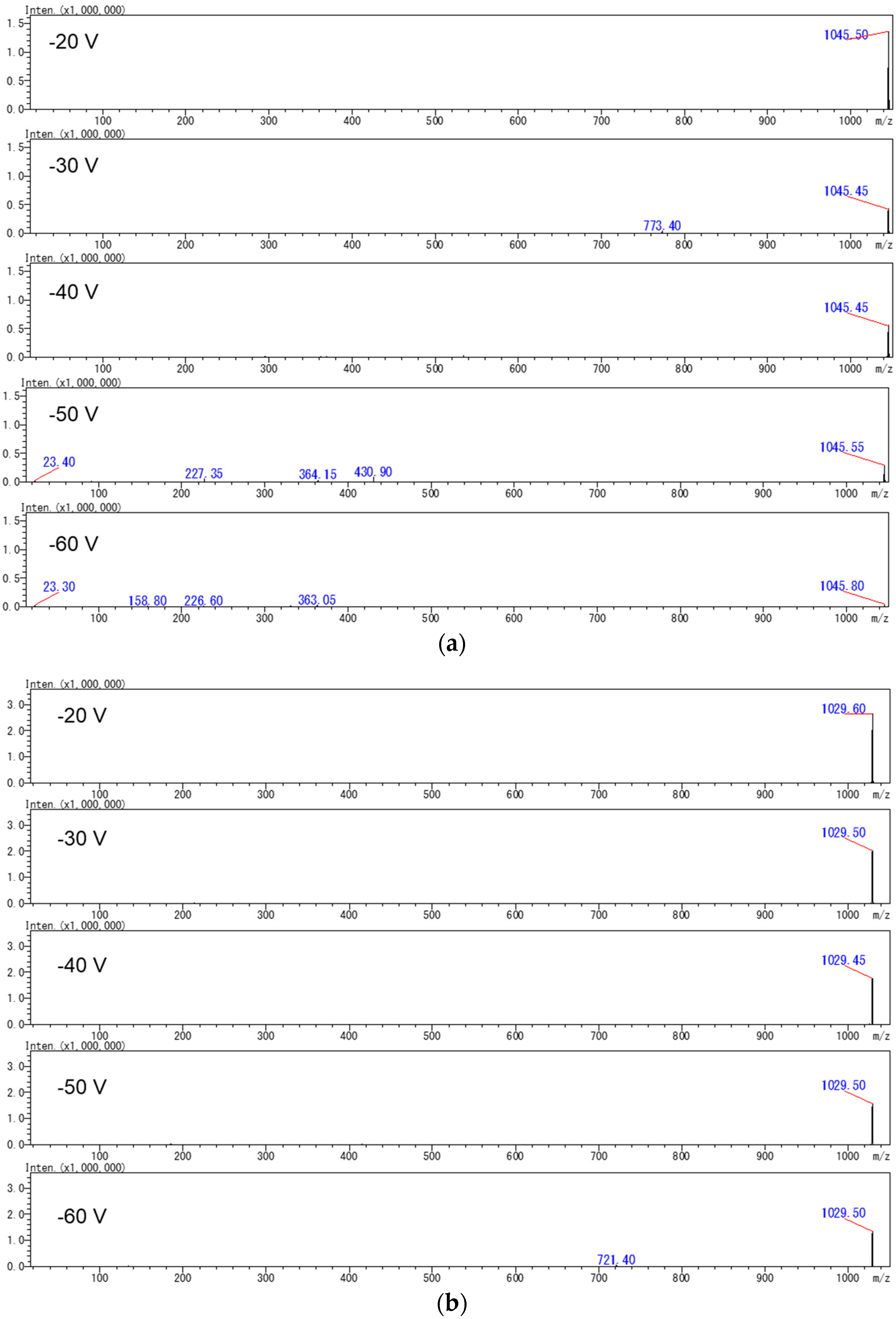
2.2. Production of [M+Na]+ or [M+Li]+ Ions Using Acetonitrile-Based Mobile Phase
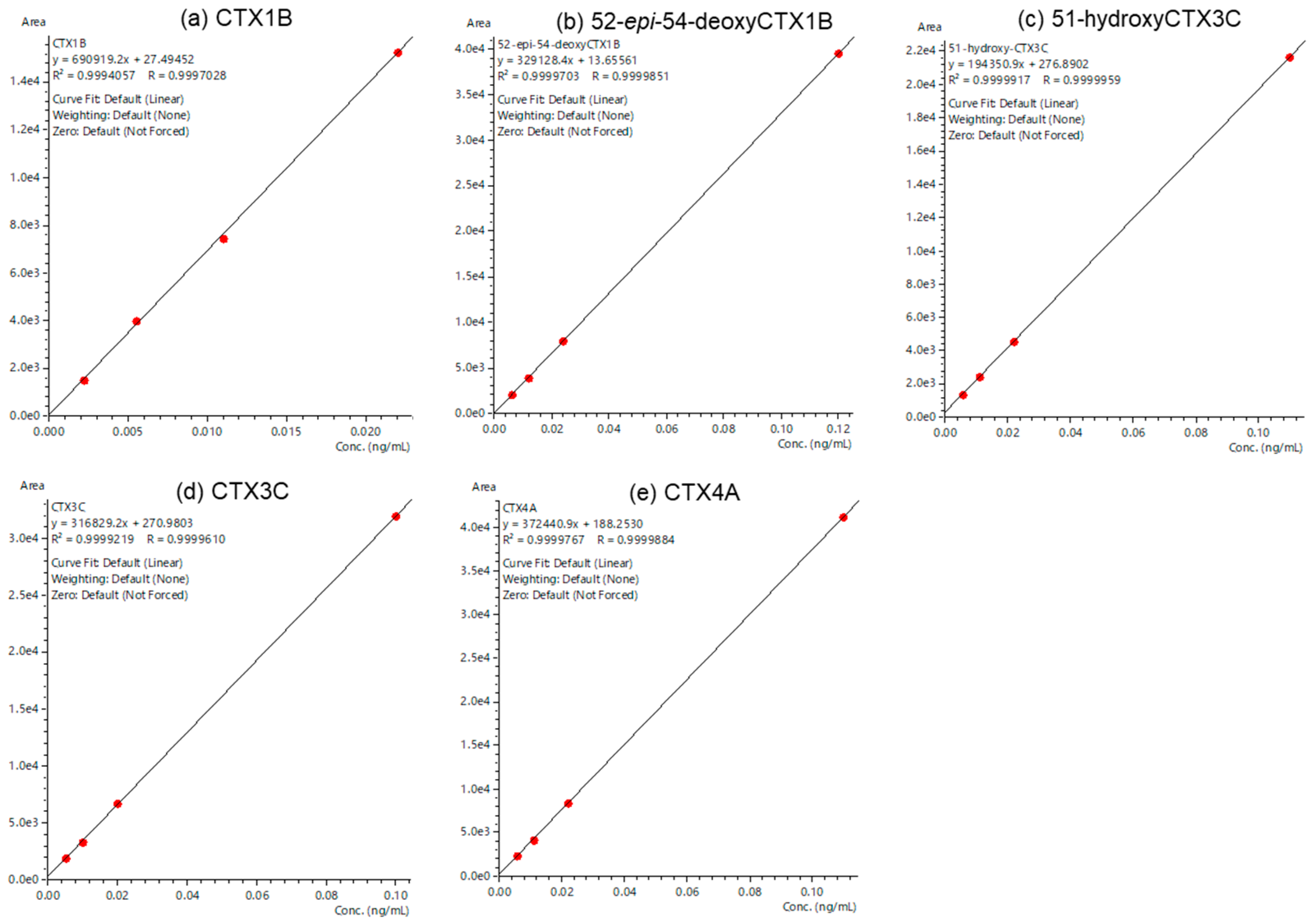
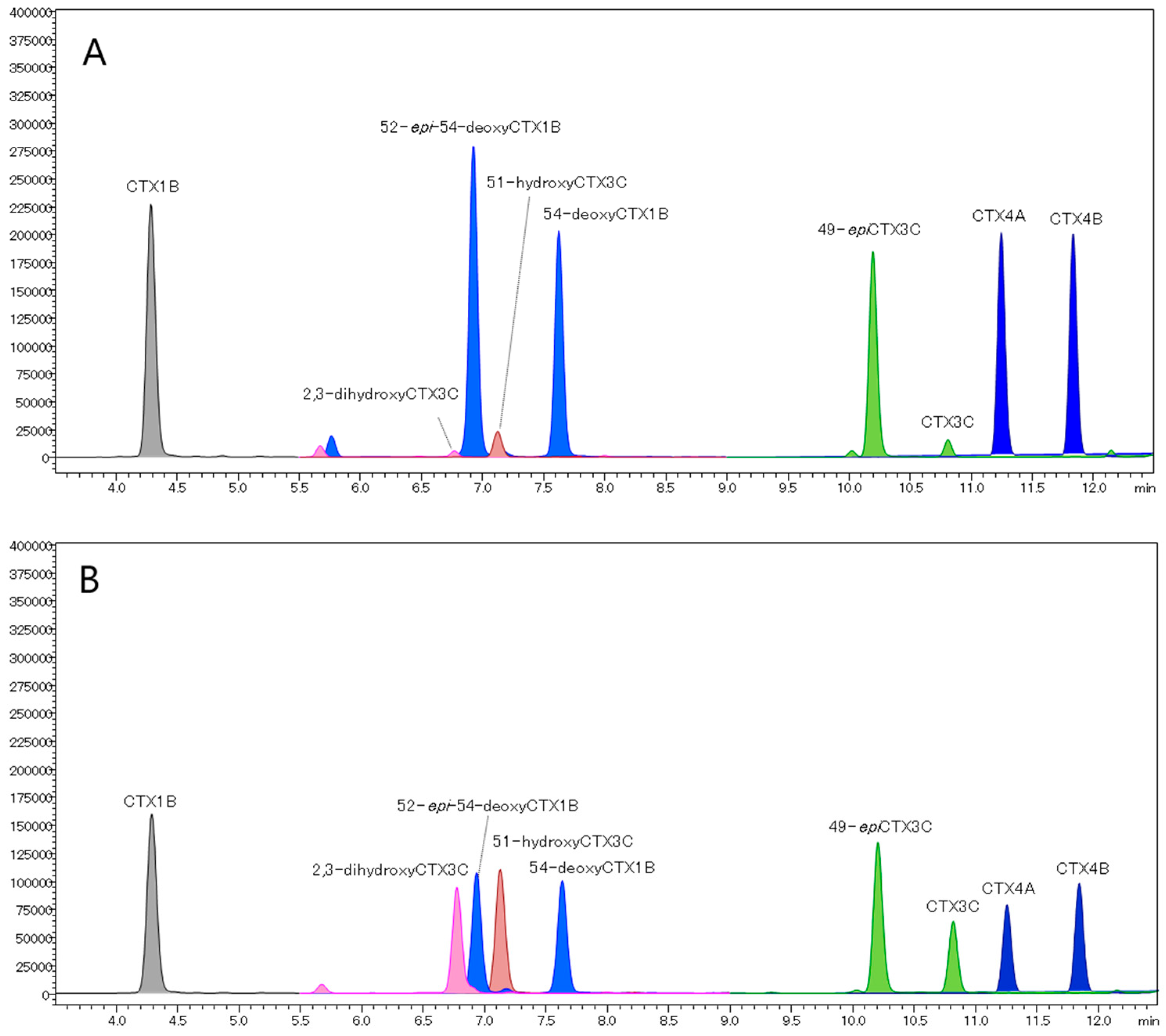
2.3. Evaluation of a Quantitative Analysis Monitoring [M+Li]+ > [M+Li]+
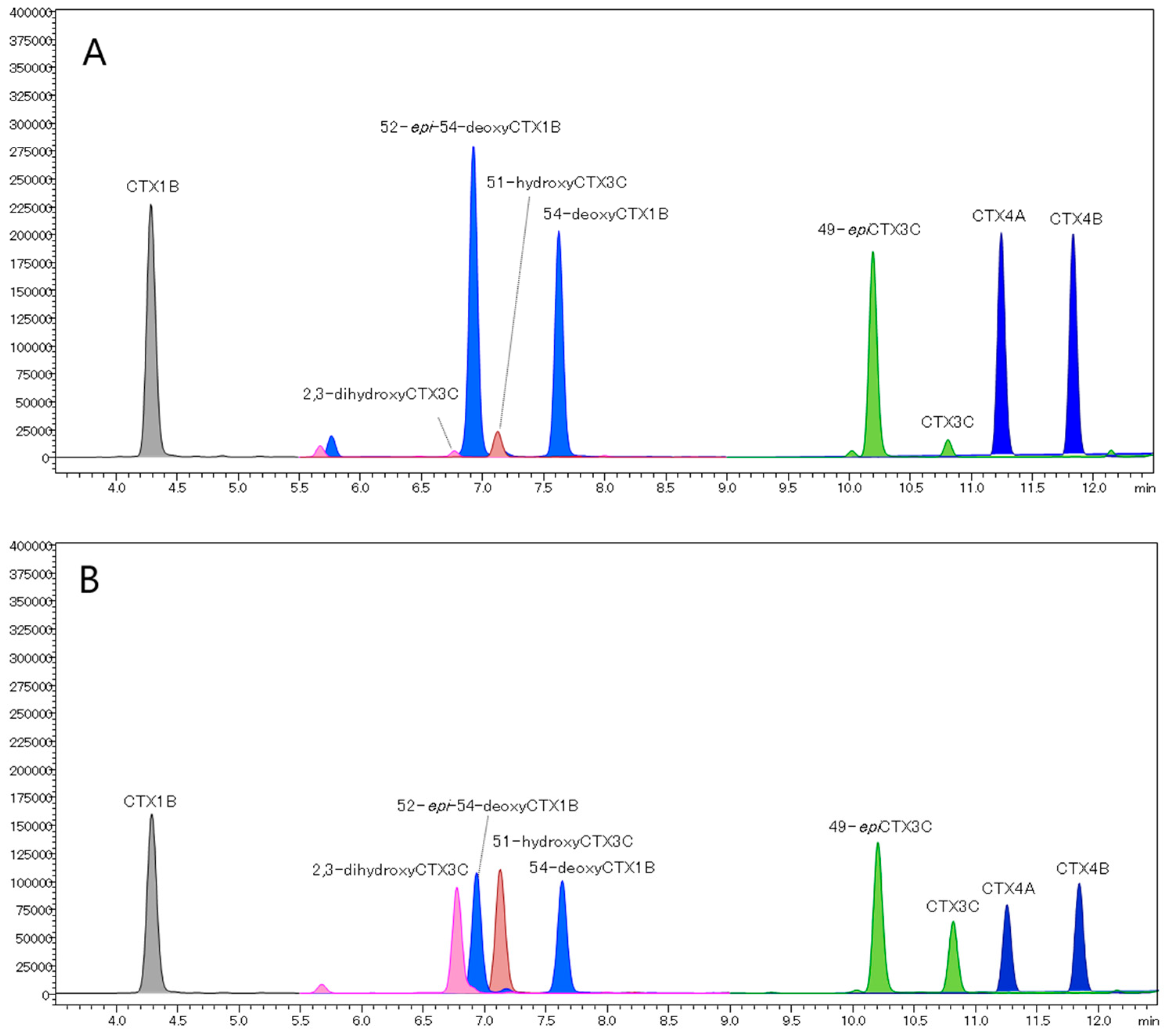
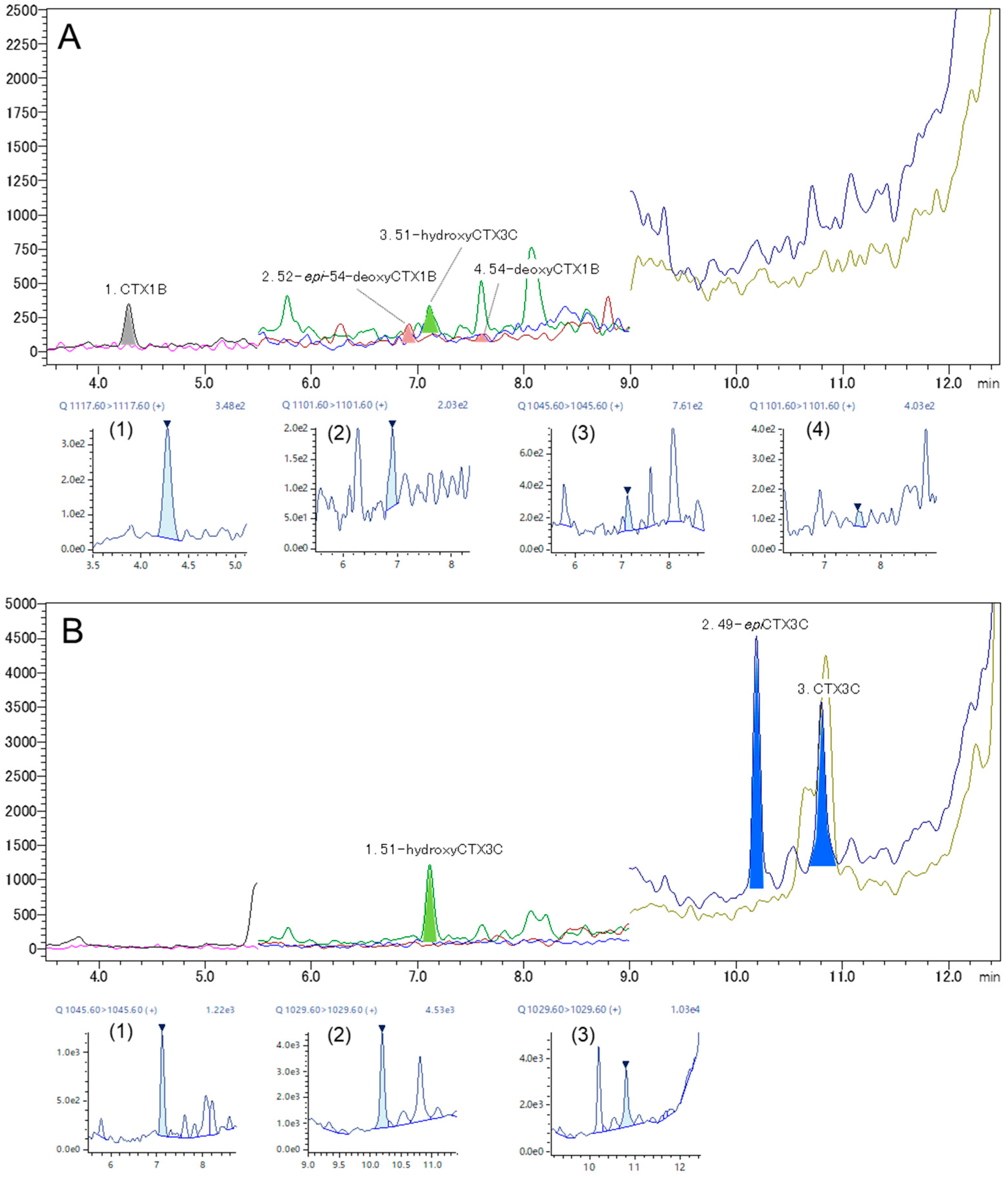
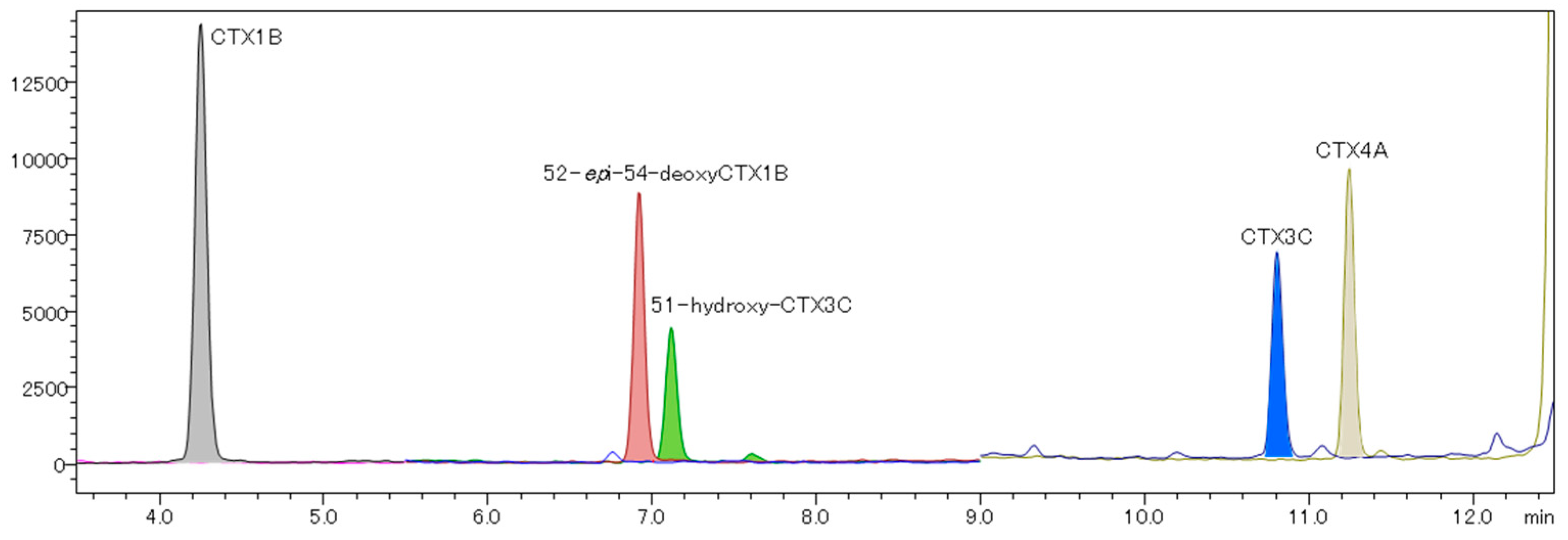
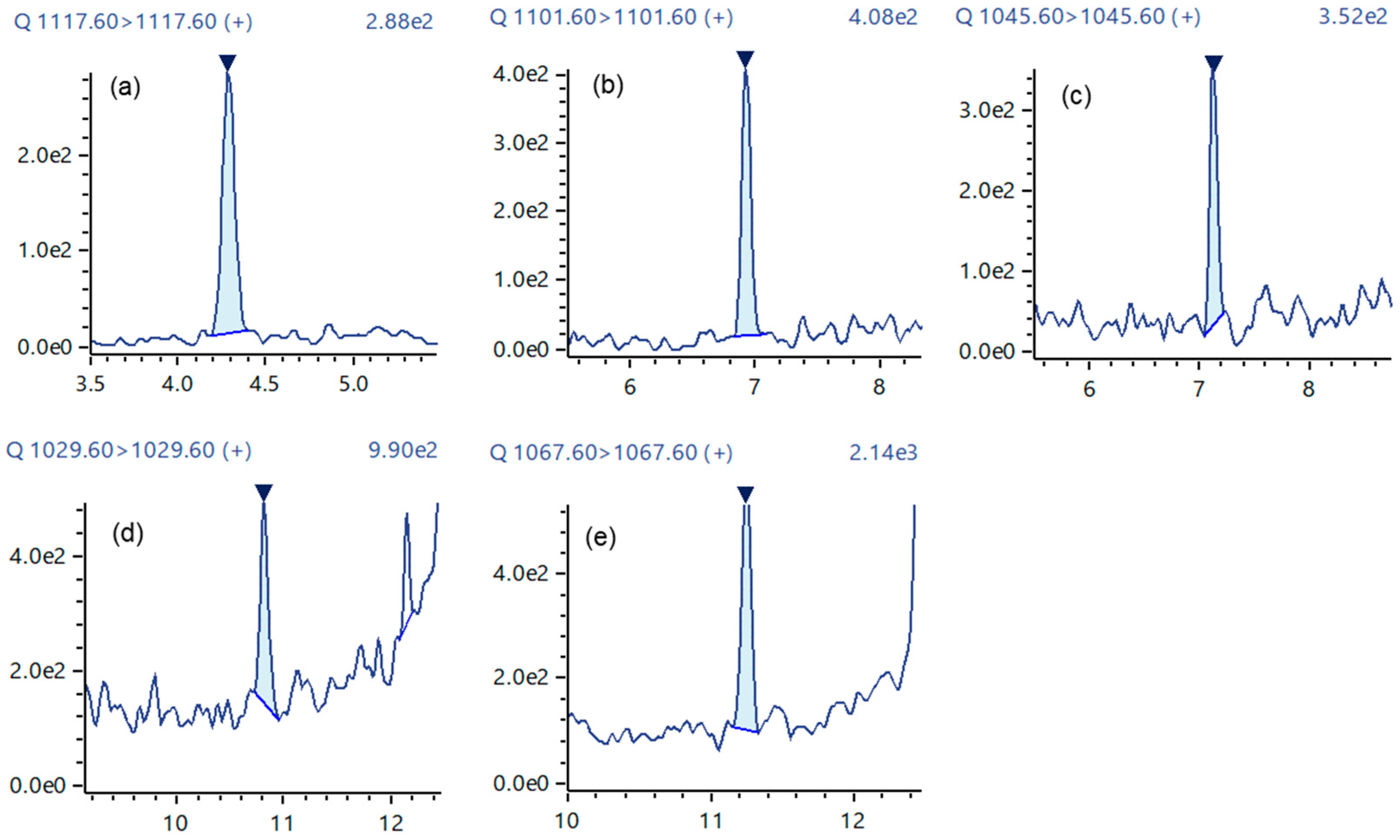
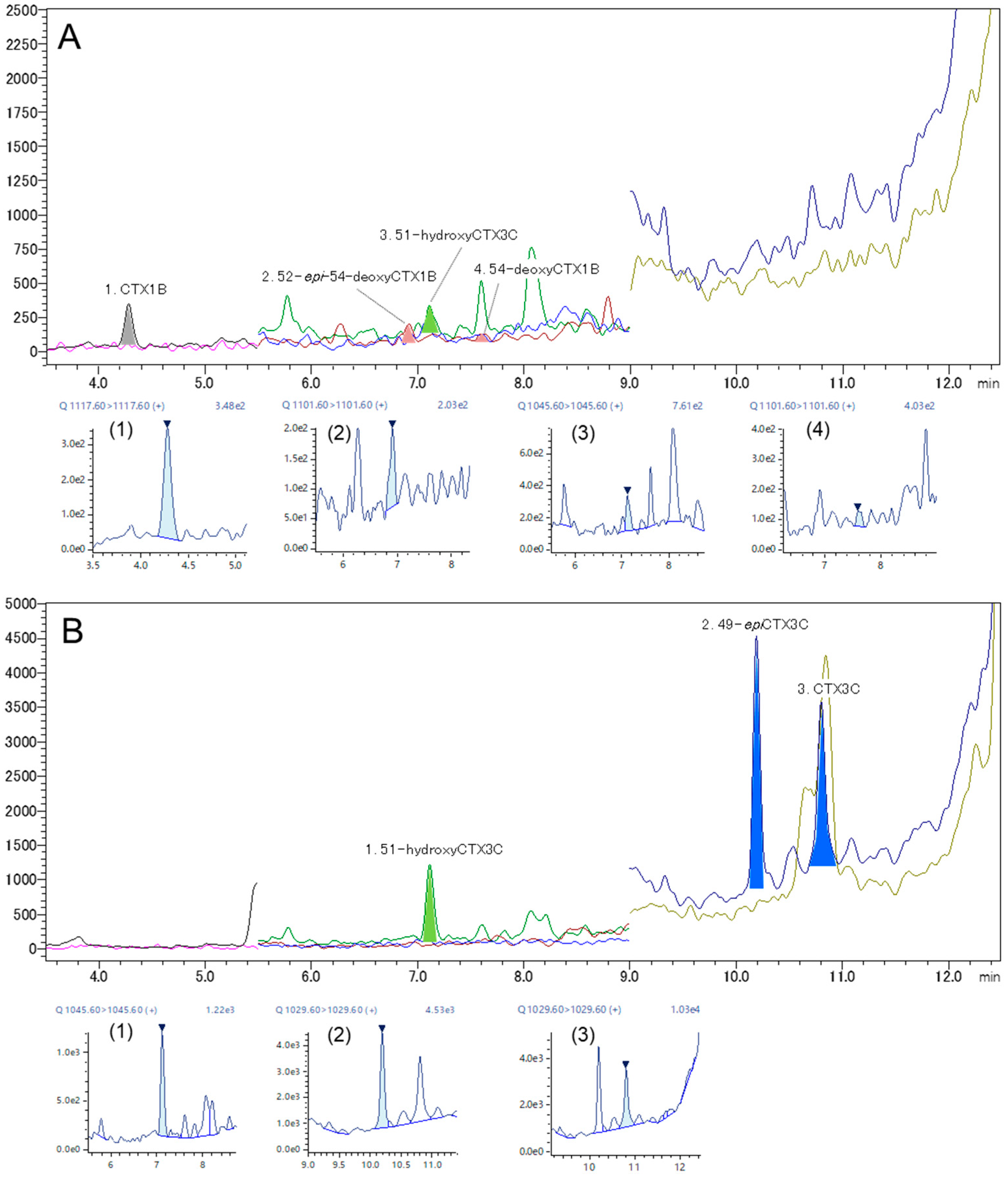
2.4. Analysis of Fish Flesh Extracts
3. Conclusions
4. Materials and Methods
4.1. CTX Reference Materials
4.2. Reagents
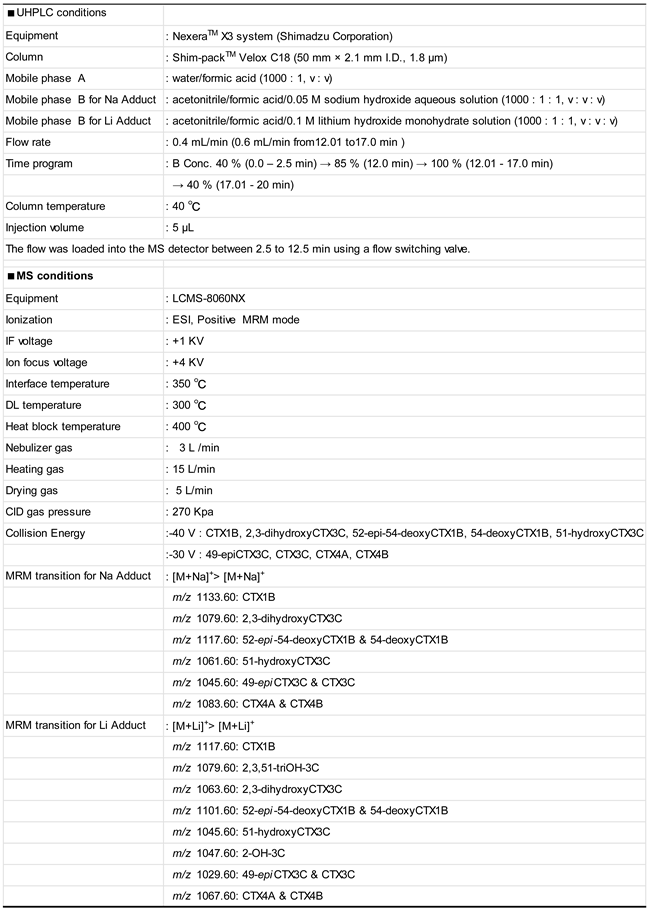
4.3. LC-MS/MS Analysis
4.3.1. Equipment
4.3.2. Analytical Conditions of Preliminary Experiments
4.3.3. Investigation into [M+Na]+ and [M+Li]+ Ions
4.3.4. Evaluation of Established LC-MS/MS Method for Monitoring [M+Li]+
4.3.5. Analysis of Fish Flesh Extract
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A
References
- Yasumoto, T. Chemistry, etiology, and food chain dynamics of marine toxins. Proc. Jpn. Acad. Ser. B 2005, 81, 43–51. [Google Scholar] [CrossRef]
- Friedman, M.; Fernandez, M.; Backer, L.; Dickey, R.; Bernstein, J.; Schrank, K.; Kibler, S.; Stephan, W.; Gribble, M.; Bienfang, P.; et al. An Updated Review of Ciguatera Fish Poisoning: Clinical, Epidemiological, Environmental, and Public Health Management. Mar. Drugs 2017, 15, 72. [Google Scholar] [CrossRef]
- Pearn, J. Neurology of ciguatera. J. Neurol. Neurosurg. Psychiatry 2001, 70, 4–8. [Google Scholar] [CrossRef]
- Chinain, M.; Gatti, C.M.I.; Darius, H.T.; Quod, J.P.; Tester, P.A. Ciguatera poisonings: A global review of occurrences and trends. Harmful Algae 2020, 102, 101873. [Google Scholar] [CrossRef]
- Yasumoto, T.; Satake, M. Chemistry, Etiology and Determination Methods of Ciguatera Toxins. J. Toxicol. Toxin Rev. 1996, 15, 91–107. [Google Scholar] [CrossRef]
- FAO; WHO. Report of the Expert Meeting on Ciguatera Poisoning. Rome, 19–23 November 2018; Food & Agriculture Org.: Rome, Italy, 2020; Volume 9, p. 156. [Google Scholar]
- Pérez-Arellano, J.-L.; Luzardo, O.P.; Brito, A.P.; Cabrera, M.H.; Zumbado, M.; Carranza, C.; Angel-Moreno, A.; Dickey, R.W.; Boada, L.D. Ciguatera Fish Poisoning, Canary Islands. Emerg. Infect. Dis. 2005, 11, 1981–1982. [Google Scholar] [CrossRef]
- Otero, P.; Pérez, S.; Alfonso, A.; Vale, C.; Rodríguez, P.; Gouveia, N.N.; Gouveia, N.; Delgado, J.; Vale, P.; Hirama, M.; et al. First Toxin Profile of Ciguateric Fish in Madeira Arquipelago (Europe). Anal. Chem. 2010, 82, 6032–6039. [Google Scholar] [CrossRef]
- Canals, A.; Martínez, C.V.; Diogène, J.; Gago-Martínez, A.; Cebadera-Miranda, L.; de Vasconcelos, F.M.; Gómez, I.L.; Sánchez, E.V.M.; Alférez, R.C.; Núñez, D.; et al. Risk characterisation of ciguatera poisoning in Europe. EFSA Support. Publ. 2021, 18, 6647E. [Google Scholar] [CrossRef]
- Yogi, K.; Oshiro, N.; Inafuku, Y.; Hirama, M.; Yasumoto, T. Detailed LC-MS/MS Analysis of Ciguatoxins Revealing Distinct Regional and Species Characteristics in Fish and Causative Alga from the Pacific. Anal. Chem. 2011, 83, 8886–8891. [Google Scholar] [CrossRef]
- Darius, H.T.; Revel, T.; Viallon, J.; Sibat, M.; Cruchet, P.; Longo, S.; Hardison, D.R.; Holland, W.C.; Tester, P.A.; Litaker, R.W.; et al. Comparative Study on the Performance of Three Detection Methods for the Quantification of Pacific Ciguatoxins in French Polynesian Strains of Gambierdiscus polynesiensis. Mar. Drugs 2022, 20, 348. [Google Scholar] [CrossRef]
- Longo, S.; Sibat, M.; Darius, H.T.; Hess, P.; Chinain, M. Effects of pH and Nutrients (Nitrogen) on Growth and Toxin Profile of the Ciguatera-Causing Dinoflagellate Gambierdiscus polynesiensis (Dinophyceae). Toxins 2020, 12, 767. [Google Scholar] [CrossRef]
- Chinain, M.; Gatti, C.M.; Roué, M.; Darius, H.T.; Subba Rao, D. Ciguatera-causing dinoflagellates in the genera Gambierdiscus and Fukuyoa: Distribution, ecophysiology and toxicology. In Dinoflagellates: Classification, Evolution, Physiology and Ecological Significance; Durvasula, S.R.V., Ed.; Nova Science Publishers: New York, NY, USA, 2020; pp. 405–457. [Google Scholar]
- Ikehara, T.; Kuniyoshi, K.; Oshiro, N.; Yasumoto, T. Biooxidation of Ciguatoxins Leads to Species-Specific Toxin Profiles. Toxins 2017, 9, 205. [Google Scholar] [CrossRef]
- Yasumoto, T.; Igarashi, T.; Legrand, A.-M.; Cruchet, P.; Chinain, M.; Fujita, T.; Naoki, H. Structural elucidation of ciguatoxin congeners by fast-atom bombardment tandem mass spectroscopy. J. Am. Chem. Soc. 2000, 122, 4988–4989. [Google Scholar] [CrossRef]
- Department of Health and Human Services, U.S. Food and Drug Administration. Fish and Fishery Products Hazards and Controls Guidance, 4th ed.; Department of Health and Human Services, U.S. Food and Drug Administration: Silver Spring, MD, USA, 2020.
- EFSA Panel on Contaminants in the Food Chain. Scientific Opinion on marine biotoxins in shellfish—Emerging toxins: Ciguatoxin group. EFSA J. 2010, 8, 1627. [Google Scholar] [CrossRef]
- Hoffman, P.A.; Granade, H.R.; McMillan, J.P. The mouse ciguatoxin bioassay: A dose-response curve and symptomatology analysis. Toxicon 1983, 21, 363–369. [Google Scholar] [CrossRef]
- Oshiro, N.; Yogi, K.; Asato, S.; Sasaki, T.; Tamanaha, K.; Hirama, M.; Yasumoto, T.; Inafuku, Y. Ciguatera incidence and fish toxicity in Okinawa, Japan. Toxicon 2010, 56, 656–661. [Google Scholar] [CrossRef]
- Caillaud, A.; De la Iglesia, P.; Darius, H.T.; Pauillac, S.; Aligizaki, K.; Fraga, S.; Chinain, M.; Diogène, J. Update on Methodologies Available for Ciguatoxin Determination: Perspectives to Confront the Onset of Ciguatera Fish Poisoning in Europe. Mar. Drugs 2010, 8, 1838–1907. [Google Scholar] [CrossRef]
- Manger, R.L.; Leja, L.S.; Lee, S.Y.; Hungerford, J.M.; Wekell, M.M. Tetrazolium-Based Cell Bioassay for Neurotoxins Active on Voltage-Sensitive Sodium Channels: Semiautomated Assay for Saxitoxins, Brevetoxins, and Ciguatoxins. Anal. Biochem. 1993, 214, 190–194. [Google Scholar] [CrossRef]
- Loeffler, C.R.; Bodi, D.; Tartaglione, L.; Dell’Aversano, C.; Preiss-Weigert, A. Improving in vitro ciguatoxin and brevetoxin detection: Selecting neuroblastoma (Neuro-2a) cells with lower sensitivity to ouabain and veratridine (OV-LS). Harmful Algae 2021, 103, 101994. [Google Scholar] [CrossRef]
- Darius, H.T.; Ponton, D.; Revel, T.; Cruchet, P.; Ung, A.; Tchou Fouc, M.; Chinain, M. Ciguatera risk assessment in two toxic sites of French Polynesia using the receptor-binding assay. Toxicon 2007, 50, 612–626. [Google Scholar] [CrossRef]
- Yokozeki, T.; Hama, Y.; Fujita, K.; Igarashi, T.; Hirama, M.; Tsumuraya, T. Evaluation of relative potency of calibrated ciguatoxin congeners by near-infrared fluorescent receptor binding and neuroblastoma cell-based assays. Toxicon 2023, 230, 107161. [Google Scholar] [CrossRef]
- Tsumuraya, T.; Sato, T.; Hirama, M.; Fujii, I. Highly Sensitive and Practical Fluorescent Sandwich ELISA for Ciguatoxins. Anal. Chem. 2018, 90, 7318–7324. [Google Scholar] [CrossRef]
- Tsumuraya, T.; Hirama, M. Rationally Designed Synthetic Haptens to Generate Anti-Ciguatoxin Monoclonal Antibodies, and Development of a Practical Sandwich ELISA to Detect Ciguatoxins. Toxins 2019, 11, 533. [Google Scholar] [CrossRef]
- Lewis, R.J.; Jones, A. Characterization of ciguatoxins and ciguatoxin congeners present in ciguateric fish by gradient reverse-phase high-performance liquid chromatography/mass spectrometry. Toxicon 1997, 35, 159–168. [Google Scholar] [CrossRef]
- Lewis, R.J.; Jones, A.; Vernoux, J.-P. HPLC/Tandem Electrospray Mass Spectrometry for the Determination of Sub-ppb Levels of Pacific and Caribbean Ciguatoxins in Crude Extracts of Fish. Anal. Chem. 1999, 71, 247–250. [Google Scholar] [CrossRef]
- Wu, J.; Mak, Y.; Murphy, M.; Lam, J.W.; Chan, W.; Wang, M.; Chan, L.; Lam, P.S. Validation of an accelerated solvent extraction liquid chromatography–tandem mass spectrometry method for Pacific ciguatoxin-1 in fish flesh and comparison with the mouse neuroblastoma assay. Anal. Bioanal. Chem. 2011, 400, 3165–3175. [Google Scholar] [CrossRef]
- Yogi, K.; Sakugawa, S.; Oshiro, N.; Ikehara, T.; Sugiyama, K.; Yasumoto, T. Determination of Toxins Involved in Ciguatera Fish Poisoning in the Pacific by LC/MS. J. AOAC Int. 2014, 97, 398–402. [Google Scholar] [CrossRef]
- Estevez, P.; Castro, D.; Manuel Leao, J.; Yasumoto, T.; Dickey, R.; Gago-Martinez, A. Implementation of liquid chromatography tandem mass spectrometry for the analysis of ciguatera fish poisoning in contaminated fish samples from Atlantic coasts. Food Chem. 2019, 280, 8–14. [Google Scholar] [CrossRef]
- Legrand, A.M.; Litaudon, M.; Genthon, J.N.; Bagnis, R.; Yasumoto, T. Isolation and some properties of ciguatoxin. J. Appl. Phycol. 1989, 1, 183–188. [Google Scholar] [CrossRef]
- Murata, M.; Legrand, A.M.; Ishibashi, Y.; Yasumoto, T. Structures of ciguatoxin and its congener. J. Am. Chem. Soc. 1989, 111, 8929–8931. [Google Scholar] [CrossRef]
- Satake, M.; Murata, M.; Yasumoto, T. The structure of CTX3C, a ciguatoxin congener isolated from cultured Gambierdiscus toxicus. Tetrahedron Lett. 1993, 34, 1975–1978. [Google Scholar] [CrossRef]
- Satake, M.; Fukui, M.; Legrand, A.-M.; Cruchet, P.; Yasumoto, T. Isolation and structures of new ciguatoxin analogs, 2,3-dihydroxyCTX3C and 51-hydroxyCTX3C, accumulated in tropical reef fish. Tetrahedron Lett. 1998, 39, 1197–1198. [Google Scholar] [CrossRef]
- Lewis, R.J.; Norton, R.S.; Brereton, I.M.; Eccles, C.D. Ciguatoxin-2 is a diastereomer of ciguatoxin-3. Toxicon 1993, 31, 637–643. [Google Scholar] [CrossRef] [PubMed]
- Vernoux, J.-P.; Lewis, R.J. Isolation and characterisation of Caribbean ciguatoxins from the horse-eye jack (Caranx latus). Toxicon 1997, 35, 889–900. [Google Scholar] [CrossRef] [PubMed]
- Mak, Y.; Wu, J.; Chan, W.; Murphy, M.; Lam, J.W.; Chan, L.; Lam, P.S. Simultaneous quantification of Pacific ciguatoxins in fish blood using liquid chromatography–tandem mass spectrometry. Anal. Bioanal. Chem. 2013, 405, 3331–3340. [Google Scholar] [CrossRef] [PubMed]
- Hirama, M. Total synthesis and related studies of large, strained, and bioactive natural products. Proc. Jpn. Acad. Ser. B 2016, 92, 290–329. [Google Scholar] [CrossRef] [PubMed]
- Kato, T.; Yasumoto, T. Quantification of Representative Ciguatoxins in the Pacific Using Quantitative Nuclear Magnetic Resonance Spectroscopy. Mar. Drugs 2017, 15, 309. [Google Scholar] [CrossRef] [PubMed]
- Oshiro, N.; Tomikawa, T.; Kuniyoshi, K.; Ishikawa, A.; Toyofuku, H.; Kojima, T.; Asakura, H. LC–MS/MS Analysis of Ciguatoxins Revealing the Regional and Species Distinction of Fish in the Tropical Western Pacific. J. Mar. Sci. Eng. 2021, 9, 299. [Google Scholar] [CrossRef]
- Oshiro, N.; Nagasawa, H.; Kuniyoshi, K.; Kobayashi, N.; Sugita-Konishi, Y.; Asakura, H.; Yasumoto, T. Characteristic Distribution of Ciguatoxins in the Edible Parts of a Grouper, Variola louti. Toxins 2021, 13, 218. [Google Scholar] [CrossRef]
- Tartaglione, L.; Loeffler, C.R.; Miele, V.; Varriale, F.; Varra, M.; Monti, M.; Varone, A.; Bodi, D.; Spielmeyer, A.; Capellacci, S.; et al. Dereplication of Gambierdiscus balechii extract by LC-HRMS and in vitro assay: First description of a putative ciguatoxin and confirmation of 44-methylgambierone. Chemosphere 2023, 319, 137940. [Google Scholar] [CrossRef]
- Moreiras, G.; Leão, J.M.; Gago-Martínez, A. Design of experiments for the optimization of electrospray ionization in the LC-MS/MS analysis of ciguatoxins. J. Mass Spectrom. 2018, 53, 1059–1069. [Google Scholar] [CrossRef] [PubMed]
- Klijnstra, M.D.; Gerssen, A. A Sensitive LC-MS/MS Method for Palytoxin Using Lithium Cationization. Toxins 2018, 10, 537. [Google Scholar] [CrossRef] [PubMed]
- Yogi, K.; Oshiro, N.; Matsuda, S.; Sakugawa, S.; Matsuo, T.; Yasumoto, T. Toxin Profiles in Fish Implicated in Ciguatera Fish Poisoning in Amami and Kakeroma Islands, Kagoshima Prefecture, Japan. Shokuhin Eiseigaku Zasshi (Food Hyg. Saf. Sci.) 2013, 54, 385–391. [Google Scholar] [CrossRef] [PubMed]
- Oshiro, N.; Nagasawa, H.; Watanabe, M.; Nishimura, M.; Kuniyoshi, K.; Kobayashi, N.; Sugita-Konishi, Y.; Asakura, H.; Tachihara, K.; Yasumoto, T. An Extensive Survey of Ciguatoxins on Grouper Variola louti from the Ryukyu Islands, Japan, Using Liquid Chromatography-Tandem Mass Spectrometry (LC-MS/MS). J. Mar. Sci. Eng. 2022, 10, 423. [Google Scholar] [CrossRef]
- Tomikawa, T.; Kuniyoshi, K.; Ito, S.; Sakugawa, S.; Ishikawa, A.; Saito, T.; Kojima, T.; Asakura, H.; Ikehara, T.; Oshiro, N. Analysis of Ciguatoxins in the Spotted Knifejaw, Oplegnathus punctatus from the Waters of Japan. Shokuhin Eiseigaku Zasshi (Food Hyg. Saf. Sci.) 2022, 63, 190–194. [Google Scholar] [CrossRef] [PubMed]
- Oshiro, N.; Nagasawa, H.; Nishimura, M.; Kuniyoshi, K.; Kobayashi, N.; Sugita-Konishi, Y.; Ikehara, T.; Tachihara, K.; Yasumoto, T. Analytical Studies on Ciguateric Fish in Okinawa, Japan (II): The Grouper Variola albimarginata. J. Mar. Sci. Eng. 2023, 11, 242. [Google Scholar] [CrossRef]
- Campàs, M.; Leonardo, S.; Oshiro, N.; Kuniyoshi, K.; Tsumuraya, T.; Hirama, M.; Diogène, J. A smartphone-controlled amperometric immunosensor for the detection of Pacific ciguatoxins in fish. Food Chem. 2022, 374, 131687. [Google Scholar] [CrossRef] [PubMed]
- Murata, M.; Legrand, A.M.; Ishibashi, Y.; Fukui, M.; Yasumoto, T. Structures and configurations of ciguatoxin from the moray eel Gymnothorax javanicus and its likely precursor from the dinoflagellate Gambierdiscus toxicus. J. Am. Chem. Soc. 1990, 112, 4380–4386. [Google Scholar] [CrossRef]
- Satake, M.; Ishibashi, Y.; Legrand, A.-M.; Yasumoto, T. Isolation and Structure of Ciguatoxin-4A, a New Ciguatoxin Precursor, from Cultures of Dinoflagellate Gambierdiscus toxicus and Parrotfish Scarus gibbus. Biosci. Biotechnol. Biochem. 1997, 60, 2103–2105. [Google Scholar] [CrossRef]
- Oshiro, N.; Tomikawa, T.; Kuniyoshi, K.; Kimura, K.; Kojima, T.; Yasumoto, T.; Asakura, H. Detection of ciguatoxins from the fish introduced to a wholesale market in Japan. Shokuhin Eiseigaku Zasshi (Food Hyg. Saf. Sci.) 2021, 62, 8–13. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CTX Analog | LOQ (ng/mL) | LOQ 1 (μg/kg) | Retention Time (min) | %RSD 2 (Area) | Accuracy 2 (%) | S/N 2 |
|---|---|---|---|---|---|---|
| CTX1B | 0.0022 | 0.00044 | 4.289 | 11.02 | 98 | 81 |
| 52-epi-54-deoxyCTX1B | 0.0060 | 0.0012 | 6.931 | 5.38 | 105 | 64 |
| 51-hydroxy-CTX3C | 0.0055 | 0.0011 | 7.121 | 8.76 | 103 | 22 |
| CTX3C | 0.0050 | 0.0010 | 10.815 | 12.32 | 102 | 22 |
| CTX4A | 0.0055 | 0.0011 | 11.250 | 17.28 | 105 | 36 |
| Eluate 1 | CTX1B | 2,3-diOH-CTX3C 2 | epi-Deoxy-CTX1B 3 | Deoxy-CTX1B 4 | 51-OH-CTX3C 5 | 49-epiCTX3C | CTX3C |
|---|---|---|---|---|---|---|---|
| MeOH 6 | 0.0005 | - | <LOQ 8 | <LOQ 8 | 0.0016 | - | - |
| ACN 7 | - | - | - | - | 0.0060 | 0.0023 | 0.0104 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kobayashi, M.; Masuda, J.; Oshiro, N. Detection of Extremely Low Level Ciguatoxins through Monitoring of Lithium Adduct Ions by Liquid Chromatography-Triple Quadrupole Tandem Mass Spectrometry. Toxins 2024, 16, 170. https://doi.org/10.3390/toxins16040170
Kobayashi M, Masuda J, Oshiro N. Detection of Extremely Low Level Ciguatoxins through Monitoring of Lithium Adduct Ions by Liquid Chromatography-Triple Quadrupole Tandem Mass Spectrometry. Toxins. 2024; 16(4):170. https://doi.org/10.3390/toxins16040170
Chicago/Turabian StyleKobayashi, Manami, Junichi Masuda, and Naomasa Oshiro. 2024. "Detection of Extremely Low Level Ciguatoxins through Monitoring of Lithium Adduct Ions by Liquid Chromatography-Triple Quadrupole Tandem Mass Spectrometry" Toxins 16, no. 4: 170. https://doi.org/10.3390/toxins16040170
APA StyleKobayashi, M., Masuda, J., & Oshiro, N. (2024). Detection of Extremely Low Level Ciguatoxins through Monitoring of Lithium Adduct Ions by Liquid Chromatography-Triple Quadrupole Tandem Mass Spectrometry. Toxins, 16(4), 170. https://doi.org/10.3390/toxins16040170