1. Introduction
Aflatoxins have been found to contaminate a variety of agricultural products, such as maize and other small-grain cereal crops, both pre-harvest and during storage.
Aspergillus spp. within the section
Flavi are principal producers of aflatoxins [
1], fungal secondary metabolites (mycotoxins) that include aflatoxin B1 (AFB1), aflatoxin B2, aflatoxin G1, and aflatoxin G2 [
2]. While these are regarded as the four most significant mycotoxins in the class [
3], AFB1 is the most potent of the group, and the effects of its consumption have been well characterised [
4]. AFB1 is passively absorbed through the gastrointestinal mucosa at a very high rate [
5,
6] and, due to its lipophilic nature, is characteristically sequestered in the liver. In the liver, AFB1 is converted to the highly reactive 8,9-epoxide-AFB1 (AFB0) primarily through the cytochrome P450 oxidative system [
7]. AFB0 is responsible for most of the toxicity of AFB1, forming adducts with DNA (AFB1 N7-guanine) and proteins (AFB1 Lys) within the cell, leading to significant macromolecular disruption [
4,
8].
Acute aflatoxicosis, a comparatively rare phenomenon, has been shown to stem from impaired mitochondrial function and the resulting oxidative stress and disrupted lipid metabolism [
9]. Through chronic exposure, AFB1 and aflatoxin M1 (AFM1, a phase I metabolite issued from the hydroxylation of AFB1) have shown teratogenic, immunotoxic, mutagenic, and hepatotoxic activity and are classified as Group I carcinogens [
9,
10,
11]. Accordingly, these aflatoxins have been implicated in the causation of hepatocellular carcinoma [
8] and extrahepatic carcinomas in both humans and animals [
12].
While commonly associated with non-industrialised countries, aflatoxins continue to be detected in grain products and animal by-products such as milk, meat, and eggs in developed nations, a significant issue given the widespread consumption of these foods by humans both young and old [
3,
13] and especially in places where access to safe food remains a significant challenge. Tolerance limits are set for many countries to mitigate much of the risk posed by the presence of mycotoxins in food and feed. Applied to animal feedstuffs in the United States, AFB1 limits range from 20 to 300 µg total aflatoxins per kg of feed material (depending on the production stage of the animal and the feed material), with a final 20 µg/kg tolerance applied to human food [
14], while the European Union stipulates comparatively tighter limits with 20 µg/kg for feed materials, 10 µg/kg for all complimentary and complete feeds, and as low as 5 µg/kg for any compound feed destined for dairy-producing or young animals [
15].
While active strategies are followed to control mycotoxin contamination in the field or during storage by implementing good agricultural practices, and despite employing chemical and/or mechanical means of reducing overall contamination (sorting, dehulling, washing, ozonation, or ammonification to chemically degrade existing mycotoxins), mycotoxin contamination is unavoidable [
16]. This task is forecast to become more difficult as we face changes in global climatic conditions and/or extreme weather patterns that stand to favour mycotoxin-producing organisms [
17], further complicated by the international trading of feed ingredients and increasing the chances of unattended patterns of contamination. While acknowledging the procedural limitations to the accurate quantification of mycotoxins in bulk feedstuffs (most often due to sampling issues rather than analytical imprecision, e.g., heterogenous distribution within a lot and/or limited sampling plans), many countries have implemented standardised methods to moderate these limitations as much as is reasonably practical [
18,
19].
Following a reduction in mycotoxin levels in feedstuffs to within acceptable tolerance, the final mitigative step should focus on reducing the bioavailability of mycotoxins to digestive absorption and decrease their interactions with animal organisms, thereby minimising potential low-dose chronic effects that could affect animal performance. The inclusion of adsorbent materials in animal feed is perhaps the most economically viable means of achieving this “final-stage-control” of low-level or unaccounted for contamination [
20]. Such supplementation can lead to the sequestration or adsorption of mycotoxins, thereby limiting the concentrations available to the animal until being excreted back into the environment [
21,
22] and/or promoting overall gut health, offsetting some of the related deleterious toxic effects [
23].
While significant bodies of work have demonstrated the sequestration of AFB1 under a range of sorbents, many of the investigations focused on efficacy in elevated mycotoxin levels with minimal evaluations in the context of the digestive system. Appropriately, fewer studies have been carried out at low, chronic, or regulation-comparable levels. The aim of this study was to improve our understanding of the efficacy and mechanisms of the protection afforded to biological systems by a yeast cell wall-based sorbent (YCW, Mycosorb
®) and a hydrated sodium calcium aluminosilicate (HSCAS) clay at comparatively low AFB1 concentrations. To achieve this, two individual in vitro and two individual ex vivo models were utilised. The first model (Model 1) is an industry standard method proposed by the FEFANA (the EU Association of Specialty Feed Ingredients and their Mixtures) for in vitro efficacy testing of mycotoxin binders [
24]. The second developed in vitro model (Model 2) was employed to assess the sequestrant sorption capabilities under physiologically relevant conditions and at low (10 µg/L) AFB1 concentrations. A third model (Model 3) was used to measure the ex vivo absorption of AFB1 into live intestinal tissue and the degree to which the sequestrants could help offset such a mechanism and, finally, a fourth model, based on earlier work [
25] (Model 4), was used again to verify the progress in ex vivo AFB1 absorption across living intestinal tissue and determine the sequestrant’s impact on the toxin bioavailability in this system.
3. Discussion
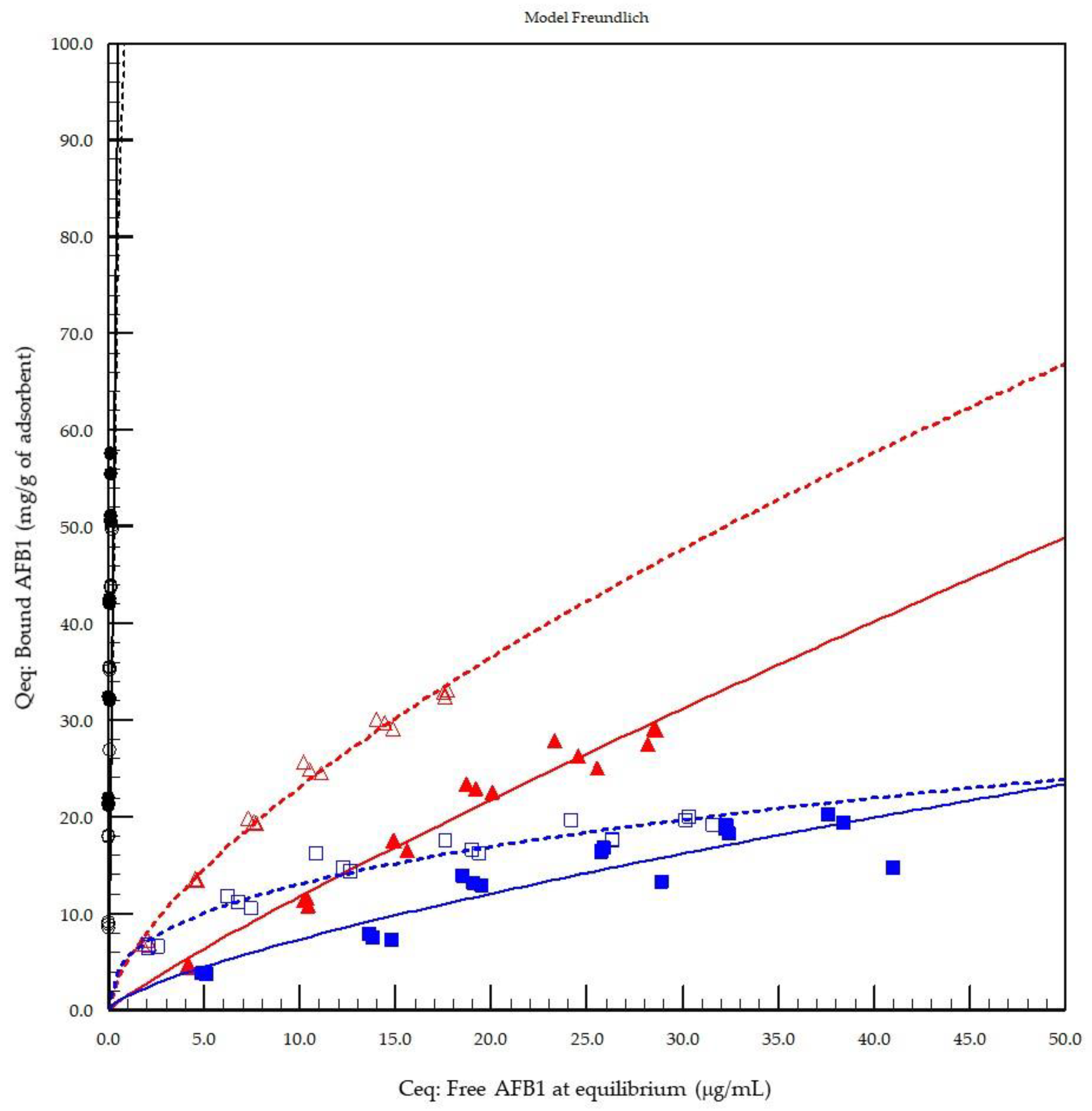
The first model of the in vitro adsorption evaluation demonstrated that activated carbon played the role of a reference material with the strongest AFB1 binding capacity at both pH 3.0 (stomach equivalent) and pH 7.0 (intestine equivalent) compared to the other test products (
Figure 1). The results from the Freundlich model were compared to those of the Langmuir and Hill’s models with
n site equations, which are other models recommended by FEFANA for the evaluation of adsorbents. The Freundlich model, although not necessarily the model providing the best fit, was able to characterise all adsorbents at the two different tested pH values (
Table 1). Hill’s equation [
26] with
n sites was particularly well-adapted to the evaluation of YCW, confirming the appropriateness of the model for organic adsorbents, as demonstrated previously [
25]; however, the model was less well-adapted to the AC reference material in the particular case of AFB1. All models tested were unable to correctly fit the reference material due to the lack of a defined experimental saturation phase with the concentrations tested. However, Freundlich was selected as an all-round model for comparing the other tested sorbent materials of a mixed nature, such as inorganic binders (HSCAS) and organic binders (YCW), thus confirming the previously observed results obtained with the same model applied to zearalenone [
25]. Interestingly, when the cooperativity factor (
n sites) of Hill’s model was examined, the results revealed that HSCAS lacks cooperativity, as expected, with a factor around a value of 1 (0.96), whereas YCW exhibited <1 at pH 3.0 (0.76) and >1 at pH 7.0 (1.63), accounting for the dynamicity of their interactions due to the flexibility and spatial reorganisation capacity of the parietal carbohydrate fraction of the yeast compared to the very static HSCAS physical organisation, thereby providing a restrictive and rigid site for interaction.
The key desirable element for in vivo mycotoxin binding is maximising the adsorption at a low pH where mycotoxins are more freely available in the digestate (chyme) and retaining the maximum quantity of bound toxins (minimising desorption) when conditions become less favourable (as pH increases in the gut). This desorption of mycotoxins when the pH increases from acidic stomach conditions to neutral/basic intestinal conditions is well established [
27,
28,
29,
30]. The larger difference observed between the pH tested in the first model and that of the HSCAS may be attributable to the greater cation exchange capacity of the latter material [
31], which is more susceptible to promotion and demotion by other ionic sources and is more greatly influenced by the pH. Conversely, the smaller differences between pH measurements observed with YCW could be attributed to the complex structure of the yeast cell wall involving non-charged polymers of glucose and mannose, which are less likely to be influenced by pH changes in their sequestration activity and, as such, can better retain bound mycotoxins during pH transitions [
27].
We acknowledge that the AFB1 concentrations in the proposed FEFANA model were markedly higher than the actual permissible levels in animal feeds. The lowest concentration used here was two hundred times higher than the recommended level for pharmacokinetic studies (50 µg/kg of feed). The purpose of this model was not to demonstrate its efficacy in the presence of low mycotoxin concentrations but to establish the degree to which the test materials would bind the target mycotoxin. Such trends were innately more evident at higher AFB1 concentrations, and the experimental fluctuations were commensurately lower. That being said, this model does not factor in the complexities and nuances of mycotoxin binding in vivo, and extrapolation of the binding efficacy at these particularly high concentrations to applications in live models would be tenuous at best. These limitations led to the development and application of novel experimental setups (Models 2–4) to more elegantly and realistically simulate the conditions of AFB1 contamination encountered, especially in animal production, thus providing a deeper understanding of the observable effects ex vivo and allowing more confident real-world inference.
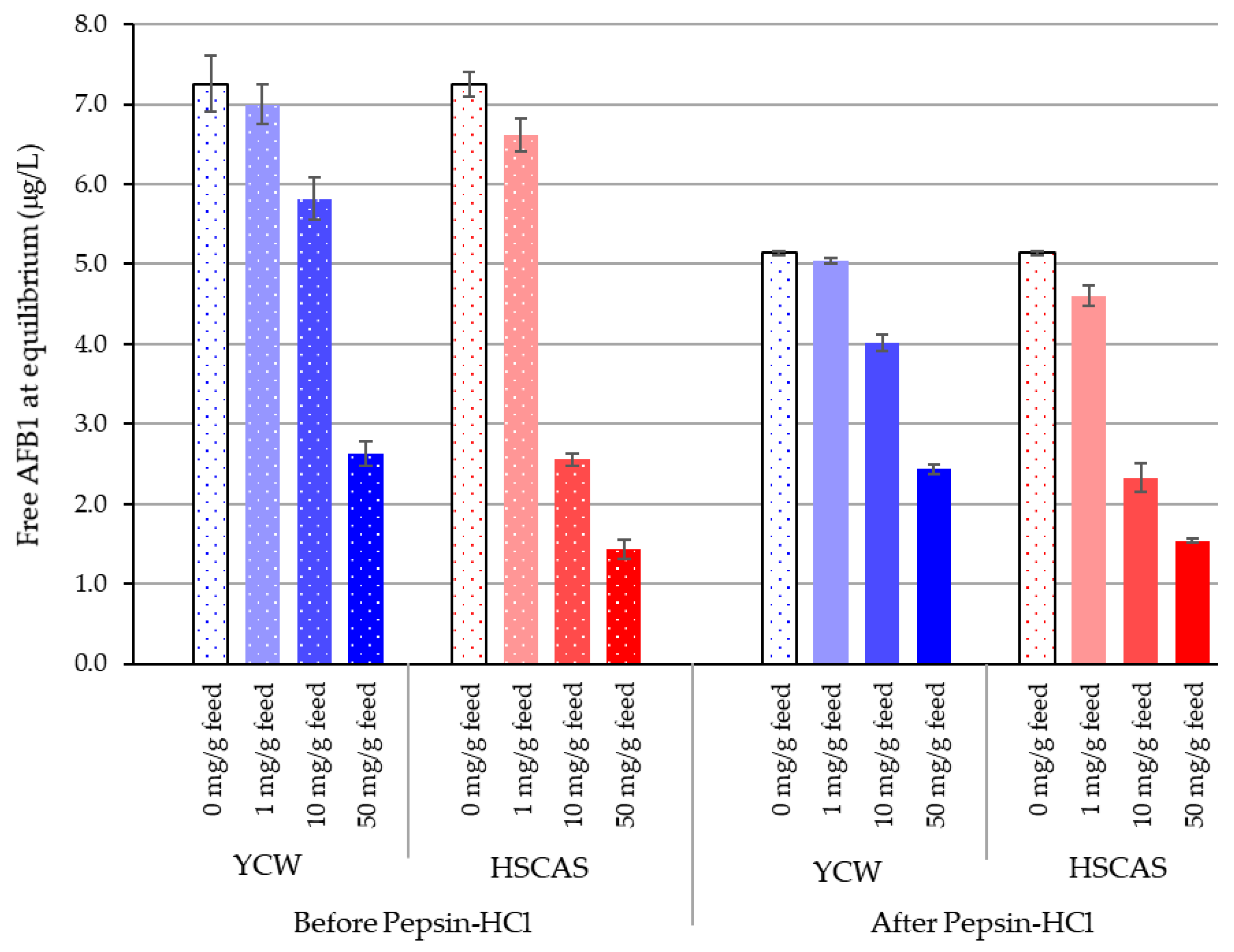
Having established that the test materials can effectively bind AFB1 at a low pH, Model 2 was designed to build upon Model 1 and demonstrate the sequestration of AFB1 at the low “chronic” levels one might expect to find in feedstuffs, using simulated upper gastrointestinal (GI) conditions. The results suggest an innately similar promotion of toxin–binder interactions with the organic materials (feed and YCW) under pepsin–HCl treatment. An inclusion of 0.1% HSCAS reduced the free AFB1 concentration from 7.25 to 6.62 µg/L in water and to 4.60 µg/L after pepsin–HCl digestion, likely due to the enhanced ionisation of clay molecules in this environment. The results obtained for HSCAS at 0.1% were not significantly different from those obtained with YCW (
p < 0.05). An increase in the inclusion of HSCAS significantly enhanced the ability of HSCAS to sequester more AFB1 than YCW. Nevertheless, the YCW feed mixture was also able to significantly reduce free AFB1 down to 2.6 and 2.4 µg/L from the initial 10 µg/L added to the reaction environment. The biggest difference between YCW and HSCAS was noted at an inclusion of 1.0%
w/
v of the tested material (
Table 3). This could be explained by the differences not only in the types of chemical interactions generated, but also because of different optima of sequestration activities that are to be expected from the differences in the composition of the two tested materials.
Interestingly, the observation that the feed can affect the availability of free AFB1 confirmed the existence of potential interactions between the toxins and the organic matrix of the feed, which is also composed of carbohydrates but of a different nature than that of the YCW. In a pre-study (data not shown), the inclusion of 3.3%, 10%, and 30% feed to water (
w/
v) was found to decrease the free AFB1 in the reaction media from 10 down to 8.9, 7.6, and 5.9 µg/L, with a respective decrease of 11.1%, 23.6%, and 41.1% of the free AFB1 after 2 h of incubation at 37 °C. In the presence of pepsin–HCl, the reduction in free AFB1 was further pronounced, with concentrations down to 7.8, 5.8, and 4.4 μg/L, representing a reduction of 22.2%, 42.3%, and 56.2%, respectively, in free AFB1. Activated vegetable fibre (AVF) is successfully used in industrial processes like brewing to systematically reduce the concentrations of mycotoxins and agricultural chemicals to mitigate their impact on waste materials often recycled into animal feed [
32]. The AVF for this application is often derived from micronized wheat envelopes (bran) comprised of a variety of carbohydrates—mainly hemicellulose molecules—composed of β-(1,4)-linked-glucose, -mannose, and -galactose [
33]. However, we hypothesise that the degradation of these molecules in the hindgut is sometimes assisted with enzymatic aids such as β-
d-glucanase but tends to liberate the mycotoxins that have interacted with this vegetal carbohydrate fraction that is then further absorbed as an energy source for the host [
34]. This degradation progressively reduces the interactions, resulting in the release of bound mycotoxins, making them bioavailable to the host once again and thus counterbalancing the competition between the feed matrix and the adsorbent for access to the mycotoxin. YCW consists of an insoluble carbohydrate fraction specifically made out of long chains of β-(1,3)-
d-glucans branched with side chains of β-(1,6)-
d-glucans, with the latter being connected to the inner chains of N-acetyl-glucosamine (forming the chitin fraction) and glycophosphatidylinositol anchors further connected to an outer layer of mannooligosaccharides linked in α-(1,3; 1,4; 1,6) and forming the parietal structure of
Saccharomyces cerevisiae [
35]. This assembly, especially that of β-
d-glucans due to its highly organised tridimensional structure consisting of a single helix further associated in a triple helix and forming fibres of glucans, as well as their high degree of polymerisation [
36,
37], resisted digestion and retained its binding capacity throughout the course of the digestive tract while still maintaining its dynamic properties and preserving cell wall integrity [
38]. Biomass density during the fermentation process also plays a role in the resistance of YCW to even complex lytic mixtures [
39]. These macromolecules interrelate with mycotoxins, such as AFB1, and sequester them into helical β-
d-glucan chains through hydroxyl group hydrogen bonding and van der Waals pi-stacking interactions, as established in previous work [
40]. By contrast, inorganic sequestrants, such as HSCAS, rely on their comparatively large cation exchange capacity, significant surface area, and structural channels and pores to facilitate binding interactions (chelation or chemisorption) with mycotoxins such as AFB1 [
31]. Differences in physical formations such as channels, pores, and interlayer spaces can restrict the sizes of molecules that can effectively be sequestered. As AFB1 is among the smallest mycotoxin molecules, binding was shown to be quite effective for this toxin, but this does not necessarily hold true for other (larger) mycotoxins [
41]. Furthermore, the very inflexible structure of HSCAS compared to the dynamic organisation and adsorption behaviour of YCW, as characterised by the latter’s cooperative and interaction nature, enables YCW to target a larger subset of mycotoxins and generate interactions with zearalenone, deoxynivalenol, patulin [
40], and ochratoxin A [
28].
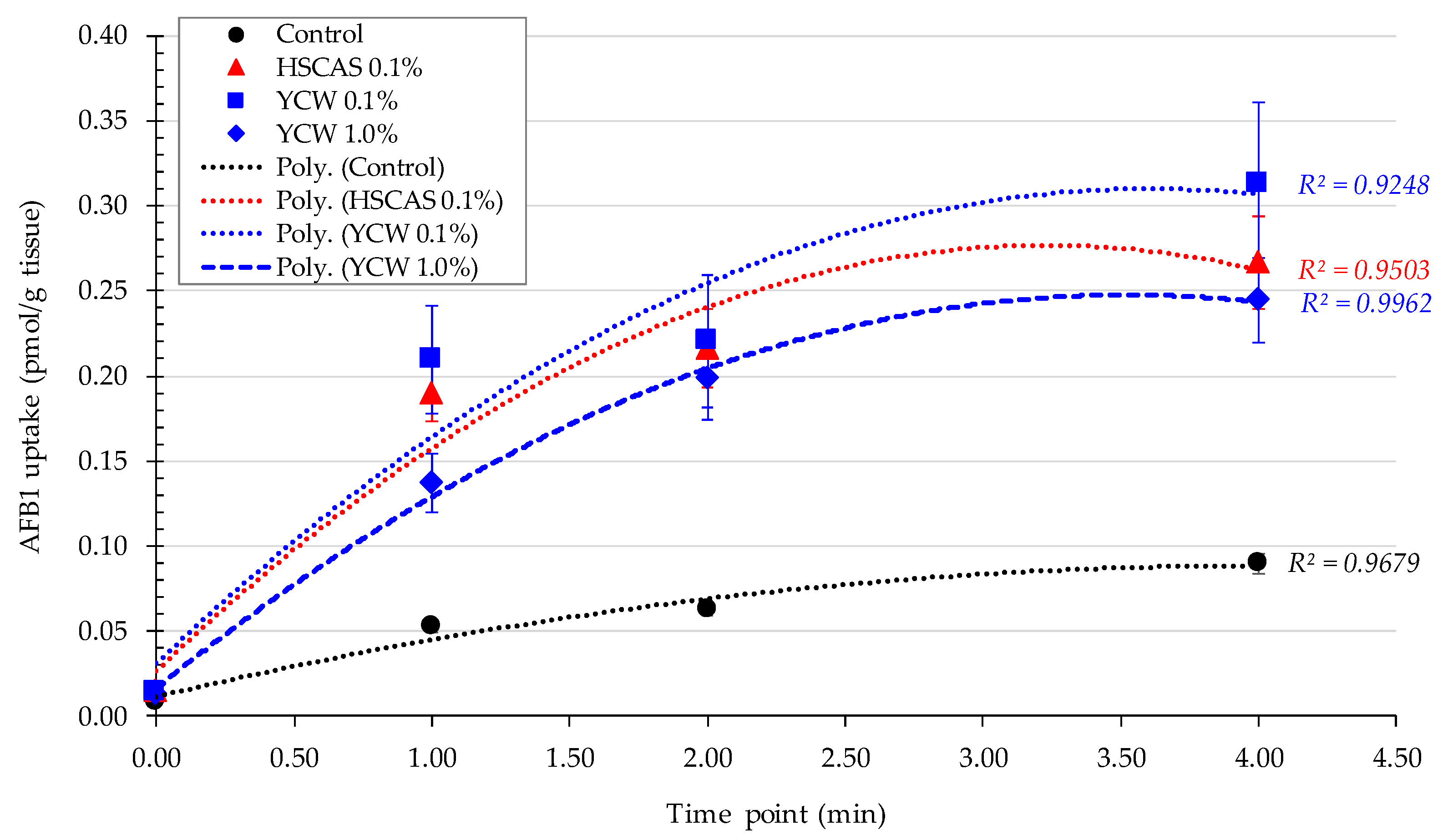
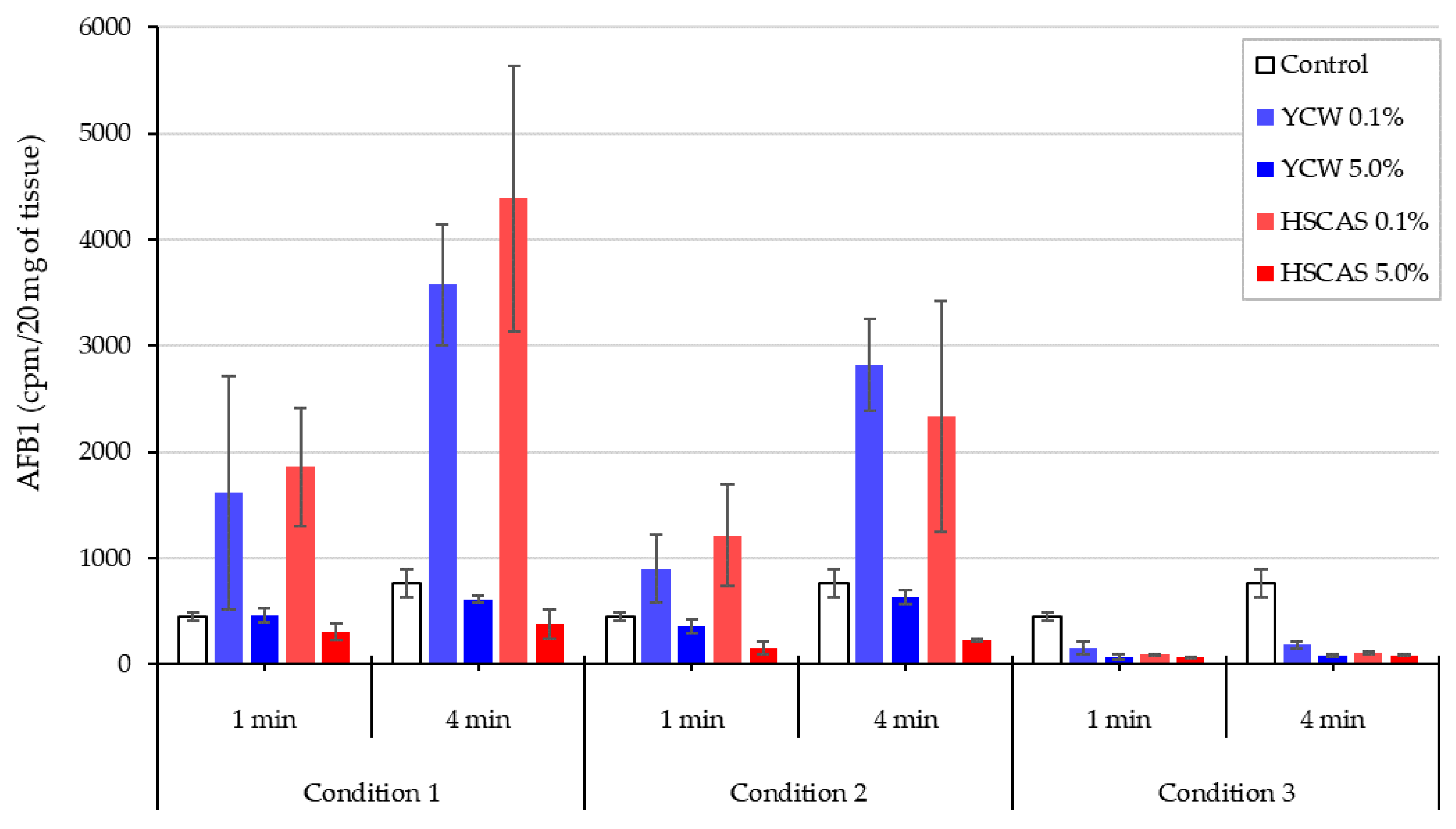
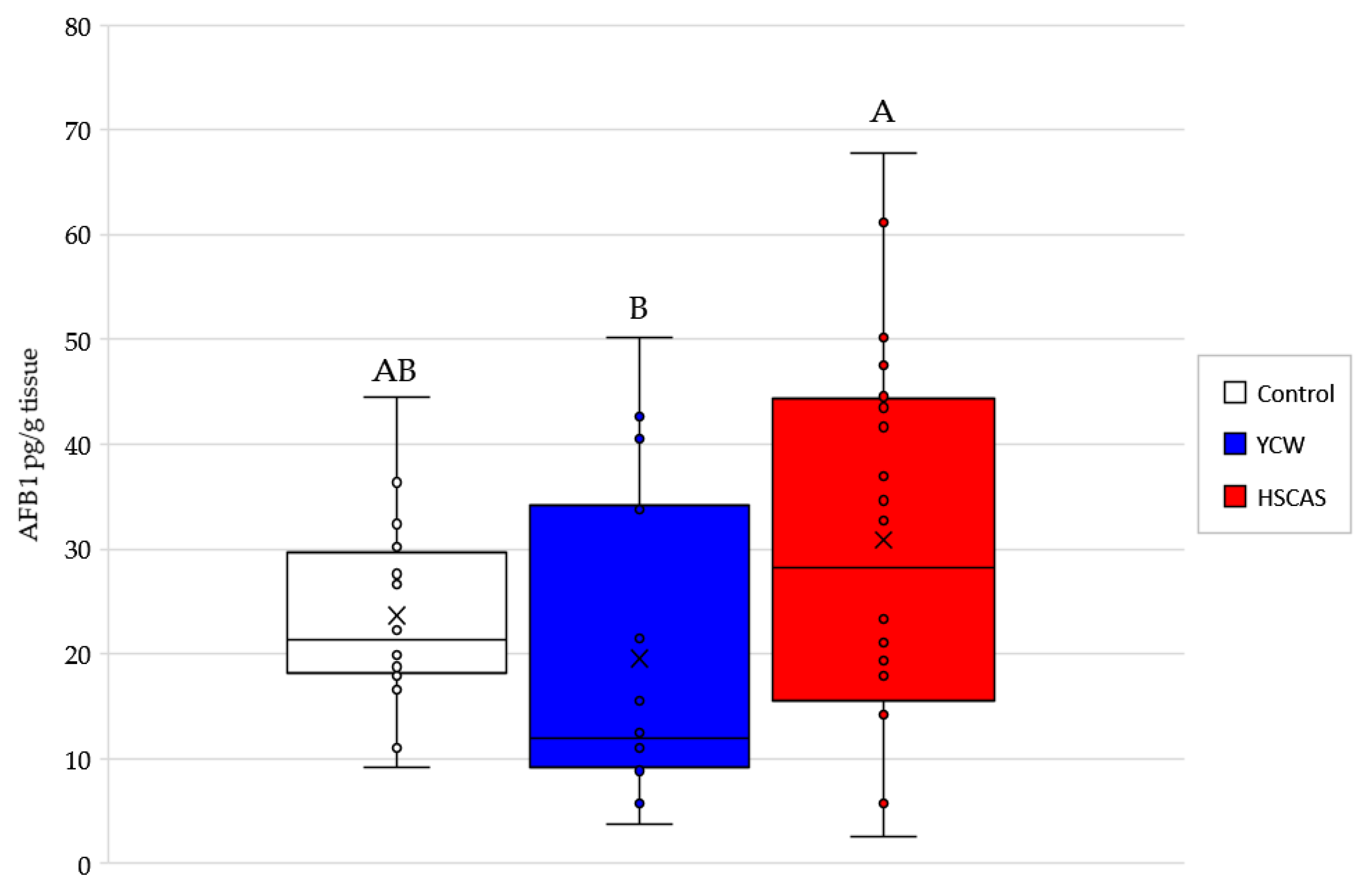
Moving into the ex vivo investigations with the third model tested, AFB1 bioavailability was further evaluated. The observed increase in AFB1 uptake in the intestinal tissue following the use of either 0.1% YCW or HSCAS was attributable to the adhesion of the (sorbent + AFB1) complex directly to the surface of the mucosal layer of the intestinal slices used in our study. Further experiments indicated that the increased inclusion levels (1.0% and 5.0%,
w/
v) of both adsorbents tended to reduce AFB1 uptake due to an increase in the ratio of the adsorbent over AFB1 (fixed) in addition to the fixed ratio of AFB1 to the mucosal surface of the intestinal slices. The adherence phenomenon did not indicate an increase in the transport of AFB1 through the epithelial membrane of the intestinal tissue, which was confirmed by the results obtained with the fourth model. As indicated, a significant factor in the sequestration of AFB1 was the net negative charge exhibited by HSCAS [
31] and the non-charged parietal element of YCW [
42] as well as the interactions that could be promoted with the polar uncharged AFB1 molecule [
40,
43]. Intestinal mucus is produced as a defense mechanism against invasion from intestinal microbiota. The mucin constituents similarly have a net negative charge that acts to draw cations towards itself, thereby facilitating the uptake of many nutrients [
44]. The interactions of significant fields of charge with intermediary, positively charged molecules (possibly dissociated elements of the elution buffer) could account for the rapid and forceful degree to which these adsorbent particles adhere to the epithelial mucus. This model indicates that at low AFB1 concentrations in simulated lower GI conditions, differences in the adsorption profiles of YCW and HSCAS are negligible. It is important to note that the interactions between sorbents (containing bound mycotoxin) and gut epithelial tissue may manifest differently in vivo. This interaction may influence commensal microbiota, the immunology of the host organism, or other physiological parameters, such as mucus exudation. It is widely appreciated that mycotoxins can have deleterious effects on bacterial populations much in the same way as they cause damage to eukaryotic cells. Indeed, penicillin is a fungal metabolite and could be considered a mycotoxin [
45]. The desorption of toxins from the adsorbent in the vicinity of the gut epithelia could conceivably disrupt commensal microbiota directly or indirectly through the host inflammatory response, potentially promoting pathogenic strains [
46]; however, “normal” microbiota typically act to acidify their local environments, thus producing acetate, lactate, butyrate, propionate, and other short chain fatty acids among other bioprotectants [
47]. Thus, within the intestinal lumen, particularly at the mucus interface, environmental conditions can enhance the pH-mediated binding of toxins with their sequestrants.
While Model 3 allowed us to determinate the YCW/HSCAS interactions with live rat intestinal tissue in the presence of a low (10 µg/L) concentration of AFB1, it did not allow us to clearly understand the absorption mechanism of the toxin by the same intestinal tissue and its transfer to the apical side and further into systemic circulation. This model also suffered from important variability due to its ex vivo nature and through the time course exposure to the toxin, notably only enabling a 4 min evaluation over a short period of the interaction. In this context, the fourth model focused on a bicameral system (an Ussing chamber) that allowed the effective measurement of AFB1 transport across live intestinal tissue and was able to “complete the journey” of AFB1 through the digestive tract. Phase separation mitigated the issues observed with Model 3, as particle interactions with the membrane in the apical chamber did not inherently lead to measurement complexities in the basolateral chamber. This model clearly demonstrated a significant reduction in the transfer rate of AFB1 into the basolateral chamber for both tested products. The linearity of AFB1 binding by both sequestrants was notable (
Figure 5). Increased incubation time (synonymous with gut residence time) lends itself to a more efficacious reduction in AFB1 bioavailability, thereby maximising the protective effect afforded by adsorbents in the feed allocation. Furthermore, the control values better fit second order polynomial kinetics, indicating the progressive acceleration of AFB1 absorption over the course of the experiment; this rate increase is of interest. Evidence from Gallo 2008 [
6] that AFB1 is absorbed through a variety of mucosal membranes (and not absorbed specifically in the gut) indicates passive diffusion to be the most likely uptake mechanism, unlike for other mycotoxins, such as deoxynivalenol or ochratoxin A [
48]. As AFB1 is known to induce apoptosis in eukaryotic cells [
49], and prokaryotic populations have been shown to be negatively affected by AFB1 in a dose-dependent manner [
46], it is conceivable that epithelial membrane permeability increased in the presence of 10 µg/L AFB1 due to cellular disruption and, perhaps, the early stage induction of apoptosis. This could also explain the observed acceleration in absorption.
The lack of differences in the accumulation of AFB1 with or without the use of an adsorbent seem to indicate the limited carrying capacity of the intestinal tissue. Contrary to the indications of Model 3, the Model 4 did not record the same adsorbent particle–gut tissue interactions. In the apical solution, the concentration of AFB1 was 30 µg/L, 1000-fold higher than that in the tissue. Moreover, throughout the experiment, the concentration of AFB1 was about 1000-fold higher in the apical than in the basolateral medium. These results suggest that AFB1 did not accumulate in the tissue and that in this system, the rate of AFB1 transport through the rat intestinal tissue was quite slow. In the live rat, AFB1 was mainly transported in the albumin fraction of the blood serum [
50]. In the present trial, this biological transport system did not exist. This may partly explain the slow rate of AFB1 transport in the Ussing chamber system. However, the adsorbent concentrations tested in this study were notably increased in Model 4, from 0.1% in earlier models to 0.3%, thus striking a balance between the results from earlier datasets and reflecting the recommended inclusion rates; this result is also consistent with the conventional 0.2% inclusion level [
41].
The overall results depicted the applicability of the YCW and HSCAS at interacting effectively with AFB1. The efficacy of the sequestration can however change according to the toxin, YCW having demonstrated significantly higher mitigation activity toward zearalenone compared to HSCAS when investigated in a previous study using the same in vitro and ex vivo evaluation [
25]. These materials could thus be of practical interest and applicability in agriculture for mitigating the mycotoxin impact in animals. Mycotoxin occurrence, variety and multiplicity in feedstuffs and corresponding sorbent efficacy should however be considered. Regulatory authorities have approved the use of HSCAS [
51] and a comprehensive toxicological and safety evaluation demonstrated no concerns with consumption of YCW products [
52], making them suitable for animal use.
5. Materials and Methods
5.1. Model 1: In Vitro (FEFANA) Protocol for Assessing AFB1 Sequestration
The FEFANA guidance document for determining the efficacy of mycotoxin inactivators formed the basis of this experiment [
24], following the concentrations established in Yiannikouris et al. [
25], with some adjustment due to the solubility of AFB1. Aflatoxin B1 was tested at six concentrations (10, 20, 30, 40, 50, and 60 µg/mL) over three replicates. An AFB1 (Fermentek, Jerusalem, Israel) stock solution was prepared in acetonitrile and diluted appropriately to obtain a stock concentration of 1.0 mg/mL. The accurate concentration of this stock solution was determined by measuring the absorbance of a 1:100 dilution via spectrophotometry at a wavelength of 350 nm and applying the following equation:
where the absorbance (
A) at 354 nm is the mean of six replicates, and the molecular weight (MW) and the molecular absorptivity (ε) are 312.3 g/mol and 20,900, respectively. Calibration curves were prepared for HPLC-FLD (high-pressure liquid chromatography with fluorescence detection) responses using the calculated AFB1 concentrations.
Treatments consisted of a control blank (water), yeast cell wall (YCW) of enzymatically hydrolysed Saccharomyces cerevisiae sp. cerevisiae (test product; Mycosorb®, Alltech Inc., Nicholasville, KY, USA), hydrated sodium calcium aluminosilicate (HSCAS) (test product containing ≥70 % smectite (dioctahedral montmorillonite); supplied by Alltech Inc., Nicholasville, KY, USA), or activated carbon (AC) (reference material; Norit CASP, Sigma-Aldrich, St. Louis, MO, USA). Treatments were prepared by pipetting 200 µL of the appropriate binding material suspension (10 mg/mL) into test tubes to obtain 0.1% w/v concentrations, the minimum administration dosage according to the supplier (Alltech) guidelines. All suspensions were mixed continuously to ensure homogeneity during pipetting. A 1600 µL volume of either phosphate buffer (pH 7.0) or citrate buffer (pH 3.0) was then added to the test tubes containing the different treatments. Acetonitrile and AFB1 stock solution (1 mg/mL), in this exact order, were added to achieve the final AFB1 10–60 µg/mL concentrations. The test tubes were then continuously mixed during the 90 min incubation at 37 °C. Following centrifugation (3000× g for 10 min), the supernatant was decanted into HPLC vials and placed in the autosampler for further HPLC analysis.
HPLC-FLD analysis was carried out on the samples using a Kinetex C18 2.6 µm (50 mm × 2.1 mm) reverse phase column (Phenomenex, Torrance, CA, USA), and a methanol/water mixture (35:65, v/v) was used for isocratic elution. The flow rate of the mobile phase was adjusted to 0.4 mL/min, with a 5 µL injection volume over a 6 min run time. AFB1 fluorescence quantification was performed at a 365 nm excitation wavelength and a 435 nm emission wavelength.
The Freundlich equation [
53] was applied to analyse the experimental data:
where
Cads is the concentration adsorbed (µg/g);
Caq is the equilibrium concentration in the solution (µg/mL);
KF is the Freundlich adsorption capacity at unit concentration (µg/g); and 1/
n is the Freundlich adsorption intensity.
5.2. Model 2: In Vitro Assessment of AFB1 Binding in Simulated Stomach Conditions
This experiment followed a similar protocol for Model 2 as per Yiannikouris et al. [
25]; however, some modifications were implemented. Tests were carried out in 20 mL silanised glass liquid scintillation vials in a 15 mL reaction volume. Here, 1, 10, or 50 mg portions of test material (YCW or HSCAS) and 1 g of feed (Alimetrics Ltd., Espoo, Finland) were suspended in water in triplicates. The experiment commenced with the addition of
3H-labelled AFB1 (Moravek Biochemicals Inc., Brea, CA, USA) at 10 µg/L, giving activity of 0.005 µCi mL
−1. The mixtures were incubated for 2 h at 37 °C with gentle shaking. Following incubation, the tubes were centrifuged at 3000×
g for 15 min, separating the test materials from the aqueous phase. A volume of 500 µL of supernatant was collected from the reaction vessels and mixed with a 3 mL scintillation cocktail (Ultima Gold, Perkin Elmer, Waltham, MA, USA) in plastic 3.5 mL liquid scintillation tubes before being analysed for tritium activity using a liquid scintillation counter (1450 MicroBeta TriLux, Perkin Elmer, MA, USA). After the first samples (before pepsin–HCl treatment) were taken, the mixtures were resuspended and homogenised, treated with 4.5 mL of 150 mM HCl plus 1.5 mL of activated pepsin solution, and incubated for a further 2 h at 37 °C with gentle shaking. Following this incubation, the tubes were again centrifuged at 3000×
g for 15 min, and 500 µL of the supernatant was drawn off and analysed as above.
5.3. Model 3: Ex Vivo Demonstration of Binding Efficiency in the Lower Gastro-Intestinal Tract
Ten 7-week-old Han/Wistar rats were utilised for this experiment (~250 g body weight). The rats were fed with a Breeding Diet 1314 Forti (Altromin, Lage, Germany) and given ad libitum access to feed and water until the time of the experiment. The rats had no exposure to aflatoxin prior to this experiment. Euthanisation was performed with CO2 asphyxiation followed by dislocation of the cervical vertebrae. A medial incision was immediately made on the peritoneal cavity post-mortem to allow excision of the proximal jejunum. This region of the small intestine (along the attachment of the mesentery) was then opened longitudinally. The digesta was gently removed, and the tissue was rinsed with ice-cold, isotonic saline solution. The live intestinal tissue was cut into 2 mm slices with a mean weight of 27 mg. Sixteen slices were prepared from each rat. The slices were kept in a carbogen-gassed (95% O2, 5% CO2) ice-cold isotonic saline solution before being transferred to warm (37 °C) medium a few minutes before the incubation treatment. Intestinal slices underwent treatment within 45 min of the animal’s death to ensure maintenance of their viability.
A pH 7.3 isotonic saline solution was prepared, as follows, for all rinsing and incubation processes: 128 mmol/L NaCl, 4.7 mmol/L KCl, 2.5 mmol/L CaCl2, 1.2 mmol/L KH2PO4, 2.6 mmol/L MgSO4, and 2.0 mmol/L NaHCO3. A solution of 5 mmol/L d-glucose was also included in the storage medium; however, treatments were conducted in a glucose-free solution.
3H-labelled AFB1 (Moravek Biochemicals Inc., Brea, CA, USA) was spiked into the AFB1 stock solution to achieve 0.15 µCi/mL, and the concentration of AFB1 was set to 10 µg/L for this experiment. Tissue slices were treated in duplicate as follows: the inclusion of YCW at 1 or 10 mg/mL, or HSCAS at 10 mg/mL. Control treatments were identical except for exclusion of the test materials. Tissue slices were gently shaken during incubation in 2 mL volumes of the test medium in glass vials maintained at 37 °C for 0 (a 1 s dip in the treatment solution), 1, 2, and 4 min, and in a pre-study experiment up to 8 min, or before being rinsed in an ice-cold isotonic saline solution to remove the 3H-labelled incubation medium from the tissue surface. Tissue paper was used to remove excess rinse solution from the tissue slice before being placed into pre-weighed 3 mL plastic liquid scintillation vials. The vials were weighed again to assess the tissue mass, and the slices were solubilised overnight in a tissue solvent (Solvable, Perkin Elmer, Waltham, MA, USA). Three millilitres of the liquid scintillation cocktail (Ultima Gold, Perkin Elmer, Waltham, MA, USA) was added, and the radioactivity of the samples was measured by liquid scintillation counting.
The amount of absorbed AFB1 was determined from the radioactivity detected in the samples, with the assumption that all the measured activity came from the 3H-labelled AFB1. The results were corrected for quenching by the tissue and tissue solvent and the efficacy of liquid scintillation counting for tritium under the used conditions.
5.4. Model 4: Impact of Adsorption Capacity on Transmission of AFB1 through Live Intestinal Tissue
Ten eight-week-old male Han/Wistar rats (mean weight of 295 g) were used for this study. The rats were fed with the Breeding Diet 1314 Forti (Altromin, Lage, Germany) and given ad libitum access to feed and water until the time of the experiment. The rats were not exposed to aflatoxin prior to the experiment. As per Model 3, the rats were euthanised with CO2 asphyxiation and cervical dislocation. Immediately after the death of the animal, the abdominal cavity was opened, and the proximal jejunum was removed and opened longitudinally along the attachment of the mesentery. The digesta was gently removed, and the tissue was rinsed with ice-cold, isotonic saline solution. Two-centimeter pieces of the intestinal tissue were cut and pinned between the apical and basolateral halves of the Ussing chambers.
An isotonic saline solution at pH 7.3 was prepared, as follows, for all rinsing and incubation processes: 128 mmol/L NaCl, 4.7 mmol/L KCl, 2.5 mmol/L CaCl2, 1.2 mmol/L KH2PO4, 2.6 mmol/L MgSO4, 2.0 mmol/L NaHCO3, and 5 mmol/L d-glucose. Ethanol was added at a level of 1.5% to increase the solubility of AFB1.
AFB1 and the test substances were only added to the apical chambers of the Ussing apparatus. As in [
25], the total AFB1 concentration in the apical medium was 30 µg/L, and activity of the
3H-labelled AFB1 was 0.5 µCi/mL.
The tissue slices were treated with the test substances (YCW or HSCAS) at 3 mg/mL for their respective treatments. No adsorbent was added to the control treatment. The incubation media were prepared a minimum of 1 h before each experiment to allow AFB1 to bind to the test substances. Both the Ussing chamber system and the incubation media were pre-warmed to 37 °C before the experiment. After pinning the intestinal slice between the chambers, 3.5 mL of apical and basolateral media were added to the respective chambers. The medium in the chambers was circulated and oxygenated via carbogen (95% O2, 5% CO2) bubbling through the chamber system.
Samples were then taken at the beginning and the end of the experiment from the apical chamber, and 40 µL volumes were taken from the basolateral chamber at 20, 40, 60, 80, 100, and 120 min time points. Each sample was placed into a liquid scintillation vial containing 3 mL liquid scintillation cocktail (Ultima Gold, Perkin Elmer, Waltham, MA, USA) and r 3H activity was measured using a liquid scintillation counter. After the incubation period, the intestinal piece was removed from the chamber. Approximately one-third of each piece was weighed, solubilised in tissue solvent, and measured for tritium activity, as for Model 3.
5.5. Statistical Analysis
5.5.1. Model 1
The free AFB1 present after the equilibrium reactions at pH 3.0 and 7.0 was plotted against the bound AFB1 per mass of product for the different adsorbent materials tested and according to the mycotoxin inactivator testing recommendations of FEFANA. In the present work, the three recommended mathematical equations were used to calculate the amount of bound mycotoxin per unit of mass of the test products, and the goodness of fit was tested for each material. The Freundlich model was ultimately chosen as it provided the best fit for the dataset (Microsoft Excel version 2019, Portland, OR, USA). The equation used was as follows: Qeq = Kf Ceq1/n where Qeq is the y-axis, and Ceq is the x-axis. Two-factor ANOVA and Tukey’s honestly significant difference (HSD) tests, with pairwise t-tests, were applied to statistically compare the measurements to the controls and other treatments (SPSS, IBM, version 22).
5.5.2. Model 2
In this model, the feed was amended (or not) with either YCW or HSCAS at three inclusion levels each, providing seven experimental treatments in total (control; 0.1%, 1.0%, and 5.0% YCW; 0.1%, 1.0%, and 5.0% HSCAS). These treatments were compared before and after treatment with pepsin–HCl via a one-way ANOVA, with Tukey’s post hoc analysis used to determine the pairwise differences between the treatment groups. Single-factor ANOVA and pairwise t-tests were applied to statistically compare these measurements to the controls and the other treatments.
5.5.3. Model 3
For each time point (0, 1, 2 and 4 min), a one-way ANOVA was conducted to determine if there was a difference between the AFB1 uptake for each of the four treatments (Control, HSCAS 0.1%, YCW 0.1% and YCW 1.0%). Tukey’s post hoc analysis was then used to determine any pairwise differences between the treatment groups. Dixon’s Q test was further applied to identify and assess statistical outliers.
5.5.4. Model 4
Data were plotted as the AFB1 uptake in the basolateral compartment against time (mins), and linear and polynomial regression analyses were applied to each variable to determine which provided the best fit. The treatments were compared statistically at 120 min using a one-way ANOVA with Tukey’s post hoc analysis used to determine the pairwise differences between the treatment groups.

 ,
, 






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}