Gliotoxin Aggravates Experimental Autoimmune Encephalomyelitis by Triggering Neuroinflammation

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Gliotoxin Aggravates EAE Evolution
2.2. EAE Aggravation by Gliotoxin Is Dose-Dependent
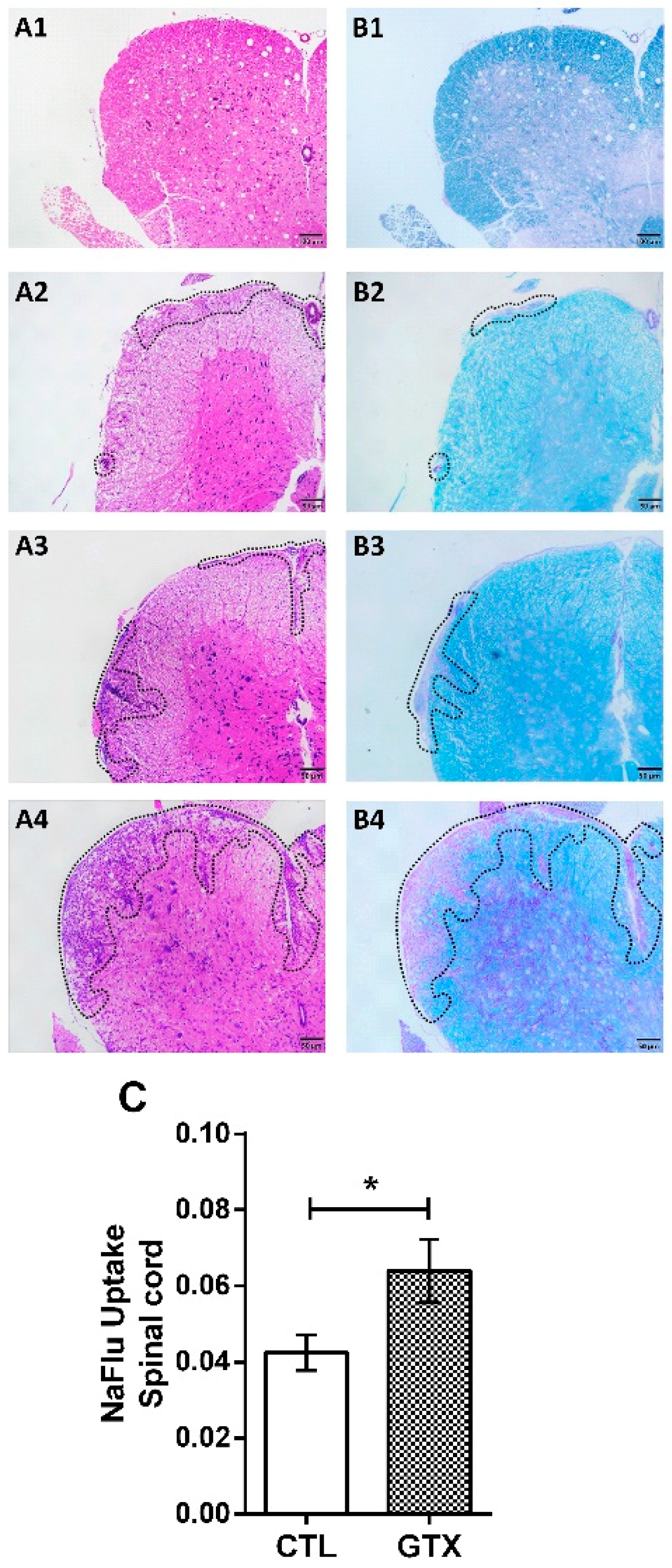
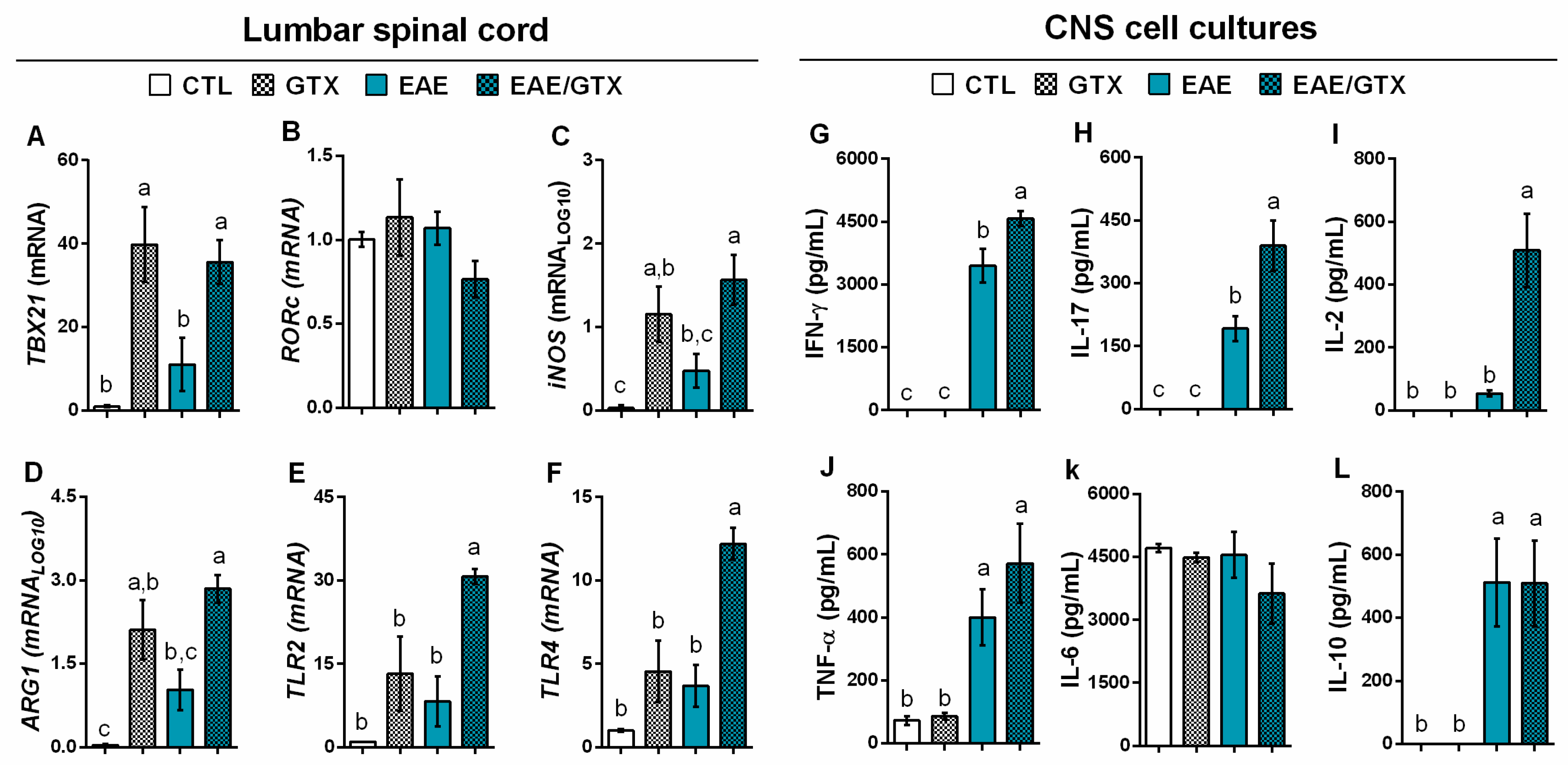
2.3. Gliotoxin Triggers Neuroinflammation and Demyelination
2.4. Gliotoxin Increases Pro-Inflammatory Cytokine Production by Splenic Cells
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. EAE Induction
4.3. Fungal Toxin and Experimental Design
4.4. Hepatic and Renal Function
4.5. Histopathology
4.6. Blood–Spinal Cord Barrier Permeability Assay
4.7. RT-qPCR Analysis
4.8. CNS-Mononuclear Cells Isolation
4.9. Cell Culture Conditions and Cytokine Quantification
4.10. Statistical Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Scharf, D.H.; Brakhage, A.A.; Mukherjee, P.K. Gliotoxin—Bane or boon? Environ. Microbiol. 2016, 18, 1096–1109. [Google Scholar] [CrossRef] [PubMed]
- Kosalec, I.; Pepeljnjak, S. Chemistry and biological effects of gliotoxin. Arch. Ind. Hyg. Toxicol. 2004, 55, 313–320. [Google Scholar]
- Stanzani, M.; Orciuolo, E.; Lewis, R.; Kontoyiannis, D.P.; Martins, S.L.R.; John, L.S.S.; Komanduri, K.V. Aspergillus fumigatus suppresses the human cellular immune response via gliotoxin-mediated apoptosis of monocytes. Blood 2005, 105, 2258–2265. [Google Scholar] [CrossRef]
- Speth, C.; Kupfahl, C.; Pfaller, K.; Hagleitner, M.; Deutinger, M.; Würzner, R.; Mohsenipour, I.; Lass-Flörl, C.; Rambach, G. Gliotoxin as putative virulence factor and immunotherapeutic target in a cell culture model of cerebral aspergillosis. Mol. Immunol. 2011, 48, 2122–2129. [Google Scholar] [CrossRef] [PubMed]
- Camire, R.B.; Beaulac, H.J.; Willis, C.L. Transitory loss of glia and the subsequent modulation in inflammatory cytokines/chemokines regulate paracellular claudin-5 expression in endothelial cells. J. Neuroimmunol. 2015, 284, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Murthy, J.M.K.; Sundaram, C. Chapter 95—Fungal Infections of the Central Nervous System, 1st ed.; Elsevier, B.V.: Amsterdam, The Netherlands, 2014; Volume 121, ISBN 9780702040887. [Google Scholar]
- Panackal, A.A.; Williamson, P.R. Fungal Infections of the Central Nervous System. Contin. Lifelong Learn. Neurol. 2015, 21, 1662–1678. [Google Scholar] [CrossRef]
- Purzycki, C.B.; Shain, D.H. Fungal toxins and multiple sclerosis: A compelling connection. Brain Res. Bull. 2010, 82, 4–6. [Google Scholar] [CrossRef]
- Ascherio, A.; Munger, K.L. Environmental risk factors for multiple sclerosis. Part I: The role of infection. Ann. Neurol. 2007, 61, 288–299. [Google Scholar] [CrossRef]
- Ascherio, A.; Munger, K.L. Environmental risk factors for multiple sclerosis. Part II: Noninfectious factors. Ann. Neurol. 2007, 61, 504–513. [Google Scholar] [CrossRef]
- Gilden, D.H. Personal View Infectious causes of multiple sclerosis. Lancet Neurol. 2005, 4, 195–202. [Google Scholar] [CrossRef]
- Mentis, A.-F.A.; Dardiotis, E.; Grigoriadis, N.; Petinaki, E.; Hadjigeorgiou, G.M.; Mentis, A.F. Viruses and endogenous retroviruses in multiple sclerosis: From correlation to causation. Acta Neurol. Scand. 2017, 136, 606–616. [Google Scholar] [CrossRef] [PubMed]
- Hänninen, A. Infections in MS: An innate immunity perspective. Acta Neurol. Scand. 2017, 136, 10–14. [Google Scholar] [CrossRef] [PubMed]
- Benito-León, J.; Laurence, M. The Role of Fungi in the Etiology of Multiple Sclerosis. Front. Neurol. 2017, 8, 535. [Google Scholar] [CrossRef] [PubMed]
- Hachim, M.Y.; Elemam, N.M.; Maghazachi, A.A. The Beneficial and Debilitating Effects of Environmental and Microbial Toxins, Drugs, Organic Solvents and Heavy Metals on the Onset and Progression of Multiple Sclerosis. Toxins 2019, 11, 147. [Google Scholar] [CrossRef] [PubMed]
- Speth, C.; Rambach, G.; Lass-Flörl, C.; Würzner, R.; Gasque, P.; Mohsenipour, I.; Dierich, M.P. Culture supernatants of patient-derived Aspergillus isolates have toxic and lytic activity towards neurons and glial cells. FEMS Immunol. Med. Microbiol. 2000, 29, 303–313. [Google Scholar] [CrossRef] [PubMed]
- Axelsson, V.; Holback, S.; Sjogren, M.; Gustafsson, H.; Forsby, A. Gliotoxin induces caspase-dependent neurite degeneration and calpain-mediated general cytotoxicity in differentiated human neuroblastoma SH-SY5Y cells. Biochem. Biophys. Res. Commun. 2006, 345, 1068–1074. [Google Scholar] [CrossRef] [PubMed]
- Frame, R.; Carlton, W. Acute toxicity of gliotoxin in hamsters. Toxicol. Lett. 1988, 40, 269–273. [Google Scholar] [CrossRef]
- Sutton, P.; Newcombe, N.R.; Waring, P.; Müllbacher, A. In vivo immunosuppressive activity of gliotoxin, a metabolite produced by human pathogenic fungi. Infect. Immun. 1994, 62, 1192–1198. [Google Scholar]
- Anselmi, K.; Stolz, D.B.; Nalesnik, M.; Watkins, S.C.; Kamath, R.; Gandhi, C.R. Gliotoxin causes apoptosis and necrosis of rat Kupffer cells in vitro and in vivo in the absence of oxidative stress: Exacerbation by caspase and serine protease inhibition. J. Hepatol. 2007, 47, 103–113. [Google Scholar] [CrossRef]
- Liu, H.; Jackman, S.; Driscoll, H.; Larsen, B. Immunologic effects of gliotoxin in rats: Mechanisms for prevention of autoimmune diabetes mellitus. Ann. Clin. Lab. Sci. 2000, 30, 366–378. [Google Scholar]
- Fitzpatrick, L.R.; Wang, J.; Le, T. In vitro and in vivo effects of gliotoxin, a fungal metabolite: Efficacy against dextran sodium sulfate-induced colitis in rats. Dig. Dis. Sci. 2000, 45, 2327–2336. [Google Scholar] [CrossRef] [PubMed]
- Lassmann, H.; Bradl, M. Multiple sclerosis: Experimental models and reality. Acta Neuropathol. (Berl.) 2017, 133, 223–244. [Google Scholar] [CrossRef] [PubMed]
- Pinheiro, M.A.L.; Kooij, G.; Mizee, M.R.; Kamermans, A.; Enzmann, G.; Lyck, R.; Schwaninger, M.; Engelhardt, B.; De Vries, H.E. Immune cell trafficking across the barriers of the central nervous system in multiple sclerosis and stroke. Biochim. Biophys. Acta Mol. Basis Dis. 2016, 1862, 461–471. [Google Scholar] [CrossRef] [PubMed]
- Schenk, G.J.; De Vries, H.E. Altered blood–brain barrier transport in neuro-inflammatory disorders. Drug Discov. Today Technol. 2016, 20, 5–11. [Google Scholar] [CrossRef] [PubMed]
- Caprariello, A.; Mangla, S.; Miller, R.H.; Selkirk, S.M. Apoptosis of oligodendroytes in the CNS results in rapid focal demyelination. Ann. Neurol. 2012, 72, 395–405. [Google Scholar] [CrossRef] [PubMed]
- Patel, R.; Hossain, M.A.; German, N.; Al-Ahmad, A.J. Gliotoxin penetrates and impairs the integrity of the human blood-brain barrier in vitro. Mycotoxin Res. 2018, 34, 257–268. [Google Scholar] [CrossRef]
- Oestreich, K.J.; Weinmann, A.S. Transcriptional mechanisms that regulate T helper 1 cell differentiation. Curr. Opin. Immunol. 2012, 24, 191–195. [Google Scholar] [CrossRef]
- Tapiero, H.; Mathé, G.; Couvreur, P.; Tew, K.D.I. Arginine. Biomed. Pharmacother. 2002, 56, 439–445. [Google Scholar] [CrossRef]
- Moghadam, Z.M.; Hosseini, H.M.; Amin, M.; Behzadi, E.; Fooladi, A.A.I. Microbial metabolite effects on TLR to develop autoimmune diseases. Toxin Rev. 2018, 1–17. [Google Scholar] [CrossRef]
- Murphy, Á.C.; Lalor, S.J.; Lynch, M.A.; Mills, K.H.G. Brain, Behavior, and Immunity Infiltration of Th1 and Th17 cells and activation of microglia in the CNS during the course of experimental autoimmune encephalomyelitis. Brain. Behav. Immun. 2010, 24, 641–651. [Google Scholar] [CrossRef]
- Lovett-racke, A.E.; Yang, Y.; Racke, M.K. Th1 Versus Th17: Are T Cell Cytokines Relevant in Multiple Sclerosis? Biochim Biophys Acta 2011, 1812, 246–251. [Google Scholar] [CrossRef] [PubMed]
- Arellano, G.; Acuña, E.; Reyes, L.I.; Ottum, P.A.; Sarno, P. Th1 and Th17 cells and associated cytokines Discriminate among clinically isolated syndrome and Multiple sclerosis Phenotypes. Front Immunol. 2017, 8, 753. [Google Scholar] [CrossRef] [PubMed]
- Müllbacher, A.; Eichner, R.D. Immunosuppression in vitro by a metabolite of a human pathogenic fungus. Proc. Natl. Acad. Sci. USA 1984, 81, 3835–3837. [Google Scholar] [CrossRef] [PubMed]
- Segal, B.H. Mouldy oldy: How fungus lives among us. Blood 2005, 105, 2239–2240. [Google Scholar] [CrossRef]
- Kupfahl, C.; Geginat, G.; Hof, H. Gliotoxin-mediated suppression of innate and adaptive immune functions directed against Listeria monocytogenes. Med. Mycol. 2006, 44, 591–599. [Google Scholar] [CrossRef] [PubMed]
- Orciuolo, E.; Stanzani, M.; Canestraro, M.; Galimberti, S.; Carulli, G.; Lewis, R.; Petrini, M.; Komanduri, K.V. Effects of Aspergillus fumigatus gliotoxin and methylprednisolone on human neutrophils: Implications for the pathogenesis of invasive aspergillosis. J. Leukoc. Biol. 2007, 82, 839–848. [Google Scholar] [CrossRef] [PubMed]
- Bondy, G.S.; Pestka, J.J. Immunomodulation by fungal toxins. J. Toxicol. Environ. Health Part B Crit. Rev. 2000, 3, 109–143. [Google Scholar]
- Schlam, D.; Canton, J.; Carreño, M.; Kopinski, H.; Freeman, S.A.; Grinstein, S. Gliotoxin Suppresses Macrophage Immune Function by Subverting. MBio 2016, 7, e02242-15. [Google Scholar] [CrossRef] [PubMed]
- Aspinall, R.; Lang, P.O. Interventions to restore appropriate immune function in the elderly. Immun. Ageing 2018, 15, 5. [Google Scholar] [CrossRef]
- Truss, C.O. The role of Candida albicans in human illness. J. Orthomol. Psychiatry 1981, 10, 228–238. [Google Scholar]
- Pisa, D.; Alonso, R.; Jiménez-Jiménez, F.J.; Carrasco, L. Fungal infection in cerebrospinal fluid from some patients with multiple sclerosis. Eur. J. Clin. Microbiol. Infect. Dis. 2013, 32, 795–801. [Google Scholar] [CrossRef] [PubMed]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fraga-Silva, T.F.d.C.; Mimura, L.A.N.; Leite, L.d.C.T.; Borim, P.A.; Ishikawa, L.L.W.; Venturini, J.; Arruda, M.S.P.d.; Sartori, A. Gliotoxin Aggravates Experimental Autoimmune Encephalomyelitis by Triggering Neuroinflammation. Toxins 2019, 11, 443. https://doi.org/10.3390/toxins11080443
Fraga-Silva TFdC, Mimura LAN, Leite LdCT, Borim PA, Ishikawa LLW, Venturini J, Arruda MSPd, Sartori A. Gliotoxin Aggravates Experimental Autoimmune Encephalomyelitis by Triggering Neuroinflammation. Toxins. 2019; 11(8):443. https://doi.org/10.3390/toxins11080443
Chicago/Turabian StyleFraga-Silva, Thais Fernanda de Campos, Luiza Ayumi Nishiyama Mimura, Laysla de Campos Toledo Leite, Patrícia Aparecida Borim, Larissa Lumi Watanabe Ishikawa, James Venturini, Maria Sueli Parreira de Arruda, and Alexandrina Sartori. 2019. "Gliotoxin Aggravates Experimental Autoimmune Encephalomyelitis by Triggering Neuroinflammation" Toxins 11, no. 8: 443. https://doi.org/10.3390/toxins11080443
APA StyleFraga-Silva, T. F. d. C., Mimura, L. A. N., Leite, L. d. C. T., Borim, P. A., Ishikawa, L. L. W., Venturini, J., Arruda, M. S. P. d., & Sartori, A. (2019). Gliotoxin Aggravates Experimental Autoimmune Encephalomyelitis by Triggering Neuroinflammation. Toxins, 11(8), 443. https://doi.org/10.3390/toxins11080443