
Etiology of Four Waves of the COVID-19 Pandemic in Ukraine according to the SARS-CoV-2 Virus Genome Sequencing Data: A Pilot Study
 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. Reverse-Transcriptase Polymerase Chain Reaction (RT-PCR)
2.3. Illumina Next-Generation Sequencing (NGS)
2.4. Bioinformatics and Data Analysis
2.5. Mutation Analysis of Refugee Situation
3. Results
3.1. COVID-19 Infection in Ukraine: March 2020 and June 2023
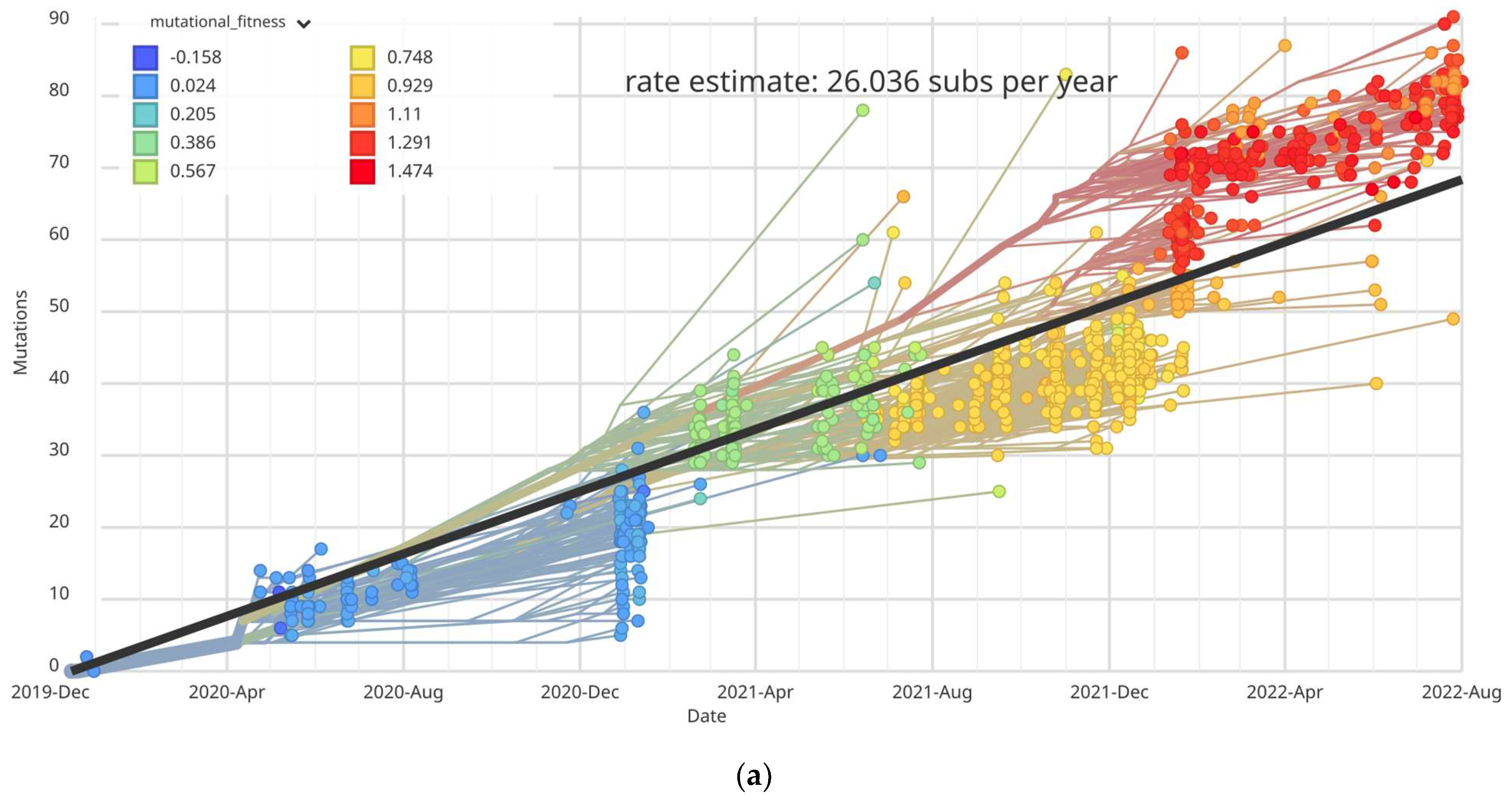
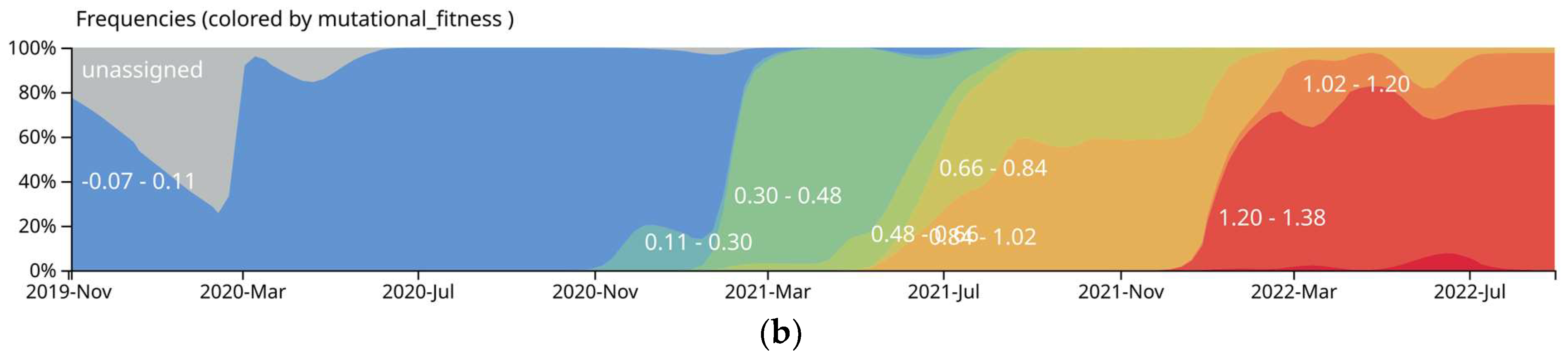
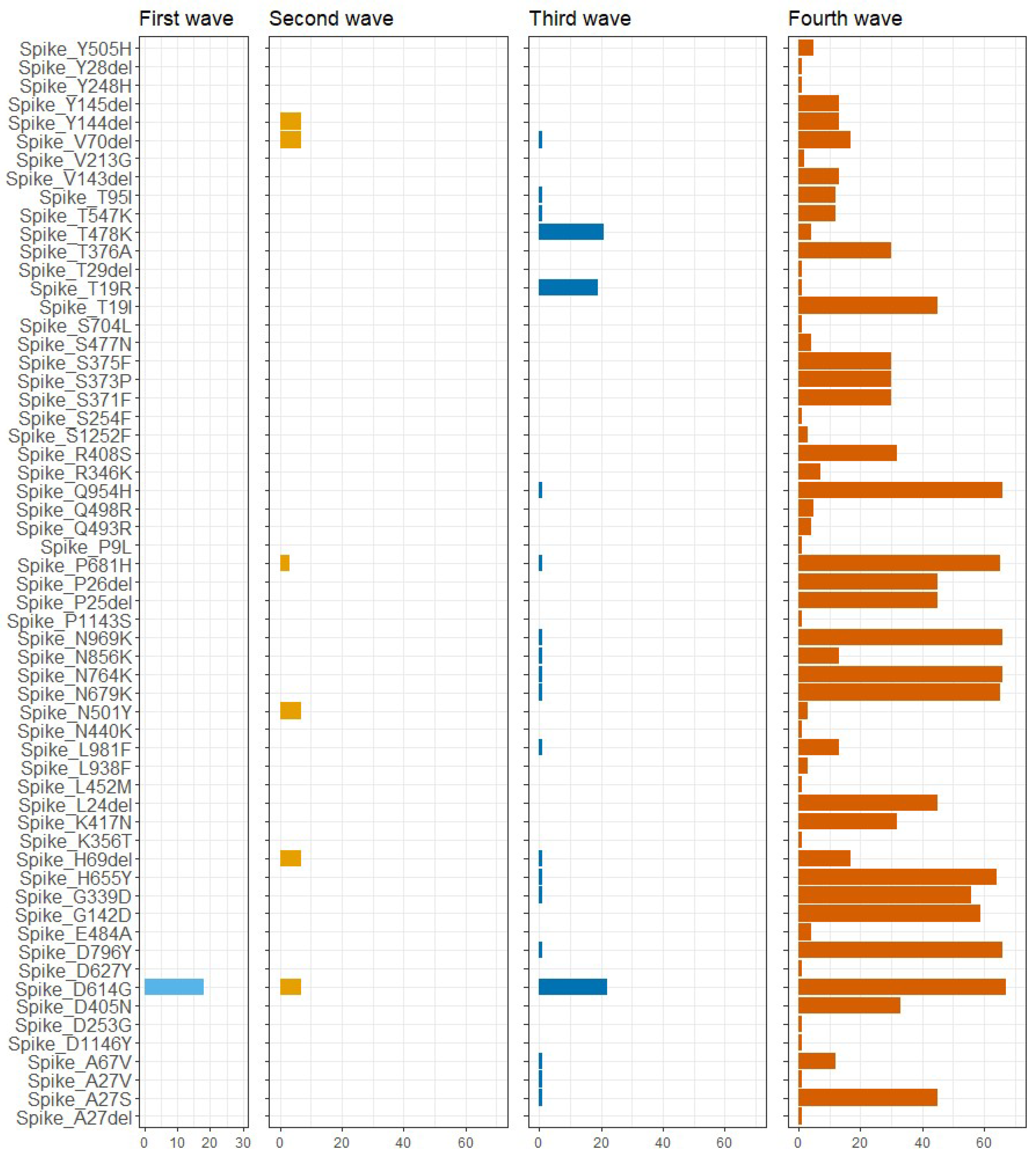
3.2. Mutational Analysis of SARS-CoV-2 in Ukraine
3.3. Cross-Border Movement of SARS-CoV-2 Variants between Ukraine and Poland in Period of Military Invasion
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- COVID-19 Epidemiological Update—19 January 2024. Available online: https://www.who.int/publications/m/item/covid-19-epidemiological-update---19-january-2024 (accessed on 5 February 2024).
- COVID-19 Data|WHO COVID-19 Dashboard. Available online: https://data.who.int/dashboards/covid19/data (accessed on 5 February 2024).
- World Health Organization. Manual for the Laboratory Diagnosis and Virological Surveillance of Influenza; World Health Organization: Geneva, Switzerland, 2011. [Google Scholar]
- Quick, J. nCoV-2019 Sequencing Protocol. 2020. Available online: https://www.protocols.io/view/ncov-2019-sequencing-protocol-bbmuik6w (accessed on 15 December 2023).
- Quick, J. nCoV-2019 Sequencing Protocol v2 (GunIt). 2020. Available online: https://www.protocols.io/view/ncov-2019-sequencing-protocol-v2-bdp7i5rn (accessed on 15 December 2023).
- Quick, J. nCoV-2019 Sequencing Protocol v3 (LoCost). 2020. Available online: https://www.protocols.io/view/ncov-2019-sequencing-protocol-v3-locost-bh42j8ye (accessed on 15 December 2023).
- Lambisia, A.W.; Mohammed, K.S.; Makori, T.O.; Ndwiga, L.; Mburu, M.W.; Morobe, J.M.; Moraa, E.O.; Musyoki, J.; Murunga, N.; Mwangi, J.N.; et al. Optimization of the SARS-CoV-2 ARTIC Network V4 Primers and Whole Genome Sequencing Protocol. Front. Med. 2022, 9, 836728. [Google Scholar] [CrossRef]
- Khare, S.; Gurry, C.; Freitas, L.; Schultz, M.B.; Bach, G.; Diallo, A.; Akite, N.; Ho, J.; Lee, R.T.; Yeo, W.; et al. GISAID’s Role in Pandemic Response. China CDC Wkly. 2021, 3, 1049–1051. [Google Scholar] [CrossRef] [PubMed]
- Huddleston, J.; Hadfield, J.; Sibley, T.; Lee, J.; Fay, K.; Ilcisin, M.; Harkins, E.; Bedford, T.; Neher, R.; Hodcroft, E. Augur: A Bioinformatics Toolkit for Phylogenetic Analyses of Human Pathogens. JOSS 2021, 6, 2906. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K. MAFFT: A Novel Method for Rapid Multiple Sequence Alignment Based on Fast Fourier Transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.-T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Sagulenko, P.; Puller, V.; Neher, R.A. TreeTime: Maximum-Likelihood Phylodynamic Analysis. Virus Evol. 2018, 4, vex042. [Google Scholar] [CrossRef]
- Nextstrain Github. Available online: https://github.com/nextstrain/ncov (accessed on 6 February 2024).
- Hadfield, J.; Megill, C.; Bell, S.M.; Huddleston, J.; Potter, B.; Callender, C.; Sagulenko, P.; Bedford, T.; Neher, R.A. Nextstrain: Real-Time Tracking of Pathogen Evolution. Bioinformatics 2018, 34, 4121–4123. [Google Scholar] [CrossRef]
- Kravchuk, I. Ikravchuk/SARS-CoV-2_Scripts. Available online: https://github.com/ikravchuk/sars-cov-2_scripts (accessed on 6 February 2024).
- Preliminary Genomic Characterisation of an Emergent SARS-CoV-2 Lineage in the UK Defined by a Novel Set of Spike Mutations. Available online: https://virological.org/t/preliminary-genomic-characterisation-of-an-emergent-sars-cov-2-lineage-in-the-uk-defined-by-a-novel-set-of-spike-mutations/563 (accessed on 14 July 2023).
- Meng, B.; Kemp, S.A.; Papa, G.; Datir, R.; Ferreira, I.A.T.M.; Marelli, S.; Harvey, W.T.; Lytras, S.; Mohamed, A.; Gallo, G.; et al. Recurrent Emergence of SARS-CoV-2 Spike Deletion H69/V70 and Its Role in the Alpha Variant B.1.1.7. Cell Rep. 2021, 35, 109292. [Google Scholar] [CrossRef]
- Bhattacharya, M.; Chatterjee, S.; Sharma, A.R.; Lee, S.-S.; Chakraborty, C. Delta Variant (B.1.617.2) of SARS-CoV-2: Current Understanding of Infection, Transmission, Immune Escape, and Mutational Landscape. Folia Microbiol. 2023, 68, 17–28. [Google Scholar] [CrossRef]
- Cherian, S.; Potdar, V.; Jadhav, S.; Yadav, P.; Gupta, N.; Das, M.; Rakshit, P.; Singh, S.; Abraham, P.; Panda, S.; et al. SARS-CoV-2 Spike Mutations, L452R, T478K, E484Q and P681R, in the Second Wave of COVID-19 in Maharashtra, India. Microorganisms 2021, 9, 1542. [Google Scholar] [CrossRef]
- Hyams, C.; Challen, R.; Marlow, R.; Nguyen, J.; Begier, E.; Southern, J.; King, J.; Morley, A.; Kinney, J.; Clout, M.; et al. Severity of Omicron (B.1.1.529) and Delta (B.1.617.2) SARS-CoV-2 Infection among Hospitalised Adults: A Prospective Cohort Study in Bristol, United Kingdom. Lancet Reg. Health Eur. 2023, 25, 100556. [Google Scholar] [CrossRef] [PubMed]
- Onyeaka, H.; Anumudu, C.K.; Al-Sharify, Z.T.; Egele-Godswill, E.; Mbaegbu, P. COVID-19 Pandemic: A Review of the Global Lockdown and Its Far-Reaching Effects. Sci. Prog. 2021, 104. [Google Scholar] [CrossRef]
- Chakkour, M.; Salami, A.; Olleik, D.; Kamal, I.; Noureddine, F.Y.; Roz, A.E.; Ghssein, G. Risk Markers of COVID-19, a Study from South-Lebanon. COVID 2022, 2, 867–876. [Google Scholar] [CrossRef]
- Wang, X.; Pasco, R.F.; Du, Z.; Petty, M.; Fox, S.J.; Galvani, A.P.; Pignone, M.; Johnston, S.C.; Meyers, L.A. Impact of Social Distancing Measures on Coronavirus Disease Healthcare Demand, Central Texas, USA. Emerg. Infect. Dis. J.—CDC 2020, 26, 2361–2369. [Google Scholar] [CrossRef]
- Parra-Lucares, A.; Segura, P.; Rojas, V.; Pumarino, C.; Saint-Pierre, G.; Toro, L. Emergence of SARS-CoV-2 Variants in the World: How Could This Happen? Life 2022, 12, 194. [Google Scholar] [CrossRef] [PubMed]
- Carabelli, A.M.; Peacock, T.P.; Thorne, L.G.; Harvey, W.T.; Hughes, J.; de Silva, T.I.; Peacock, S.J.; Barclay, W.S.; de Silva, T.I.; Towers, G.J.; et al. SARS-CoV-2 Variant Biology: Immune Escape, Transmission and Fitness. Nat. Rev. Microbiol. 2023, 21, 162–177. [Google Scholar] [CrossRef]
- Flores-Vega, V.R.; Monroy-Molina, J.V.; Jiménez-Hernández, L.E.; Torres, A.G.; Santos-Preciado, J.I.; Rosales-Reyes, R. SARS-CoV-2: Evolution and Emergence of New Viral Variants. Viruses 2022, 14, 653. [Google Scholar] [CrossRef]
- Xia, S.; Wang, L.; Jiao, F.; Yu, X.; Xu, W.; Huang, Z.; Li, X.; Wang, Q.; Zhu, Y.; Man, Q.; et al. SARS-CoV-2 Omicron Subvariants Exhibit Distinct Fusogenicity, but Similar Sensitivity, to Pan-CoV Fusion Inhibitors. Emerg. Microbes Infect. 2023, 12, 2178241. [Google Scholar] [CrossRef]
- Noureddine, F.Y.; Chakkour, M.; El Roz, A.; Reda, J.; Al Sahily, R.; Assi, A.; Joma, M.; Salami, H.; Hashem, S.J.; Harb, B.; et al. The Emergence of SARS-CoV-2 Variant(s) and Its Impact on the Prevalence of COVID-19 Cases in the Nabatieh Region, Lebanon. Med. Sci. 2021, 9, 40. [Google Scholar] [CrossRef]
- Announcement on the Suspension of the Airspace of Ukraine. Available online: https://uksatse.ua/index.php?act=Part&CODE=247&id=772&lang=en (accessed on 18 July 2023).
- Situation Ukraine Refugee Situation—Poland. Available online: https://data2.unhcr.org/en/situations/ukraine/location/10781 (accessed on 17 July 2023).
- Ukraine Situation: Regional Refugee Response Plan—March–December 2022. Available online: https://data2.unhcr.org/en/documents/details/92257 (accessed on 18 July 2023).
- Kardas, P.; Babicki, M.; Krawczyk, J.; Mastalerz-Migas, A. War in Ukraine and the Challenges It Brings to the Polish Healthcare System. Lancet Reg. Health Eur. 2022, 15, 100365. [Google Scholar] [CrossRef]
- Rzymski, P.; Falfushynska, H.; Fal, A. Vaccination of Ukrainian Refugees: Need for Urgent Action. Clin. Infect. Dis. 2022, 75, 1103–1108. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.R.; Choe, Y.J.; Jang, E.J.; Kim, J.; Lee, J.J.; Lee, H.Y.; Park, H.; Lee, S.E.; Kim, M.; Kim, S.; et al. Time from Exposure to Diagnosis among Quarantined Close Contacts of SARS-CoV-2 Omicron Variant Index Case-Patients, South Korea. Emerg. Infect. Dis. 2022, 28, 901–903. [Google Scholar] [CrossRef] [PubMed]
- Galmiche, S.; Cortier, T.; Charmet, T.; Schaeffer, L.; Chény, O.; von Platen, C.; Lévy, A.; Martin, S.; Omar, F.; David, C.; et al. SARS-CoV-2 Incubation Period across Variants of Concern, Individual Factors, and Circumstances of Infection in France: A Case Series Analysis from the ComCor Study. Lancet Microbe 2023, 4, e409–e417. [Google Scholar] [CrossRef] [PubMed]
- Zeng, K.; Santhya, S.; Soong, A.; Malhotra, N.; Pushparajah, D.; Thoon, K.C.; Yeo, B.; Ho, Z.J.M.; Cheng, M.C.I. Serial Intervals and Incubation Periods of SARS-CoV-2 Omicron and Delta Variants, Singapore. Emerg. Infect. Dis. 2023, 29, 814–817. [Google Scholar] [CrossRef] [PubMed]
- COVID-19 Omicron Variant: Infectious Period and Asymptomatic and Symptomatic Transmission. Available online: https://www.gov.uk/government/publications/covid-19-omicron-variant-infectious-period-and-asymptomatic-and-symptomatic-transmission (accessed on 25 July 2023).





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1st Wave (March 2020–January 2021) | 2nd Wave (February–August 2021) | 3rd Wave (September–December 2021) | 4th Wave (January–June 2022) | |
|---|---|---|---|---|
| WHO name | Wuhan and its first variants | Alpha | Delta | Omicron |
| Nextstrain clade | 19A, 20A, 20B, 20D | 20I | 21A, 21I, 21J | 21K, 21L, 21M, 22A, 22B, 22C |
| GISAID clade | G, GR, GV, O, V | GRY | GK | GRA |
| Pangolin lineage | B.1, B.1.131, B.1.527, B.1.1, B.1.1.1, B.1.1.243, B.1.1.325, B.1.1.351, B.1.1.374, B.1.1.398, B.1.1.485 | B.1.1.7 | B.1.617 + AY | B.1.1.529 + BA |
| Overall samples from Ukraine used in bioinformatics analysis (n = 1081) | 246 | 69 | 440 | 326 |
| Samples collected and sequenced by authors of this research (n = 167) | 25 | 37 | 28 | 77 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mironenko, A.; Kravchuk, I.; Radchenko, L.; Teteriuk, N.; Holubka, O.; Bolotova, L.; Pydiura, M.; Goy, A. Etiology of Four Waves of the COVID-19 Pandemic in Ukraine according to the SARS-CoV-2 Virus Genome Sequencing Data: A Pilot Study. Microbiol. Res. 2024, 15, 994-1006. https://doi.org/10.3390/microbiolres15020065
Mironenko A, Kravchuk I, Radchenko L, Teteriuk N, Holubka O, Bolotova L, Pydiura M, Goy A. Etiology of Four Waves of the COVID-19 Pandemic in Ukraine according to the SARS-CoV-2 Virus Genome Sequencing Data: A Pilot Study. Microbiology Research. 2024; 15(2):994-1006. https://doi.org/10.3390/microbiolres15020065
Chicago/Turabian StyleMironenko, Alla, Ihor Kravchuk, Larysa Radchenko, Nataliia Teteriuk, Olha Holubka, Liudmyla Bolotova, Mykola Pydiura, and Andriy Goy. 2024. "Etiology of Four Waves of the COVID-19 Pandemic in Ukraine according to the SARS-CoV-2 Virus Genome Sequencing Data: A Pilot Study" Microbiology Research 15, no. 2: 994-1006. https://doi.org/10.3390/microbiolres15020065
APA StyleMironenko, A., Kravchuk, I., Radchenko, L., Teteriuk, N., Holubka, O., Bolotova, L., Pydiura, M., & Goy, A. (2024). Etiology of Four Waves of the COVID-19 Pandemic in Ukraine according to the SARS-CoV-2 Virus Genome Sequencing Data: A Pilot Study. Microbiology Research, 15(2), 994-1006. https://doi.org/10.3390/microbiolres15020065