Abstract
The Sasso Fratino Integral Nature Reserve (Italy) aims to protect nature and territory. Since no anthropic activities are allowed, it represents a good model to study the bacterial community of a wild environment. The aim of this work was to characterise the cultivable and the total bacterial community of soil samples from the reserve in terms of taxonomy, composition, and structure. Seven soil samples were collected at different altitudes, and the chemical composition, the total and the cultivable microbiota, and the antibiotic resistance profiles of isolates were investigated. Total bacterial communities, studied through Next Generation Sequences analysis, included 390 genera. Samples differed in terms of microbial composition basing on the different altitude/vegetation of collection points. Random Amplified Polymorphic DNA Analysis (RAPD) allowed to identify 82 haplotypes out of 158 bacterial isolates. The taxonomic identification through 16S rDNA sequencing revealed that the strains were affiliated to 21 genera. Antibiotic resistance profiles of bacteria were also investigated, highlighting a high resistance against streptomycin and kanamycin. This work represents the first description of the soil bacterial community from the Natural Reserve of Sasso Fratino, and it is the first study considering the soil microbiota of an Italian integral nature reserve.
1. Introduction
The Sasso Fratino Integral Nature Reserve, located within the Parco Nazionale Foreste Casentinesi, Falterona e Campigna National Park, is an important conservation area harbouring one of the few remnants of old-growth forests in Italy. According to the classification of the International Union for Conservation of Nature (IUCN), Sasso Fratino is a “Strict Nature Reserve”, a “protected area that is strictly set aside to protect biodiversity and also possibly geological/geomorphological features, where human visitation, use and impacts are strictly controlled and limited to ensure the protection of the conservation values” [1]. It was the first of this kind to be established in Italy [2]. The term “integral” means that no anthropic activities are allowed (except for the scientific research); therefore, any intervention on the reserve is forbidden, leaving it in its natural state. The Sasso Fratino Reserve was established in 1959, aiming to protect a forest, although not a virgin one, low-intensively disturbed in the past by comparison with other neighbouring stands, thanks to the highly limited accessibility of the area due to the very steep slopes characterising the site morphology, as well as historical features [3,4]. From 2017, Sasso Fratino Reserve is part of the UNESCO World Heritage List, as part of the Ancient and Primeval Beech Forests of the Carpathians and Other Regions of Europe [5].
Forests provide important ecosystem services [6], and the soil is an essential, although often overlooked, component of these ecosystems, since it mediates fundamental nutrient and energy flow patterns that ensure forest productivity, sustain biodiversity, and regulate climate stability [7]. Within the soil, many of the most important services are provided by the microbiome [8], which contributes to nutrient cycling, maintenance of soil fertility, soil carbon sequestration [9], and modulation of resistance and resilience to perturbations [10]. Bacteria represent the most abundant group of soil microorganisms, and soil is considered to harbour the most diverse populations of bacteria of any other environment on Earth [11]. Here, bacteria play important roles such as biogeochemical cycling of carbon, nitrogen, sulfur, phosphorus, and metals, and biodegradation or stabilisation of environmental contaminants [12]. Moreover, many soil microorganisms led to the discovery of novel antibiotics and unique microbial metabolic pathways [9]. In forest soils, the physicochemical properties of soil can influence bacterial diversity and richness [13] and, on the other hand, changes in microbial communities may directly affect ecosystem processes [14].
Following the establishment of Sasso Fratino Integral Nature Reserve, 34 more integral reserves were instituted in Italy, along the whole peninsula (according to the official list of protected areas, last updated in 2010 and published on the Official Gazette n. 125 of 31 May 2010). However, very few studies have been undertaken in these areas, and among these no research on the soil bacterial communities has been conducted. Moreover, even considering the nature reserves spread around the planet, studies on soil microbiome are extremely scarce [15,16,17]. Since Sasso Fratino Reserve is almost not contaminated by human activities, and given the scarcity of scientific research on these areas, it represents a good model to study the bacterial community of a wild environment. Thus, the aim of this work was to characterise the cultivable and the total bacterial community of soil samples from Sasso Fratino Reserve from different viewpoints, in terms of taxonomy, composition, structure of bacterial communities, and antibiotic resistance profile. To the best of our knowledge, this study is among the first taking into consideration the soil bacterial communities of an integral nature reserve, and could be a springboard for further research in this field.
2. Materials and Methods
2.1. Study Area
The Integral Nature Reserve of Sasso Fratino (National Park of Casentine Forests, Mount Falterona and Campigna, northern Apennines, FC, Italy) is located in Central Italy in the northern portion of the Tuscany and Romagna Apennines (43°50′ N; 11°47′ E). The reserve covers an area of 764 ha. The altitude ranges between 650 and 1520 m a.s.l, with a prevailing north-east exposure. Morphology is irregular, with an average slope higher than 65% (and often 100%) and frequent rock crags and overhanging. It presents an oceanic temperate bioclimate, with an average annual temperature of 9 °C and an average annual rainfall of 1750 mm, 255 mm of which falls during summer. Atmospheric moisture is high throughout the year and foggy days are quite frequent. Snow cover lasts about three months, from December to February. The prevailing winds blow from SW and NE and can be blustery along the ridges. The forest is a pure beech stand from 1250 m a.s.l. upwards, while below this altitude is a mixed beech and silver fir forest [2,3].
2.2. Biological Samples Collection
In October 2018, sampling of soil from Sasso Fratino Integral Nature Reserve was performed. Seven soil samples were collected with 50 mL sterile falcon tubes, far from the usual path of the Forest Rangers. Samples were taken proceeding from the highest point of the reserve (Poggio Scali, 1520 m a.s.l.) to La Lama (699 m a.s.l.), to cover all the different microhabitats in the reserve. The list of samples is reported in Table 1, and the geographic localisation of the collection points is represented in Figure S1.

Table 1.
List of samples collected in Sasso Fratino Reserve. For the soil samples, coordinates and altitude of the sampling points are reported. Samples IDs are organised as follows: SF are the initials of Sasso Fratino, followed by the number of the site on the basis of the collection order, while the last letter stands for soil.
2.3. Chemical Characterisation of the Soil Samples
After sampling, the seven soil samples were brought to the laboratory for chemical analyses, performed by an external service (Idro-Consult Laboratori Riuniti s.r.l., Calenzano, Italy). Several parameters were measured, according to the methods reported in Table S1; concentrations of nitrates, sulphates, fluorides, nitrous nitrogen, chemical oxygen demand (COD), total organic carbon (TOC), total iron content, metal(loid)s, and non-metals (i.e., nickel, chrome, zinc, cadmium, arsenic, manganese, selenium, boron, aluminium, copper, lead) were evaluated.
2.4. Genomic DNA Extraction and Next Generation Sequencing
Genomic DNA was extracted from each sample using PowerLyzer® PowerSoil® DNA Isolation Kit (MO BIO Laboratories, Inc., Carlsbad, CA, USA) following the manufacturer instructions. The V3-V4 hypervariable regions of bacterial 16S rRNA gene were sequenced using the primers 341F and 805R [18] according to the protocol reported in the 16S Metagenomic Sequencing Library Preparation protocol from Illumina [19]. Library preparation and demultiplexing were performed following Illumina standard pipeline [20]. Libraries were sequenced in a single run using Illumina MiSeq technology with pair-end sequencing strategy ( bp) with MiSeq Reagent Kit v3. PCR amplification, library construction, and sequencing were performed by an external company (IGA Technology Services, Udine, Italy). Raw sequence files were deposited in the NCBI Sequence Read Archive (SRA) under the accession PRJNA672352.
2.5. Amplicon Sequence Variant (ASV) Inference
PCR primers were removed from all the sequences with Cutadapt [21] (version 2.9) with a maximum error rate of 0.2. Sequences were discarded if the software did not detect one or both primers (‘--discard-untrimmed’ option) and, to ensure that sequences were paired after the trimming process, the ‘--pair-filter=any’ option was used. Then, sequences were clustered into ASVs following the DADA2 pipeline (version 1.16) described at https://benjjneb.github.io/dada2/tutorial.html (accessed on 14 January 2021) [22], using the R software version 3.6.3 [23]. Quality of the reads was checked, then sequences were filtered with the ‘filterAndTrim’ function of DADA2, with a maximum error rate of 2 and a fixed length of 280 and 210 bp for forward and reverse reads, respectively. Trimmed sequences were used for error rate estimation (‘learnErrors’ function) (Figure S2). Finally, sequences were denoised, forward and reverse reads were merged, and an ASV table was obtained [22]; then, chimeras were removed, and taxonomic annotation was performed using the Silva database version 138 [24]. Tables produced by the DADA2 pipeline were then analysed through the phyloseq R package (version 1.30.0) [25]. Representative sequences that were not assigned to domain Bacteria were removed from the dataset (Table S2).
2.6. Count and Isolation of Cultivable Bacteria
Soil samples (1 g) were treated separately at room temperature for 1 h with 20 mL of 10 mM Mg2SO4 in a 50 mL sterile tube under horizontal shaking at 150 rpm, to allow the detachment of bacteria from soil particles. Soil suspensions were diluted (10−1, 10−3, 10−5, 10−7) and 100 µL of each dilution were plated on tryptic soy agar (TSA) medium (Biolife) in triplicate. Plates were incubated at 30 °C, and the CFU (colony forming units) were counted after 48 h. For each compartment about 25 different colonies were randomly chosen, then they were streaked on TSA medium and grown at 30 °C for 48 h. Thus, a panel of 158 isolates from different soil samples (25, 24, 23, 25, 21, 17, 23 from sites 1–7, respectively) was obtained.
2.7. Bacterial Strains and Growth Conditions
The 158 isolates are referred to as “SF” followed by the number of the site and “S” indicating soil samples, followed by the number of the isolate. Bacterial isolates were grown on TSA medium for 48 h at 30 °C.
2.8. Random Amplified Polymorphic DNA (RAPD) Analysis
Cell lysates were prepared by processing a single colony resuspended in 20 μL of sterile dH2O, by thermal lysis (95 °C for 10 min), followed by cooling on ice for 5 min. RAPD profiles of the bacterial isolates were obtained using the primer 1253 (5′-GTTTCCGCCC-3′) [26]. The reaction mix was performed in a 25 μL volume with 1X DreamTaq Buffer, 200 mM dNTPs, 500 ng of primer 1253, 1 U of DreamTaq DNA Polymerase (Thermo Fisher Scientific, Waltham, MA, USA), and 2 μL of thermal lysate used as template. The PCR cycling adopted was set up in a Bio-Rad T100 thermal cycler as follows: 90 °C for 1 min, 95 °C for 95 s followed by 45 cycles of 95 °C for 30 s, 36 °C for 1 min, 75 °C for 2 min, then 75 °C for 10 min, and finally 60 °C for 10 min. Amplicons were visualised through a 2% w/v agarose gel electrophoresis. The assignment of different isolates to the same haplotype group was determined on the basis of the fingerprint pattern of each RAPD product, comparing them for the presence/absence of bands. For haplotypes represented by more than one isolate derived from the same site, a single bacterial isolate was randomly chosen as representative strain for that RAPD haplotype.
2.9. S rRNA Gene Sequences
For each established RAPD haplotype, the 16S rRNA coding gene sequence was used for the taxonomic affiliation of the bacterial isolates. Amplification of 16S rRNA gene was performed in a total volume of 20 μL, containing 1X DreamTaq Buffer, 250 μM dNTPs, 0.6 μM of primers P0 and P6 (5′-GAGAGTTTGATCCTGGCTCAG-3′ and 5′-CTACGGCTACCTTGTTACGA-3′, respectively) [27], 2 U of Dream Taq DNA Polymerase (Thermo Scientific), and 1 μL of thermal lysate used as template. Samples were incubated for 3 min in a thermal cycler (Bio-Rad T100) at 95 °C, then amplification was achieved with 30 cycles of 30 s at 95 °C, 30 s at 50 °C, and 1 min at 72 °C, with a final extension at 72 °C for 10 min. Amplicons were analysed through a 0.8% w/v agarose gel electrophoresis. Sequencing of the 16S rDNA amplicons was performed by the Microsynth Seqlab (Germany). Each sequence was submitted to GenBank, and the accession numbers are from No. MW181140 to No. MW181225 (Table S3).
2.10. Phylogenetic Tree Analysis
Taxonomic affiliation of the strains was determined through the analysis of 16S rRNA gene sequences obtained, as previously described [28]. Briefly, they were aligned to those of type strains retrieved from the Ribosomal Database Project (RDP) [29] using BioEdit [30]. The obtained alignments were then used to build a phylogenetic tree through MEGA X [31] for each genus, applying the Neighbour-Joining algorithm with a 1000-bootstrap resampling.
2.11. Antibiotics Resistance Profile of Cultivable Bacteria
According to Mengoni et al. [32], evaluation of antibiotic resistance of the strains was performed by streaking each of them on TSA medium containing a different concentration of selected antibiotics. The chosen antibiotics were ciprofloxacin, rifampicin, streptomycin, kanamycin, and tetracycline (Table 2). After growth on TSA medium for 48 h at 30 °C, a colony of each strain was suspended in 100 μL of saline solution (0.9% w/v NaCl), streaked on TSA medium supplemented with each antibiotic and then incubated at 30 °C for 48 h. The tested antibiotic concentrations (expressed in μg/mL) were as follows: chloramphenicol (1–2.5–5–10–25–50); ciprofloxacin, streptomycin, and kanamycin (0.5–1–2.5–5–10–50); rifampicin (5–10–25–50–100); tetracycline (0.5–1.25–2.5–5–12.5–25). The different growth levels were ranked as follows: complete (++), strong (+), weak (+−), very weak (+−−), and absence of growth (−); furthermore, MIC (Minimal Inhibitory Concentration) values were determined.
Table 2.
Class and target of the antibiotics used in this work.
2.12. Statistical Analysis
Rarefaction curves of the detected ASVs across samples were visualised. Good’s coverage estimator was calculated using the formula: , where is the number of sequences found only one time in a specimen (singletons) and is the total number of sequences assigned to an ASV in that specimen [33]. Bacterial diversity was estimated using the Shannon index (H), and species evenness (J) was estimated as a function of the Shannon diversity and the number of ASVs detected in the sample (S), according to the Pielou’s formula: [34]. The correlation between soil samples bacterial diversity, and the altitude of the sampling points was estimated using the Pearson coefficient. Correspondence analysis [35] was performed using the ASVs abundance matrix as input, highlighting the different samples on the basis of the vegetation of the sampling points. Different community structures in function of the vegetation were tested using permutational multivariate analysis of variance with 1000 permutations and Bray–Curtis method to calculate pairwise distances [36]. The chemical composition of soil samples was then fitted onto the previously produced ordination analysis. All statistical analyses were performed in the R environment, version 3.6.3 [23], using the vegan package 2.5.6 [37]. Plots were obtained using the ggplot2 package 3.3.0 [38] and the pheatmap package 1.0.12 [39].
3. Results
3.1. Chemical Features of Soil Samples
The complete data set is reported in Table S4: in general, soil samples showed a similar chemical composition. However, the sample SF5S showed reduced concentrations of iron and aluminium, in comparison with the other samples, and the highest TOC; the clustering of the soil samples on the basis of the chemical features showed SF5S as the most divergent in terms of chemical composition (Figure 1). SF7T was the only sample presenting a detectable selenium concentration, and SF6S showed a reduced concentration of nitrates ranging from about 100 to 200-fold in comparison to the others.
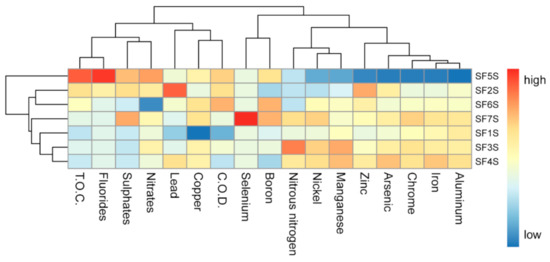
Figure 1.
Heatmap showing the scaled measures of detected soil chemical compounds. Samples are clustered on the basis of their chemical composition similarity. T.O.C.: total organic carbon. C.O.D.: chemical oxygen demand.
3.2. Characterisation of Total Bacterial Communities
The total bacterial communities of soil samples were studied through NGS analysis. Sequencing yielded a total of 576,241 reads. More than 70% of the initial pairs were correctly merged (423,935 sequences); quality filtering steps produced 417,565 high-quality sequences that were correctly mapped into 10,499 ASVs, with an average of 59,652 reads per sample (Figure 2). All the rarefaction curves for the samples reached the plateau (Figure S3), suggesting a good representation of each microbial community. Samples reported a Good’s coverage estimator higher than 99.9%, indicating that singletons accounted only for the 0.1% of reads.
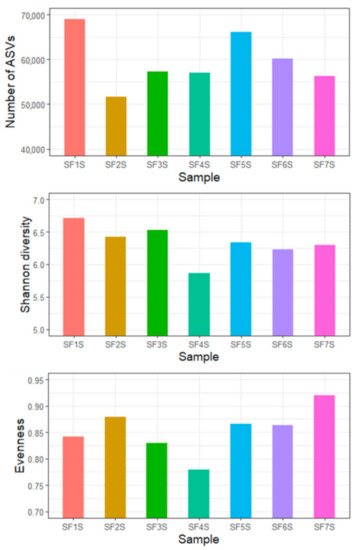
Figure 2.
Indices of the diversity of the soil bacterial community from the Sasso Fratino Integral Nature Reserve. Number of ASVs, Shannon diversity indices, and evenness indices are reported.
Samples showed similar Shannon diversity and evenness, with SF4S resulting less diverse than the others (Figure 2). No correlation was detected between diversity and the altitude of sampling sites (Pearson’s r = 0.32, p-value = 0.48) (Figure S4).
ASVs were classified into 390 genera belonging to 34 phyla, revealing that more than 80% of the ASVs were classified into seven phyla, i.e., Proteobacteria (25.53%), Patescibacteria (15.1%), Verrucomicrobiota (12.84%), Bacteroidota (10.7%), Acidobacteriota (7.26%), Planctomycetota (5.45%), and Actinobacteriota (5.24%). Sample SF1S harboured a total of 32 phyla, SF2S 23, SF3S 29, SF4S 32, SF5S 25, SF6S 27, and SF7S 26 phyla, with 17 out of the 34 phyla shared by all the seven samples. The relative abundances of phyla detected appeared to be similar in all samples (Figure 3a). The ASVs, ordered based on their abundance, were split in five groups, each having 2000 ASVs. From the most abundant 2000 (i.e., ASV1-ASV2000) to the least (i.e., ASV8001-ASV10499), the abundance trend in samples changes (Figure 3b). In particular, there is a shift from an increase in abundance, moving from the first collected soil sample (SF1S) to the last (SF7S), to a progressive decrease.
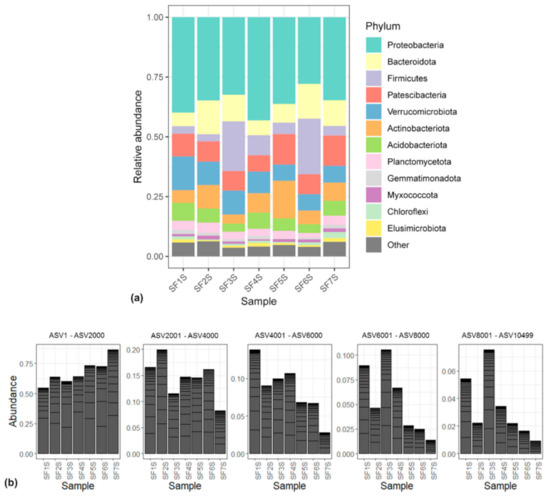
Figure 3.
(a) Bar plot showing the relative abundance of bacterial phyla in each sample. ASVs representing <5% of the whole community have been reported as “Other”; (b) bar plots showing the ASVs abundances in each sample, merged at phylum taxonomic rank, considering 2000 ASVs at a time. From left to right, bar plots considering from ASV1 to ASV2000, from ASV2001 to ASV4000, from ASV4001 to ASV6000, from ASV6001 to ASV8000, from ASV8001 to ASV10499.
The Sasso Fratino forest is a pure beech stand from 1250 m a.s.l. upwards, while below this altitude it is a mixed beech and silver fir forest. Samples SF6S and SF7S are the only ones that were collected below 1250 m, specifically at 1034 and 969 m a.s.l. Correspondence analysis (CA) showed a clear separation between samples collected at different altitudes; samples from SF1S to SF5S, appeared to be very similar to each other and to form a distinct cluster, while SF6S and SF7S resulted separated from the others, along the CA1 (Figure 4). Moreover, SF6S and SF7S appeared to be well separated from each other on the CA2. Different community structures, on the basis of the different vegetation, were tested using permutational multivariate analysis of variance, resulting in a coefficient of determination R2 = 0.26 and in a p-value = 0.048.
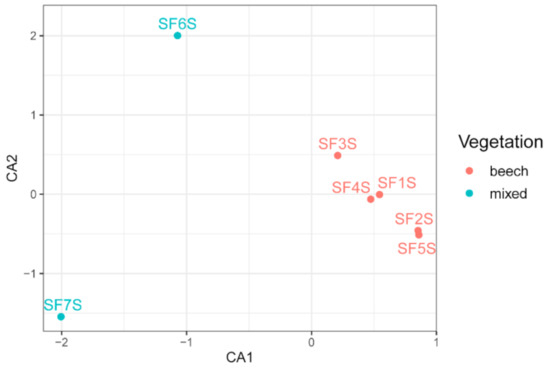
Figure 4.
Correspondence analysis (CA) of soil samples based on bacterial community composition. Sample names are coloured based on the vegetation present in the sampling points.
To assess the possibility that one (or more) chemical compound could affect the taxonomic distribution, the fitting of the chemical composition into the ordination analysis was performed, highlighting that none of the analysed chemical compounds seem to have a statistically significant influence on the bacterial communities distribution among samples (Table S5).
3.3. Isolation of Cultivable Bacteria
Soil samples collected from the different sites were treated and plated as described in Section 2. Data obtained revealed that the highest bacterial titres were detected in samples SF1S, SF2S, SF3S, and SF4S (Table 3), while the lowest ones were represented by the sites SF6S and SF7S. It is possible to notice that there is a decrease of the bacterial titres found in the different soil sites that parallels the order of sampling (i.e., the altitude at which samples were collected).
Table 3.
Titres detected in the different soil samples.
3.4. Structure and Composition of Cultivable Bacterial Communities: RAPD and 16S rRNA Gene Analysis
To investigate the structure of the culturable microbiota and to define the strains (i.e., individual genotypes) present, a RAPD analysis was performed on 158 bacterial isolates. Assuming that isolates sharing the same RAPD fingerprinting correspond to the same strain [40], 82 different RAPD haplotypes were identified, corresponding to at least 82 different bacterial strains. The distribution of the RAPD haplotypes within the different samples is shown in Table S6. The lowest number of haplotypes was detected in the SF1S and SF2S sites (7 haplotypes out of 25 and 24 bacterial isolates, respectively), while the site exhibiting the highest number of haplotypes was SF5S (20 haplotypes out of 21 isolates, respectively), as shown in Table 4. Hence, the first four soil samples collected (i.e., SF1S, SF2S, SF3S, and SF4S) presented high clonal communities, unlike the others. Notably, a low number of strains was shared between the different sites: indeed, only 3 strains were shared among different soil samples.
Table 4.
Distribution of bacterial strains, species, and genera detected in the seven soil samples.
The taxonomic affiliation of representative strains belonging to each RAPD haplotype was determined through 16S rDNA sequencing, as reported in Table S6 and summarised in Figure 5. This analysis revealed that (i) strains were affiliated to 21 different bacterial genera (52.6% Gram negative, 47.6% Gram positive); (ii) the main represented phyla were Proteobacteria and Actinobacteria with 9 and 8 genera, respectively; (iii) the majority of the 16S rDNA sequences was affiliated to the genus Bacillus (16 strains); (iv) Agromyces and Micrococcus were the second most represented genera (11 strains each) followed by Pseudomonas (8 strains); (v) 12 genera (57.14%) were shared among the different sites, however, just the genus Bacillus was shared by all the seven samples; (vi) the number of detected genera for each site seems to increase proceeding from the first collection point to the last one; (vii) Shannon diversity index, calculated on the basis of the distribution of genera for each site, resulted higher for SF6S and SF7S samples in comparison to the others; (viii) moreover, a total of 11 genera were represented exclusively in a single district, such as Agrococcus, Arthrobacter, and Stenotrophomonas genera which were detected only in SF7S (Figure 5).
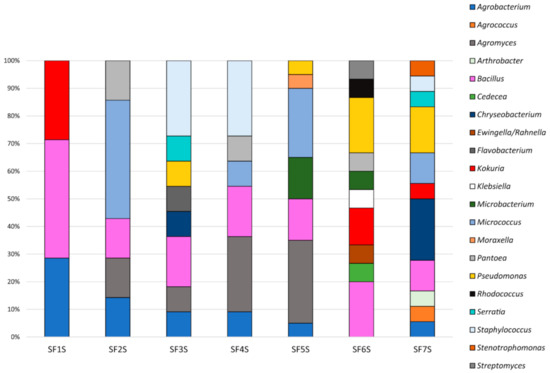
Figure 5.
Composition of cultivable bacterial communities of soil samples collected in Sasso Fratino.
For each genus a 16S rDNA-based phylogenetic tree was constructed as described in Section 2. The analysis of the obtained phylogenetic trees (Figures S5–S23) allowed to affiliate the strains to 37 (possible) different species. The 32.43% of species resulted to be shared by at least two sites. The sample with the highest number of species identified was SF7S, in which 14 different possible species were identified.
3.5. Antibiotics Resistance Profile of Cultivable Bacteria
All RAPD haplotype representatives for each sample (i.e., 89 isolates) were tested. The Minimal Inhibitory Concentration (MIC) of each antibiotic was evaluated and the values are reported in Table 5. The 71.96% of the tested isolates showed a very high MIC value for the antibiotic streptomycin (≥50 μg/mL, i.e., isolates were able to grow even in the presence of the highest antibiotic concentration used in the assay). Analogously, regarding the antibiotic kanamycin, the 57.94% revealed a MIC value ≥50 μg/mL. A few strains (14.01%) demonstrated to tolerate low concentrations (up to 25 μg/mL) of rifampicin, and only 3 strains (2.80%) tolerated rifampicin at the highest concentration (50 μg/mL). Moreover, 45.79% of strains were very sensitive to the antibiotic ciprofloxacin, having a MIC value of 0.5 μg/mL or being completely unable to grow at this antibiotic concentration.
Table 5.
Antibiotic resistance assay of RAPD haplotype representatives. Digits indicate the number of strains having the corresponding minimal inhibitory concentration (MIC) value. MIC values are reported in μg/mL.
4. Discussion
In this work, the cultivable and the total bacterial community from seven soil samples, collected at different altitudes, from the Integral Nature Reserve of Sasso Fratino (Central Italy) were characterised, in terms of taxonomy, composition, and structure of bacterial communities, as well as the culturable bacteria antibiotic resistance profiles.
The total bacterial communities were studied through Next Generation Sequencing (NGS) analysis. Overall, samples showed a similar alpha-diversity and detected Amplicon Sequence Variants (ASVs) were classified into 390 genera belonging to 34 phyla, with more than 80% of the ASVs classified into the seven phyla Proteobacteria, Patescibacteria, Verrucomicrobiota, Bacteroidota, Acidobacteriota, Planctomycetota, and Actinobacteriota. Six out of these seven phyla are reported to be dominant phylum groups in all soils [15]. Otherwise, Patescibacteria is a newly defined superphylum, with unique features as small genome size, ultra-small cell size, and lacking CRISPR, found to be prevalent in aquifer environments [41] and frequently neglected in the soil microbial surveys [42]. The presence of this phylum could be explained by the high air humidity of Sasso Fratino forest throughout the year.
Considering both the abundance and the distribution of ASVs, differences among samples were highlighted in function of the altitude of the sampling points: more abundant ASVs were more represented in the samples collected at the lowest altitudes than at the higher ones. On the contrary, the less abundant ASVs were more represented at high than low altitudes. This uneven distribution of ASVs may suggest that altitude, and thus also vegetation, could have a role in selecting soil microorganisms. Indeed, ordination analysis showed a clear separation between the bacterial communities of samples belonging to sites characterised by different altitude and vegetation. Samples SF6S and SF7S are the only ones that were collected at the altitude corresponding to the mixed beech and silver fir forest; on the contrary, the other samples, collected at a higher altitude, belonged to a pure beech forest. The testing of different community structures, on the basis of the different vegetation, resulted in a statistically significant p-value. Nonetheless, the reliability of this data is limited, due to the small number of samples and the uneven distribution of them among the different altitude/vegetation ranges. Since altitude and vegetation are closely related, the effect of the two factors is not easily distinguishable. However, a change in vegetation/altitude could be responsible for the difference in terms of bacterial communities between the samples; although soil characteristics have been frequently reported as strong drivers of microbial diversity, tree species have been shown to exhibit a stronger impact on community structure than the soil environment. Forest trees exert a stronger influence on soil than other perennial vegetation due to their longevity and the lack of soil management in forests [43], and plant litter is a critical nutrient input source for forest soil and can shape different soil microbial communities [44]. Moreover, also soil temperature and moisture, which are spatially variable in temperate regions [45], are important in shaping microbial communities at both taxonomic and functional gene levels [46]. The ordination analysis showed a clear separation also between samples SF6S and SF7S, whilst the others appeared to be more similar to each other in terms of bacterial composition. This aspect may suggest that besides the two considered macro-factors (altitude and vegetation) other aspects should be taken into account. Thus, to assess the possibility that soil chemical compounds could affect the bacterial taxonomic distribution, the chemical composition was fitted into the ordination analysis. Results showed that none of the analysed chemical compounds seemed to have an influence on the bacterial community distribution; however, also in this case the reliability of this data is limited, due to the small number of samples.
Regarding the titres of the culturable bacterial communities, it is possible to notice that there is a progressive decrease of the titre values found in the different sites, starting from the first sample collected to the last one. Moreover, Random Amplified Polymorphic DNA (RAPD) analysis revealed that the first two sites (i.e., SF1S and SF2S) exhibited the lowest number of haplotypes, thus having a higher degree of clonal diversity in respect to the others. Notably, only three strains were shared between the different sites and 16S rDNA sequencing revealed that the most represented genera were Bacillus, Agromyces, Micrococcus, and Pseudomonas. The 57.14% of genera were shared between more than one sample, although just the genus Bacillus was shared by all the seven sites. This is in line with the fact that members of the Bacillus genus are generally found in soil [47,48]. Notably, the number of detected genera for each site seems to increase proceeding from the first collection point to the last and Shannon diversity index resulted higher for SF6S and SF7S samples in comparison to the others. Altogether, the trends regarding bacterial load and alpha-diversity distribution outlined that sites collected at a lower altitude could have more complex bacterial communities, even though a lower number of culturable bacteria was isolated: this highlights a different bacterial composition in function of the altitude also considering the culturable fraction of soil bacteria, and might be related to the medium used for isolation that exerted a more relevant selective effect on these samples.
In addition, the antibiotic resistance profiles of bacterial isolates were investigated highlighting that most strains showed high resistance against streptomycin and kanamycin, while the most effective antibiotics were rifampicin and ciprofloxacin. Increasing antibiotic resistance, due to an overuse of antibiotics, and emergence of multidrug-resistant pathogens are aspects of great interest nowadays. However, the presence of resistant phenotypes in an uncontaminated area such as Sasso Fratino Reserve is not necessarily a reason for concern; it is known that antibiotic resistance is observed even in bacteria isolated from totally uninhabited places, where they could not come in contact with antibiotics. Antibiotic resistance is an outcome of evolution and resistant genes evolved in natural environments much before these had been used in human therapy [49]. The obtained resistance profiles are in line with previous results regarding bacteria isolated from soil and/or vegetal matrices [28,50].
5. Conclusions
In conclusion, the major differences between samples might be due to the different altitude/vegetation of the collection points, since SF6S and SF7S appear to be different from the others in terms of bacterial composition, lower vital titre (Table 3), lower cultivable clonal communities, and higher cultivable diversity. We are aware that these results are influenced by the small number of analysed samples: this was due to the strict rules regarding the natural reserve and its preservation. However, this work represents the first description of the bacterial community present in soil samples of the Natural Reserve of Sasso Fratino, and it is the first study taking into account the soil bacterial communities of an Italian integral nature reserve. Although it is often difficult to identify specific links between the taxa found in soil and the functional capabilities of the soil microbiome, future works might have this aim and results could be used to compare properties of polluted and anthropized soils with those of uncontaminated environments, and to evaluate the effects of management practices or environmental pollution on the soil bacterial communities [51]. These kinds of studies might allow the prediction of the rates at which a given biogeochemical process occurs, or how they might shift in response to perturbations such as climate change or land-use change [9].
Supplementary Materials
The following are available online at https://www.mdpi.com/article/10.3390/microbiolres12040063/s1, Figure S1: Map of the sampling points, Figure S2: Plots of the estimated error rates of the amplicon dataset, Figure S3: Rarefaction curves for the samples, Figure S4: Absence of correlation between soil sample bacterial diversity and altitude of samples collection points, Figure S5: Phylogenetic tree for the genera Rhizobium and Agrobacterium, Figure S6: Phylogenetic tree for the genus Agrococcus, Figure S7: Phylogenetic tree for the genus Agromyces, Figure S8: Phylogenetic tree for the genus Arthrobacter, Figure S9: Phylogenetic tree for the genus Bacillus, Figure S10: Phylogenetic tree for the genera Cedecea and Klebsiella, Figure S11: Phylogenetic tree for the genus Chryseobacterium, Figure S12: Phylogenetic tree for the genus Flavobacterium, Figure S13: Phylogenetic tree for the genus Kokuria, Figure S14: Phylogenetic tree for the genus Microbacterium, Figure S15: Phylogenetic tree for the genus Micrococcus, Figure S16: Phylogenetic tree for the genus Moraxella, Figure S17: Phylogenetic tree for the genus Pantoea, Figure S18: Phylogenetic tree for the genus Pseudomonas, Figure S19: Phylogenetic tree for the genus Rhodococcus, Figure S20: Phylogenetic tree for the genus Staphylococcus, Figure S21: Phylogenetic tree for the genus Stenotrophomonas, Figure S22: Phylogenetic tree for the genus Streptomyces, Figure S23: Phylogenetic tree for the family Yersiniaceae, Table S1: Soil samples chemical parameters measured, and methods used, Table S2: Reads pair processing throughout the filtering steps, Table S3: Accession numbers of 16S rRNA gene sequences submitted to Gene Bank, Table S4: Values of measured soil chemical compounds, Table S5: Fitting of chemical composition on the ordination analysis of soil samples based on bacterial community composition, Table S6: Distribution of the RAPD haplotypes and taxonomy of bacteria associated to the soil samples in Sasso Fratino.
Author Contributions
Conceptualisation, P.C., G.S., M.Z. and R.F.; data curation, S.D.D.; formal analysis, L.M.C., S.D.D., S.C., A.V. and R.F.; funding acquisition, R.F.; investigation, L.M.C., S.D.D., S.C., A.V., D.F. and E.C.; methodology, L.M.C., S.D.D. and R.F.; project administration, R.F.; resources, P.C. and R.F.; software, S.D.D.; supervision, R.F.; validation, L.M.C. and S.D.D.; visualisation, S.D.D. and A.V.; writing—original draft, L.M.C. and S.D.D.; writing—review and editing, L.M.C., S.D.D., S.C., A.V., D.F., E.C., P.C., G.S., M.Z. and R.F. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
All sequences have been submitted online. Metagenomic sequences were deposited in the NCBI Sequence Read Archive (SRA) under the accession PRJNA672352. 16S rDNA sequences of the bacterial isolates were submitted to GenBank and the accession numbers are from No. MW181140 to No. MW181225.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Category Ia: Strict Nature Reserve|IUCN. Available online: https://www.iucn.org/theme/protected-areas/about/protected-areas-categories/category-ia-strict-nature-reserve (accessed on 29 January 2021).
- Gonnelli, V.; Bottacci, A.; Quilghini, G.; Zoccola, A. Contributo alla conoscenza della flora della Riserva Naturale Integrale di Sasso Fratino. Quad. Stud. Nat. Romagna 2006, 23, 27–75. [Google Scholar]
- Bianchi, L.; Bottacci, A.; Calamini, G.; Maltoni, A.; Mariotti, B.; Quilghini, G.; Salbitano, F.; Tani, A.; Zoccola, A.; Paci, M. Structure and dynamics of a beech forest in a fully protected area in the northern Apennines (Sasso Fratino, Italy). IForest 2011, 4, 136–144. [Google Scholar] [CrossRef] [Green Version]
- Arma dei Carabinieri-Home. Available online: http://www.carabinieri.it/ (accessed on 5 February 2021).
- Ancient and Primeval Beech Forests of the Carpathians and Other Regions of Europe-UNESCO World Heritage Centre. Available online: http://whc.unesco.org/en/list/1133/ (accessed on 29 January 2021).
- Chazdon, R.L. Beyond deforestation: Restoring forests and ecosystem services on degraded lands. Science 2008, 320, 1458–1460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartmann, M.; Niklaus, P.A.; Zimmermann, S.; Schmutz, S.; Kremer, J.; Abarenkov, K.; Lüscher, P.; Widmer, F.; Frey, B. Resistance and resilience of the forest soil microbiome to logging-associated compaction. ISME J. 2014, 8, 226–244. [Google Scholar] [CrossRef]
- Barrios, E. Soil biota, ecosystem services and land productivity. Ecol. Econ. 2007, 64, 269–285. [Google Scholar] [CrossRef]
- Fierer, N. Embracing the unknown: Disentangling the complexities of the soil microbiome. Nat. Rev. Microbiol. 2017, 15, 579–590. [Google Scholar] [CrossRef] [PubMed]
- Allison, S.D.; Martiny, J.B.H. Resistance, resilience, and redundancy in microbial communities. Proc. Natl. Acad. Sci. USA 2008, 105, 11512–11519. [Google Scholar] [CrossRef] [Green Version]
- Roesch, L.F.W.; Fulthorpe, R.R.; Riva, A.; Casella, G.; Hadwin, A.K.M.; Kent, A.D.; Daroub, S.H.; Camargo, F.A.O.; Farmerie, W.G.; Triplett, E.W. Pyrosequencing enumerates and contrasts soil microbial diversity. ISME J. 2007, 1, 283–290. [Google Scholar] [CrossRef] [PubMed]
- He, Z.; Van Nostrand, J.D.; Deng, Y.; Zhou, J. Development and applications of functional gene microarrays in the analysis of the functional diversity, composition, and structure of microbial communities. Front. Environ. Sci. Eng. China 2011, 5, 1–20. [Google Scholar] [CrossRef]
- Fierer, N.; Jackson, R.B. The diversity and biogeography of soil bacterial communities. Proc. Natl. Acad. Sci. USA 2006, 103, 626–631. [Google Scholar] [CrossRef] [Green Version]
- López-Lozano, N.E.; Heidelberg, K.B.; Nelson, W.C.; García-Oliva, F.; Eguiarte, L.E.; Souza, V. Microbial secondary succession in soil microcosms of a desert oasis in the Cuatro Cienegas Basin, Mexico. PeerJ 2013, 2013, e47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, J.; Yin, Y.; Zhu, W.; Zhou, Y. Variations in soil bacterial community diversity and structures among different revegetation types in the Baishilazi nature reserve. Front. Microbiol. 2018, 9, 2874. [Google Scholar] [CrossRef] [Green Version]
- Fonseca, J.P.; Hoffmann, L.; Cabral, B.C.A.; Dias, V.H.G.; Miranda, M.R.; de Azevedo Martins, A.C.; Boschiero, C.; Bastos, W.R.; Silva, R. Contrasting the microbiomes from forest rhizosphere and deeper bulk soil from an Amazon rainforest reserve. Gene 2018, 642, 389–397. [Google Scholar] [CrossRef] [PubMed]
- Priya, G.; Lau, N.S.; Furusawa, G.; Dinesh, B.; Foong, S.Y.; Amirul, A.A.A. Metagenomic insights into the phylogenetic and functional profiles of soil microbiome from a managed mangrove in Malaysia. Agri Gene 2018, 9, 5–15. [Google Scholar] [CrossRef]
- Herlemann, D.P.R.; Labrenz, M.; Jürgens, K.; Bertilsson, S.; Waniek, J.J.; Andersson, A.F. Transitions in bacterial communities along the 2000 km salinity gradient of the Baltic Sea. ISME J. 2011, 5, 1571–1579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kozich, J.J.; Westcott, S.L.; Baxter, N.T.; Highlander, S.K.; Schloss, P.D. Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the miseq illumina sequencing platform. Appl. Environ. Microbiol. 2013, 79, 5112–5120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caporaso, J.G.; Lauber, C.L.; Walters, W.A.; Berg-Lyons, D.; Huntley, J.; Fierer, N.; Owens, S.M.; Betley, J.; Fraser, L.; Bauer, M.; et al. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J. 2012, 6, 1621–1624. [Google Scholar] [CrossRef] [Green Version]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 10. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [Green Version]
- R: The R Project for Statistical Computing. Available online: https://www.r-project.org/ (accessed on 29 January 2021).
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590. [Google Scholar] [CrossRef] [PubMed]
- McMurdie, P.J.; Holmes, S. phyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef] [Green Version]
- Mori, E.; Liò, P.; Daly, S.; Damiani, G.; Perito, B.; Fani, R. Molecular nature of RAPD markers from Haemophilus influenzae Rd genome. Res. Microbiol. 1999, 150, 83–93. [Google Scholar] [CrossRef]
- Di Cello, F.; Fani, R. A molecular strategy for the study of natural bacterial communities by PCR-based techniques. Minerva Biotecnol. 1996, 8, 126–134. [Google Scholar]
- Castronovo, L.M.; Calonico, C.; Ascrizzi, R.; Del Duca, S.; Delfino, V.; Chioccioli, S.; Vassallo, A.; Strozza, I.; De Leo, M.; Biffi, S.; et al. The Cultivable Bacterial Microbiota Associated to the Medicinal Plant Origanum vulgare L.: From Antibiotic Resistance to Growth-Inhibitory Properties. Front. Microbiol. 2020, 11, 862. [Google Scholar] [CrossRef] [PubMed]
- Cole, J.R.; Wang, Q.; Fish, J.A.; Chai, B.; McGarrell, D.M.; Sun, Y.; Brown, C.T.; Porras-Alfaro, A.; Kuske, C.R.; Tiedje, J.M. Ribosomal Database Project: Data and tools for high throughput rRNA analysis. Nucleic Acids Res. 2014, 42, D633–D642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hall, T.A. BioEdit: A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. In Proceedings of the Nucleic Acids Symposium Series; Information Retrieval Ltd.: London, UK, 1999; Volume 41, pp. 95–98. [Google Scholar]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Mengoni, A.; Maida, I.; Chiellini, C.; Emiliani, G.; Mocali, S.; Fabiani, A.; Fondi, M.; Firenzuoli, F.; Fani, R. Antibiotic resistance differentiates Echinacea purpurea endophytic bacterial communities with respect to plant organs. Res. Microbiol. 2014, 165, 686–694. [Google Scholar] [CrossRef] [PubMed]
- Good, I.J. The Population Frequencies of Species and the Estimation of Population Parameters. Biometrika 1953, 40, 237. [Google Scholar] [CrossRef]
- Magurran, A.E. Measuring Biological Diversity; John Wiley and Sons: Hoboken, NJ, USA, 2013; ISBN 1118687922. [Google Scholar]
- Borcard, D.; Gillet, F.; Legendre, P. Numerical Ecology with R; Springer: New York, NY, USA, 2011. [Google Scholar]
- Anderson, M.J. A new method for non-parametric multivariate analysis of variance. Austral. Ecol. 2001, 26, 32–46. [Google Scholar] [CrossRef]
- Dixon, P. VEGAN, a package of R functions for community ecology. J. Veg. Sci. 2003, 14, 927–930. [Google Scholar] [CrossRef]
- Wickham, H. ggplot2; Use R!; Springer International Publishing: Cham, Switzerland, 2016; ISBN 978-3-319-24275-0. [Google Scholar]
- Kolde, R.; Kolde, M.R. Package ‘pheatmap’. R Packag. 2015, 1, 790. [Google Scholar]
- Chiellini, C.; Maida, I.; Emiliani, G.; Mengoni, A.; Mocali, S.; Fabiani, A.; Biffi, S.; Maggini, V.; Gori, L.; Vannacci, A.; et al. Endophytic and rhizospheric bacterial communities isolated from the medicinal plants echinacea purpurea and echinacea angustifolia. Int. Microbiol. 2015, 17, 165–174. [Google Scholar] [CrossRef]
- Tian, R.; Ning, D.; He, Z.; Zhang, P.; Spencer, S.J.; Gao, S.; Shi, W.; Wu, L.; Zhang, Y.; Yang, Y.; et al. Small and mighty: Adaptation of superphylum Patescibacteria to groundwater environment drives their genome simplicity. Microbiome 2020, 8, 51. [Google Scholar] [CrossRef] [Green Version]
- Lemos, L.; Manoharan, L.; Mendes, L.; Venturini, A.; Pylro, V.; Tsai, S.M. Metagenome assembled-genomes reveal similar functional profiles of CPR/Patescibacteria phyla in soils. Environ. Microbiol. Rep. 2020, 12, 651–655. [Google Scholar] [CrossRef] [PubMed]
- Dukunde, A.; Schneider, D.; Schmidt, M.; Veldkamp, E.; Daniel, R. Tree species shape soil bacterial community structure and function in temperate deciduous forests. Front. Microbiol. 2019, 10, 1519. [Google Scholar] [CrossRef] [PubMed]
- Vuong, T.M.D.; Zeng, J.Y.; Man, X.L. Soil fungal and bacterial communities in southern boreal forests of the Greater Khingan Mountains and their relationship with soil properties. Sci. Rep. 2020, 10, 22025. [Google Scholar] [CrossRef]
- Tripathi, B.M.; Lee-Cruz, L.; Kim, M.; Singh, D.; Go, R.; Shukor, N.A.A.; Husni, M.H.A.; Chun, J.; Adams, J.M. Spatial Scaling Effects on Soil Bacterial Communities in Malaysian Tropical Forests. Microb. Ecol. 2014, 68, 247–258. [Google Scholar] [CrossRef]
- Cong, J.; Yang, Y.; Liu, X.; Lu, H.; Liu, X.; Zhou, J.; Li, D.; Yin, H.; Ding, J.; Zhang, Y. Analyses of soil microbial community compositions and functional genes reveal potential consequences of natural forest succession. Sci. Rep. 2015, 5, 10007. [Google Scholar] [CrossRef] [Green Version]
- Amin, M.; Rakhisi, Z.; Zarei Ahmady, A. Isolation and Identification of Bacillus Species From Soil and Evaluation of Their Antibacterial Properties. Avicenna J. Clin. Microbiol. Infect. 2015, 2, 23233. [Google Scholar] [CrossRef]
- Aslim, B.; Beyatli, Y. Determination of Some Properties of Bacillus Isolated from Soil. Turk. J. Biol. 2002, 26, 41–48. [Google Scholar]
- Sengupta, S.; Chattopadhyay, M.K.; Grossart, H.-P. The multifaceted roles of antibiotics and antibiotic resistance in nature. Front. Microbiol. 2013, 4, 47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vassallo, A.; Miceli, E.; Fagorzi, C.; Castronovo, L.M.; Del Duca, S.; Chioccioli, S.; Venditto, S.; Coppini, E.; Fibbi, D.; Fani, R. Temporal Evolution of Bacterial Endophytes Associated to the Roots of Phragmites australis Exploited in Phytodepuration of Wastewater. Front. Microbiol. 2020, 11, 1652. [Google Scholar] [CrossRef] [PubMed]
- Hackl, E.; Zechmeister-Boltenstern, S.; Bodrossy, L.; Sessitsch, A. Comparison of diversities and compositions of bacterial populations inhabiting natural forest soils. Appl. Environ. Microbiol. 2004, 70, 5057–5065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).