
Gastrointestinal Pathologies Associated with Thalassemia: A Systematic Review

 and
and
Abstract
1. Introduction
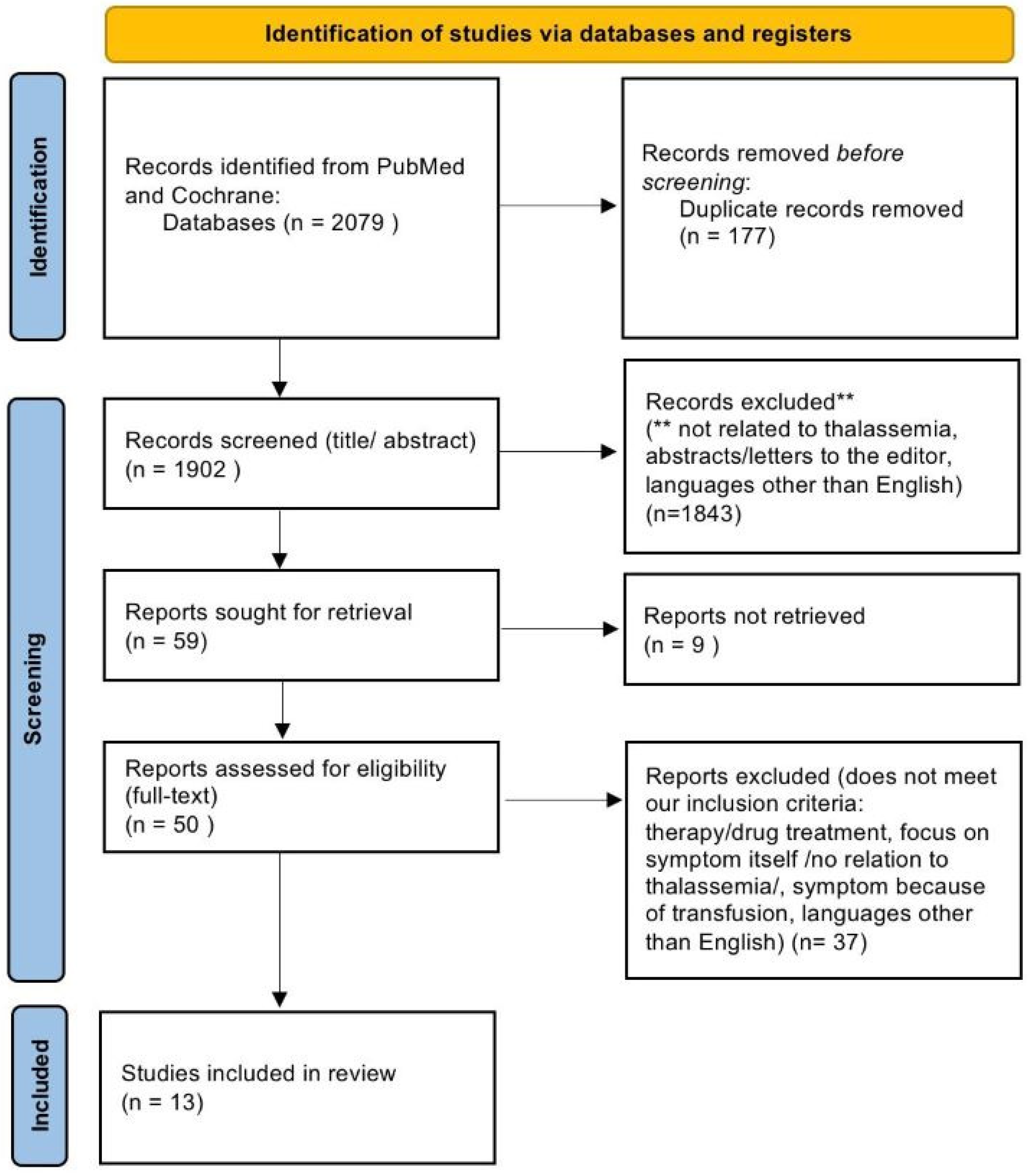
2. Methodology
3. Results
3.1. H. pylori and Thalassemia
3.2. Inflammatory Bowel Disease (IBD) and Thalassemia
3.3. Malabsorption in Thalassemia Including Celiac Disease
3.3.1. Celiac Disease and Thalassemia
3.3.2. Malabsorption and Nutrient Deficiency in Thalassemia
3.4. Immunological Changes in Thalassemia MajorW
3.5. Alpha-Thalassemia X-Linked Intellectual Disability (ATR-X) Syndrome and GI Complications
3.6. Anomalies in Homozygous Alpha-Thalassemia Patients
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Rao, E.; Kumar Chandraker, S.; Misha Singh, M.; Kumar, R. Global distribution of β-thalassemia mutations: An update. Gene 2024, 896, 148022. [Google Scholar] [CrossRef] [PubMed]
- Angastiniotis, M.; Lobitz, S. Thalassemias: An Overview. Int. J. Neonatal Screen. 2019, 5, 16. [Google Scholar] [CrossRef] [PubMed]
- Bajwa, H.; Basit, H. Thalassemia; StatPearls: Treasure Island, FL, USA, 2023. Available online: https://www.ncbi.nlm.nih.gov/books/NBK545151/ (accessed on 24 February 2025).
- MedlinePlus Genetics. Alpha Thalassemia; National Library of Medicine: Bethesda, MD, USA, 2020. Available online: https://medlineplus.gov/genetics/condition/alpha-thalassemia/ (accessed on 19 February 2025).
- MedlinePlus Genetics. Beta Thalassemia; National Library of Medicine: Bethesda, MD, USA, 2020.
- Baird, D.C.; Batten, S.H.; Sparks, S.K. Alpha- and Beta-thalassemia: Rapid Evidence Review. Am. Fam. Physician 2022, 105, 272–280. [Google Scholar] [PubMed]
- Origa, R. Beta-Thalassemia. Genet. Med. 2017, 19, 609–619. [Google Scholar] [CrossRef]
- Musallam, K.M.; Rivella, S.; Vichinsky, E.; Rachmilewitz, E.A. Non-transfusiondependent thalassemias. Haematologica 2013, 98, 833–844. [Google Scholar] [CrossRef]
- Farashi, S.; Harteveld, C.L. Molecular basis of alpha-thalassemia. Blood Cells Mol. Dis. 2018, 70, 43–53. [Google Scholar] [CrossRef]
- El Hasbani, G.; Musallam, K.M.; Uthman, I.; Cappellini, M.D.; Taher, A.T. Thalassemia and autoimmune diseases: Absence of evidence or evidence of absence? Blood Rev. 2022, 52, 100874. [Google Scholar] [CrossRef]
- Hodroj, M.H.; Bou-Fakhredin, R.; Nour-Eldine, W.; Noureldine, H.A.; Noureldine, M.H.A.; Taher, A.T. Thalassemia and malignancy: An emerging concern? Blood Rev. 2019, 37, 100585. [Google Scholar] [CrossRef]
- Cappellini, M.D.; Cohen, A.; Eleftheriou, A.; Piga, A.; Porter, J.; Taher, A. Chapter 8, The Liver in Thalassaemia. In Guidelines for the Clinical Management of Thalassaemia, 2nd Revised ed.; Thalassaemia International Federation: Nicosia, Cyprus, 2008. Available online: https://www.ncbi.nlm.nih.gov/books/NBK173968/ (accessed on 24 February 2025).
- Cappellini, M.D.; Cohen, A.; Eleftheriou, A.; Piga, A.; Porter, J.; Taher, A. Chapter 18, Outline of Diagnostic Dilemmas in Thalassaemia. In Guidelines for the Clinical Management of Thalassaemia, 2nd Revised ed.; Thalassaemia International Federation: Nicosia, Cyprus, 2008. Available online: https://www.ncbi.nlm.nih.gov/books/NBK173977/ (accessed on 24 February 2025).
- Chauhan, D.; Kilic, Y.; Segal, J.; Patel, N.; Koizia, L. An Unusual Cause of Gastrointestinal Perforation in an Adolescent Patient With Beta-Thalassemia on Deferasirox and SARS-CoV-2 Infection. J. Hematol. 2021, 10, 76–79. [Google Scholar] [CrossRef]
- Drugs.com. Deferasirox Side Effects; c2000–2025. Available online: https://www.drugs.com/sfx/deferasirox-side-effects.html (accessed on 19 February 2025).
- Schnedl, W.J.; Mangge, H.; Schenk, M.; Enko, D. β-thalassemia minor, carbohydrate malabsorption and histamine intolerance. Hematol. Rep. 2017, 9, 7043. [Google Scholar] [CrossRef]
- Taher, A.T.; Musallam, K.M.; Karimi, M.; El-Beshlawy, A.; Belhoul, K.; Daar, S.; Saned, M.S.; El-Chafic, A.H.; Fasulo, M.R.; Cappellini, M.D. Prevalence and distribution of iron overload in patients with β-thalassemia major: A multicenter study. Eur. J. Haematol. 2014, 93, 520–527. [Google Scholar] [CrossRef]
- Ezer, U.; Gulderen, F.; Culha, V.K.; Akgul, N.; Gurbuz, O. Immunological status of thalassemia syndrome. Pediatr. Hematol. Oncol. 2002, 19, 51–58. [Google Scholar] [CrossRef] [PubMed]
- McColl, K.E. Clininal practice Helicobacter pylori infection. N. Engl. J. Med. 2010, 362, 1597–1604. [Google Scholar] [CrossRef] [PubMed]
- Mentis, A.; Lehours, P.; Mégraud, F. Epidemiology and diagnosis of Helicobacter pylori infection. Helicobacter 2015, 20, 1–7. [Google Scholar] [CrossRef]
- Balci, Y.I.; Aral, Y.Z.; Covut, I.E.; Polat, Y.; Turk, M.; Acimis, N. The frequency of Helicobacter pylori infection in Beta Thalassemia major Patients with recurrent abdominal pain. Pak. J. Med. Sci. 2011, 27, 316–319. [Google Scholar]
- Woods, K.F.; Onuoha, A.; Schade, R.R.; Kutlar, A. Helicobacter pylori infection in sickle cell disease. J. Natl. Med. Assoc. 2000, 92, 361–365. [Google Scholar]
- Drossman, D.A. Functional Gastrointestinal Disorders: History, Pathophysiology, Clinical Features, and Rome IV. Gastroenterology 2016, 150, 1262–1279.e2. [Google Scholar] [CrossRef]
- Karimi, M.; Imanieh, M.H.; Ghiam, A.F.; Hashemi, Z. Investigation of Helicobacter pylori infection in β- thalassaemia major patients with recurrent abdominal pain. Eur. J. Gastroenterol. Hepatol. 2005, 17, 1363–1367. [Google Scholar] [CrossRef]
- Filmann, N.; Rey, J.; Schneeweiss, S.; Ardizzone, S.; Bager, P.; Bergamaschi, G.; Koutroubakis, I.; Lindgren, S.; de la Morena, F.; Moum, B.; et al. Prevalence of anemia in inflammatory bowel diseases in European countries: A systematic review and individual patient data meta-analysis. Inflamm. Bowel Dis. 2014, 20, 936–945. [Google Scholar] [CrossRef]
- Gomollón, F.; Gisbert, J.P. Anemia and inflammatory bowel diseases. World J. Gastroenterol. 2009, 15, 4659–4665. [Google Scholar] [CrossRef]
- Reinisch, W.; Staun, M.; Bhandari, S.; Muñoz, M. State of the iron: How to diagnose and efficiently treat iron deficiency anemia in inflammatory bowel disease. J. Crohns. 2013, 7, 429–440. [Google Scholar] [CrossRef] [PubMed]
- Testa, A.; Rispo, A.; Romano, M.; Riegler, G.; Selvaggi, F.; Bottiglieri, E.; Martorano, M.; Rea, M.; Gravina, A.; Nardone, O.M.; et al. The burden of anaemia in patients with inflammatory bowel diseases. Dig. Liver Dis. 2016, 48, 267–270. [Google Scholar] [CrossRef] [PubMed]
- Bank, I.; Busari, J.O. Crohn’s disease, autoimmune thyroiditis, and beta-thalassemia trait in an adolescent: An unusual combination of diseases. Eur. J. Pediatr. 2008, 167, 1343–1346. [Google Scholar] [CrossRef]
- Dubcenco, E.; Jeejeebhoy, K.N.; Streutker, C.J.; Zalev, A.H.; Garvey, M.B.; Kim, Y.-I.; Baker, J.P. A patient with anemia of obscure origin: Crohn’s disease in disguise. Nat. Clin. Pract. Gastroenterol. Hepatol. 2006, 3, 229–233. [Google Scholar] [CrossRef]
- Valizadeh, N.; Shateri, K. Association of celiac disease with inflammatory bowel disease and colonic cancer and liver involvement in a case of β-thalassemia minor. Shiraz E-Med. J. 2009, 10, 90–92. [Google Scholar]
- Oxentenko, A.S.; Rubio-Tapia, A. Celiac Disease. Mayo Clin. Proc. 2019, 94, 2556–2571. [Google Scholar] [CrossRef]
- Nijhawan, S.; Katiyar, P.; Nagaich, N.; Saradava, V.; Nijhawan, M.; Gupta, G.; Mathur, A.; Sharma, R.; Nepalia, S. Prevalence of associated disorders in Indian patients with celiac disease. Indian J. Gastroenterol. 2013, 32, 330–334. [Google Scholar] [CrossRef]
- Shahramian, I.; Dehghani, S.M.; Haghighat, M.; Noori, N.M.; Teimouri, A.R.; Sharafi, E.; Kalili, M. Serologic evaluation of celiac disease in patients with beta thalassemia major and control. Gastroenterol. Hepatol. Bed Bench 2015, 8, 153–159. [Google Scholar]
- Honar, N.; Kamali, S.; Karimi, M. Frequency of Celiac Disease in Children with Beta Thalassemia major. Iran. J. Ped. Hematol. Oncol. 2014, 4, 48–52. [Google Scholar]
- Noori, N.M.; Shahramian, I.; Teimouri, A.; Haghighat, M.; Dehghani, S.M.; Sharafi, E. Evaluation of Tissue Transglutaminase IgA in Thalassemia Minor Patients. Asian J. Med. Pharm. Res. 2017, 7, 19–24. [Google Scholar]
- Parakh, A.; Sudha, S.; Dubey, A.P.; Gupta, A. Celiac disease in a child with beta-thalassemia major: A need for improved screening and awareness. J. Pediatr. Hematol. Oncol. 2008, 30, 913–914. [Google Scholar] [CrossRef] [PubMed]
- Acquaviva, A.; Municchi, G.; Marconcini, S.; D’Ambrosio, A.; Morgese, G. Celiac disease in a patient with beta-thalassemia major. J. Pediatr. Gastroenterol. Nutr. 2003, 36, 489–491. [Google Scholar] [CrossRef] [PubMed]
- Montuori, M.; Smacchia, M.P.; Iorfida, D.; Leoni, S.; Trovato, C.M.; Gatti, S.; Celletti, I.; Valitutti, F.; Capogna, M.; Anania, C.; et al. An uncommon diagnosis of celiac disease in a thalassemic girl. Dig. Liver Dis. 2014, 46, e117. [Google Scholar] [CrossRef]
- Ciccocioppo, R.; Bernardo, M.E.; Russo, M.L.; Vanoli, A.; Franco, C.; Martinetti, M.; Catenacci, L.; Giorgiani, G.; Zecca, M.; Piralla, A.; et al. Allogeneic hematopoietic stem cell transplantation may restore gluten tolerance in patients with celiac disease. J. Pediatr. Gastroenterol. Nutr. 2013, 56, 422–427. [Google Scholar] [CrossRef]
- Oerter, K.E.; Kamp, G.A.; Munson, P.J.; Nienhuis, A.W.; Cassorla, F.G.; Manasco, P.K. Multiple hormone deficiencies in children with hemochromatosis. J. Clin. Endocrinol. Metab. 1993, 76, 357–361. [Google Scholar] [CrossRef]
- Hashemi, A.; Ghilian, R.; Golestan, M.; Akhavan, G.M.; Zare, Z.; Dehghani, M.A. The study of growth in thalassemic patients and its correlation with serum ferritin level. Iran. J. Pediatr. Hematol. Oncol. 2011, 1, 147–151. [Google Scholar]
- Pironi, L.; Arends, J.; Bozzetti, F.; Cuerda, C.; Gillanders, L.; Jeppesen, P.B.; Joly, F.; Kelly, D.; Lal, S.; Staun, M.; et al. ESPEN guidelines on chronic intestinal failure in adults. Clin. Nutr. 2016, 35, 247–307. [Google Scholar] [CrossRef]
- Pironi, L. Definitions of intestinal failure and the short bowel syndrome. Best Pract. Res. Clin. Gastroenterol. 2016, 30, 173–185. [Google Scholar] [CrossRef]
- Doseděl, M.; Jirkovský, E.; Macáková, K.; Krčmová, L.K.; Javorská, L.; Pourová, J.; Mercolini, L.; Remião, F.; Nováková, L.; Mladěnka, P.; et al. Vitamin C-Sources, Physiological Role, Kinetics, Deficiency, Use, Toxicity, and Determination. Nutrients 2021, 13, 615. [Google Scholar] [CrossRef]
- Liakakos, D.; Matsaniotis, N.; Karpouzas, J.; Morphis, L.; Agathopoulos, A. Clinica Chimica Acta 197 Ascorbic Acid Malabsorption In Thalassaemia. Clin. Chim. Acta 1969, 26, 197–202. [Google Scholar] [CrossRef]
- Yusof, J.; d’Arqom, A.; Andriani, A.P.; Nasution, M.Z.; Fatimah, N.; Mustika, A.; Handayani, S. Dietary Supplement Consumption and Mental Health in Indonesian Adults During Second Wave of COVID-19 Pandemic. Patient Prefer. Adherence 2023, 17, 1799–1811. [Google Scholar] [CrossRef] [PubMed]
- Chatterjea, B.; Maitra, A.; Banerjee, D.K.; Basu, A.K. Status of Ascorbic Acid in Iron Deficiency Anaemia and Thalassaemia. Acta Haematol. 1980, 64, 271–275. [Google Scholar] [CrossRef] [PubMed]
- Cleveland Clinic. Herpes Simplex. Available online: https://my.clevelandclinic.org/health/diseases/22855-herpes-simplex (accessed on 28 October 2024).
- Atefi, A.; Binesh, F.; Hashemi, A.; Atefi, A.; Aminorroaya, M.M. Seroprevalence of herpes simplex 1, 2 IgG antibodies in patients with beta-thalassemia in a major tertiary care hospital located in Yazd, Iran. Iran. J. Ped. Hematol. Oncol. 2014, 4, 64–67. [Google Scholar] [PubMed]
- National Organization for Rare Disorders (NORD). Alpha-Thalassemia X-Linked Intellectual Disability Syndrome. Available online: https://rarediseases.org/rare-diseases/alpha-thalassemia-x-linked-intellectual-disability-syndrome/ (accessed on 28 October 2024).
- Gibbons, R.J.; Suthers, G.K.; Wilkie, A.O.; Buckle, V.J.; Higgs, D.R. X-linked a-Thalassemia/Mental Retardation (ATR-X) Syndrome: Localization to XqI2-q21.31 by X Inactivation and Linkage Analysis. Am. J. Hum. Genet. 1992, 51, 1136–1149. [Google Scholar] [PubMed]
- Thienpont, B.; de Ravel, T.; Van Esch, H.; Van Schoubroeck, D.; Moerman, P.; Vermeesch, J.R.; Fryns, J.-P.; Froyen, G.; Lacoste, C.; Badens, C.; et al. Partial duplications of the ATRX gene cause the ATR-X syndrome. Eur. J. Hum. Genet. 2007, 15, 1094–1097. [Google Scholar] [CrossRef]
- Gibbons, R.J. Molecular-clinical spectrum of the ATR-X syndrome. Am. J. Med. Genet. 2000, 97, 204–212. [Google Scholar] [CrossRef]
- Martucciello, G. Gastrointestinal phenotype of ATR-X syndrome. Am. J. Med. Genet. A 2006, 140, 1172–1176. [Google Scholar] [CrossRef]
- Watanabe, T. Esophago-gastric motility and nutritional management in a child with ATR-X syndrome. Pediatr. Int. 2014, 56, e48–e51. [Google Scholar] [CrossRef]
- Lee, S.Y.; Chow, C.B.; Li, C.K.; Chiu, M.C. Outcome of intensive care of homozygous alpha-thalassaemia without prior intra- uterine therapy. J. Paediatr. Child. Health 2007, 43, 546–550. [Google Scholar] [CrossRef]
- Lee, S.Y.R. Survival of homozygous alpha-thalassemia with aplasia/hypoplasia of phalanges and jejunal atresia. J. Matern. Fetal Neonatal Med. 2009, 22, 711–713. [Google Scholar] [CrossRef]

{kind=link}
| Key Findings | |
|---|---|
| H. pylori in thalassemia | H. pylori infection is more prevalent in β-thalassemia patients than controls, potentially contributing to recurrent abdominal pain (RAP). Studies suggest a link between H. pylori and iron deficiency anemia in thalassemia, with older, transfusion-dependent, or splenectomized patients being at higher risk. Gastritis was found in 72% of β-thalassemia patients in one study. |
| IBD and thalassemia | Limited evidence links IBD with thalassemia. A cross-sectional study showed a 6.7% prevalence of thalassemia traits among anemic IBD patients, with Crohn’s disease being more common than UC. Case reports highlight cases of Crohn’s disease and ulcerative colitis in thalassemia patients, often presenting with anemia. Evidence does not indicate a significant association. |
| Celiac disease and thalassemia | Conflicting evidence links celiac disease with thalassemia. Case–control studies report varying prevalence rates, and celiac disease symptoms often overlap with thalassemia symptoms (e.g., delayed puberty and fatigue). The shared genetic and immunological factors may suggest a possible association. |
| Malabsorption and nutrient deficiency in thalassemia | Thalassemia is associated with malabsorption, including vitamin C deficiency, as shown by lower plasma ascorbic acid levels in thalassemia patients compared to controls. This deficiency can lead to anemia, impaired wound healing, and muscle weakness. |
| GI infections in thalassemia | Immunological changes in thalassemia patients could predispose them to infections like HSV. Thalassemia major patients show a higher prevalence (88.8%) of HSV-1 and HSV-2 IgG antibodies than controls (77.7%). These findings suggest a high prevalence of HSV antibodies in both thalassemia patients and healthy individuals, with no significant disparity between the two groups. |
| ATR-X syndrome and GI pathologies | ATR-X syndrome presents with GI problems such as feeding difficulties, chronic constipation, and severe conditions like upper GI bleeding and intestinal malrotation. |
| GI anomalies in thalassemia | A case of Hb Bart’s disease highlighted GI complications, such as jejunal atresia, in a newborn with aplastic digits, hepatosplenomegaly, and pulmonary hypertension. This case suggests an association between homozygous alpha-thalassemia and GI abnormalities. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fakeh, S.; Masoud, A.; Abuqtaish, R.; Salman, B.; Al-Ramahi, L.; AlWahkyan, O.; Abuquteish, D. Gastrointestinal Pathologies Associated with Thalassemia: A Systematic Review. Gastroenterol. Insights 2025, 16, 8. https://doi.org/10.3390/gastroent16010008
Fakeh S, Masoud A, Abuqtaish R, Salman B, Al-Ramahi L, AlWahkyan O, Abuquteish D. Gastrointestinal Pathologies Associated with Thalassemia: A Systematic Review. Gastroenterology Insights. 2025; 16(1):8. https://doi.org/10.3390/gastroent16010008
Chicago/Turabian StyleFakeh, Sara, Ahmad Masoud, Raneem Abuqtaish, Bayan Salman, Layth Al-Ramahi, Omar AlWahkyan, and Dua Abuquteish. 2025. "Gastrointestinal Pathologies Associated with Thalassemia: A Systematic Review" Gastroenterology Insights 16, no. 1: 8. https://doi.org/10.3390/gastroent16010008
APA StyleFakeh, S., Masoud, A., Abuqtaish, R., Salman, B., Al-Ramahi, L., AlWahkyan, O., & Abuquteish, D. (2025). Gastrointestinal Pathologies Associated with Thalassemia: A Systematic Review. Gastroenterology Insights, 16(1), 8. https://doi.org/10.3390/gastroent16010008