1. Introduction
Dominguez et al. (1993) [
1] first defined caudal duplication syndrome (CDS) as an association of hindgut duplication, lower urogenital tract duplication, spinal anomalies (myelomeningocele, sacral duplication, diplomyelia, hemivertebrae), and abdominal wall defects (ventral herniation) at heterogeneous degrees of severity resulting from afetal insult at different stages of embryogenesis. Its association with caudal regression syndrome (CRS) has been recognized recently [
2] due to its rare encounter as separate syndromes and very wide spectrum of clinical presentation. In 1961, Duhamel [
3] described caudal regression syndrome as a defect of formation of the caudal region with: (1) flexion and inversion in the external rotation of the lower limbs, club foot, atrophy of the limbs, and defective motion of the joints; (2) anomalies of the lumbar and sacral spine/vertebra—sacral agenesis, vertebral dysmorphia, and hemivertebra; (3) imperforated anus; (4) agenesis of the kidneys and urinary tract; and(5) agenesis of the Wolffian or Mullerian duct structures, except the gonads.
We describe a novel case resembling the clinical features of both caudal duplicationand caudal regression syndrome in a newborn and discuss its multiple stages surgical treatment throughout infancy and early childhood and the results regarding urinary and fecal continence and the perineum cosmetic appearance.
2. Case Presentation
A 13-day-old female, born at 37 weeks of gestation via normal delivery was admitted to the surgical department for evaluation of an apparently plurimalfomative syndrome diagnosed at birth. The pregnancy was uneventful, and no risk factors could be identified.
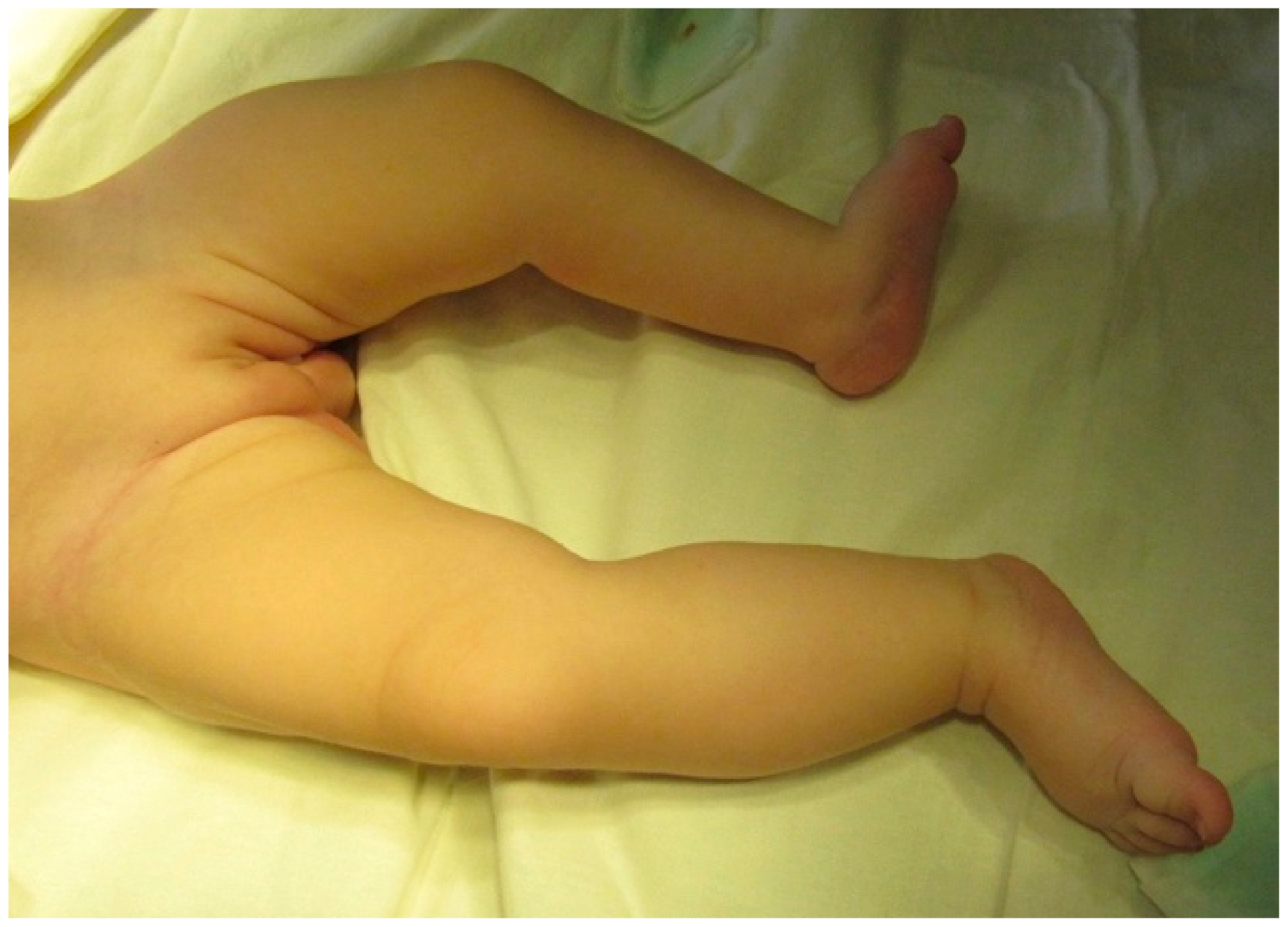
Physical examination revealed abdominal distension, especially after breastfeeding, duplication of the external genital organs and anus, lumbar kyphosis, a lumbosacral mass on the median line covered by normal skin, deviation of the gluteal fold to the right, and a shorter, hypoplastic left lower limb with no neurologic deficit on either side. The orthopedic clinical exam of the lower limbs showed a left leg that was 3 cm shorter than the other (
Figure 1) with muscle hypotrophy, limited abduction of the hip, and valgus deformity of the ankle. The X-Ray exam and ultrasound of the hip showed hypotrophy and asymmetry of the left hip jointbut no signs of hip dysplasia.
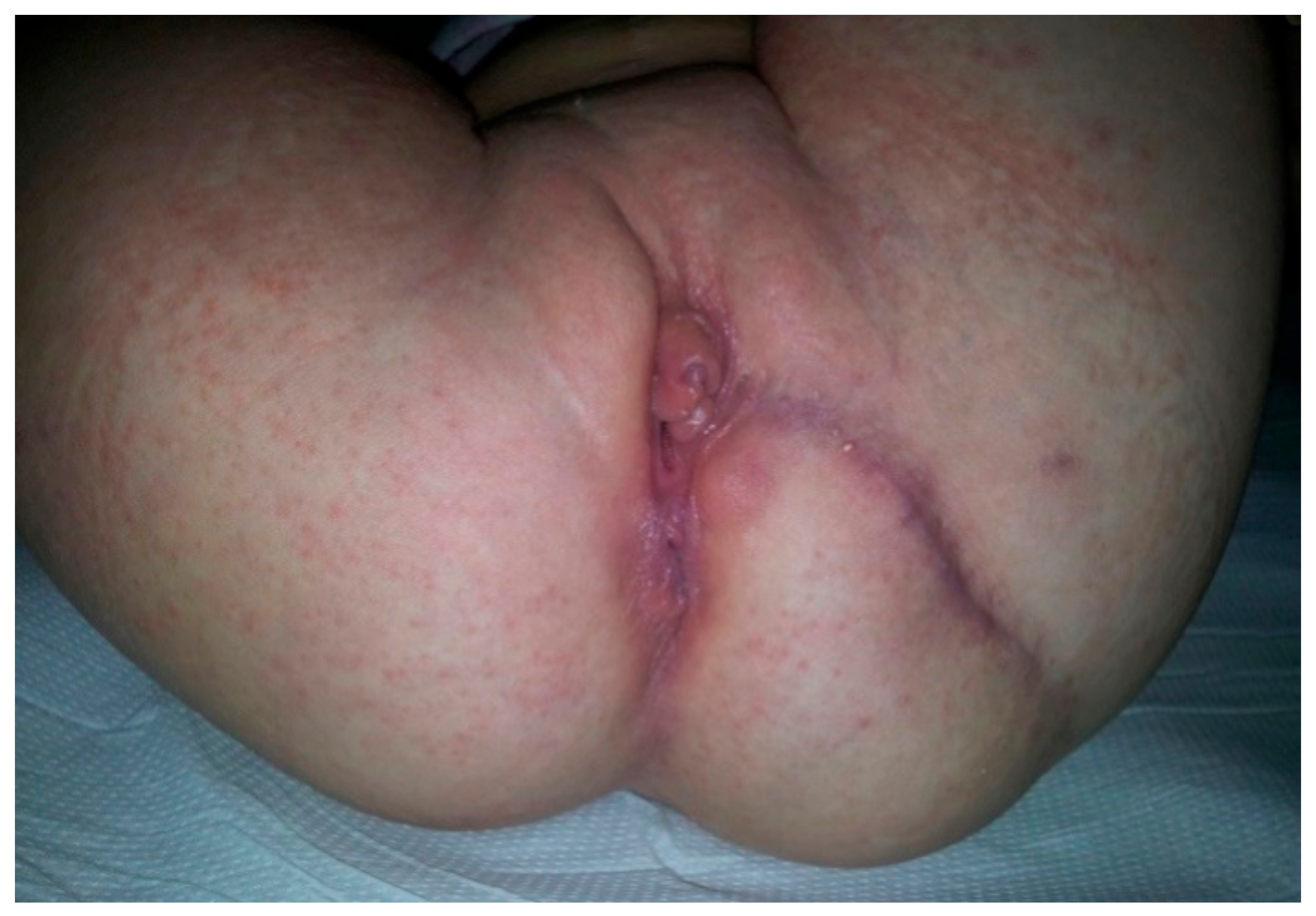
Perineum examination (
Figure 2) revealed a duplication of the external genital organs: two vaginal vestibules located side-by-side with a well-defined minor labia and clitoris on each side but hypoplastic and displaced duplicated medial major labia. Each vestibule had one vaginal and urethral opening, with the normal passing of urine through the right urethral orifice, but an incontinent, continuous stream of urine through the left one. Urine samplesdid not show a urinary tract infection (UTI). Below the two vestibules in the sagittal plane was a normal anal orifice on the right side that was able to pass stool normally, and on the left, an anal dimple with stool evidence at the level of the left vestibule near the posterior wall—the left anovestibular fistula.
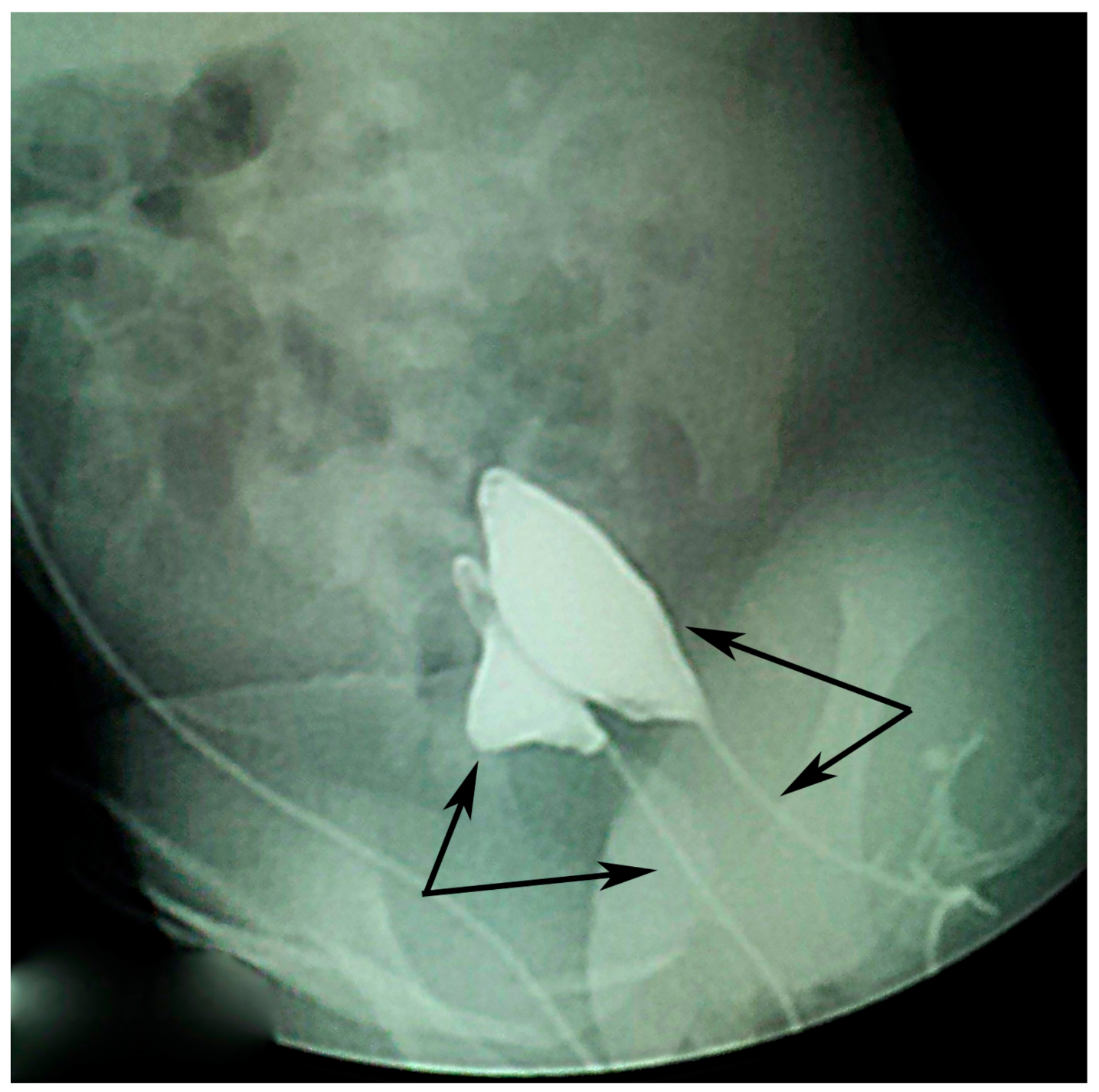
Voiding cystourethrogram done on both urethral openings (
Figure 3) showed no signs of vesicoureteral reflux (VUR) but showed one smaller duplicated bladder and the duplicated urethras.
Barium enema done simultaneously through both anal orifices—the right anus at the perineum and the left anovestibular fistula, highlighted two digestive tracts that were completely separated for at least 20 cm with no evidence of communication up to the splenic flexure.
Computed tomography (CT) scan and magnetic resonance imaging (MRI) of the abdomen and pelvis showed a normally structured liver located on the left side together with the portal and superior mesenteric vein. There was malrotation of the left kidney with a horizontal lay, but the right kidney was of normal structure. There was complete duplication of the bladder, with the left on being of a smaller size and each having a well-defined muscular wall. Each kidney and ureter were draining into the ipsilateral bladder. There was an abnormal terminal aorta branching into 3 arteries at the L4 level: the right common iliac artery, left iliac artery, and a median branch with a posterior median pathway. Evaluation of the lumbosacral and pelvic bones revealed type I sacral agenesis: the abnormal fusion of the vertebral bodies was obvious on the anterior side of the lumbar spine region (from L2 to S1 with vertebral body, L1 fused with the L2 hemivertebra, L3 hemivertebra, L4 with two non-fused nuclei, and partial unilateral left agenesis of L5 and sacrum); asymmetry of the hip was due to the higher insertion of the left ilium bone at the malformed lumbosacral spine with a normal coxofemoral left joint. Posterior vertebral fusion abnormalities—lipomyelomeningocele and a tethered spinal cord—were also noted (
Figure 4).
Informed consent was signed by the legal representative prior the surgical treatment. At 4 months of age, the type III lipomyelomeningocele was surgically treated. Dethetering of the spinal cord was also achieved. Small remnants of the lipoma and some levels of cauda equinawere deliberately left attached in order to preserve the function of the sacral nerves. The dura mater was repaired using the watertight suture technique.
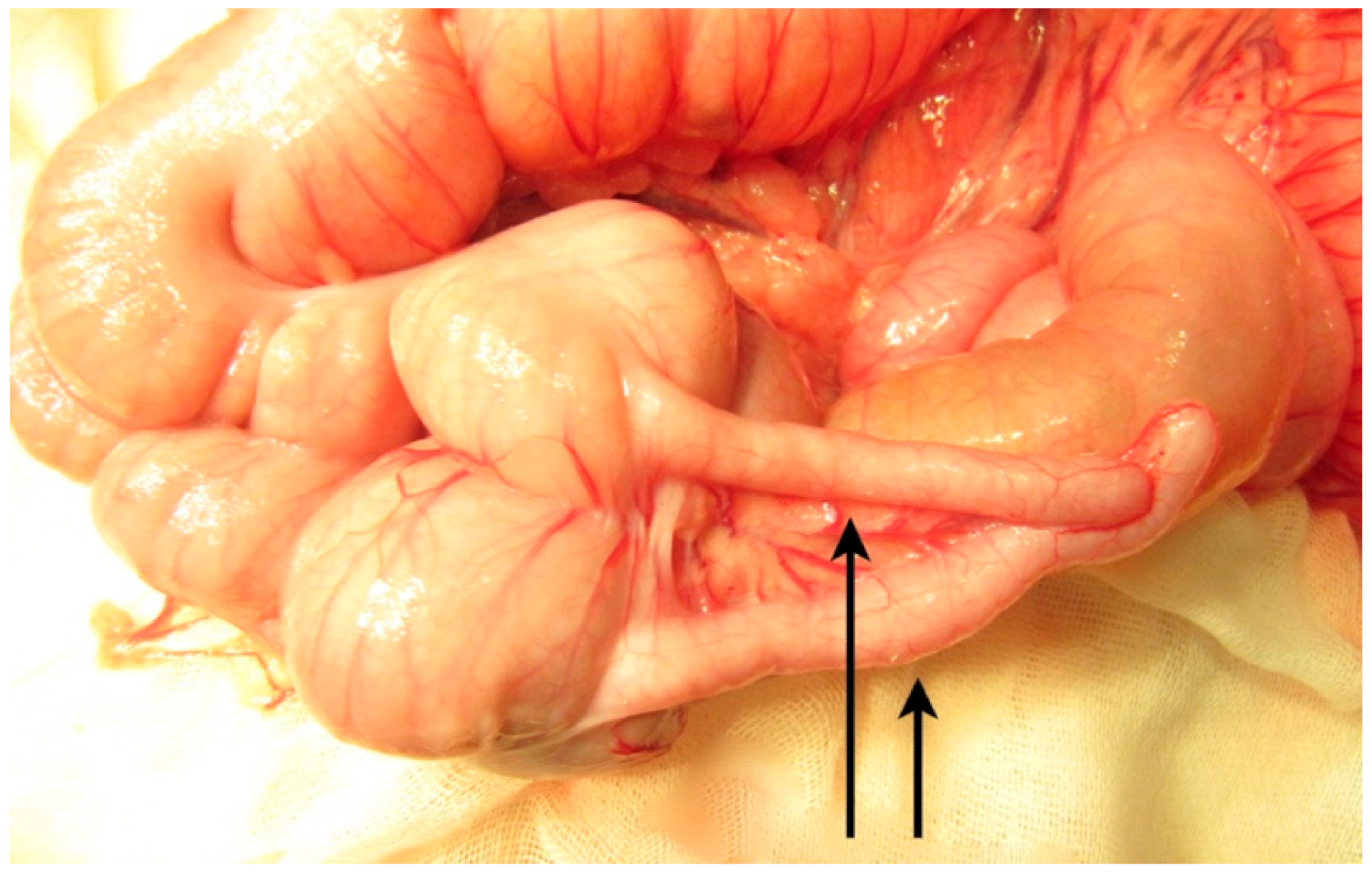
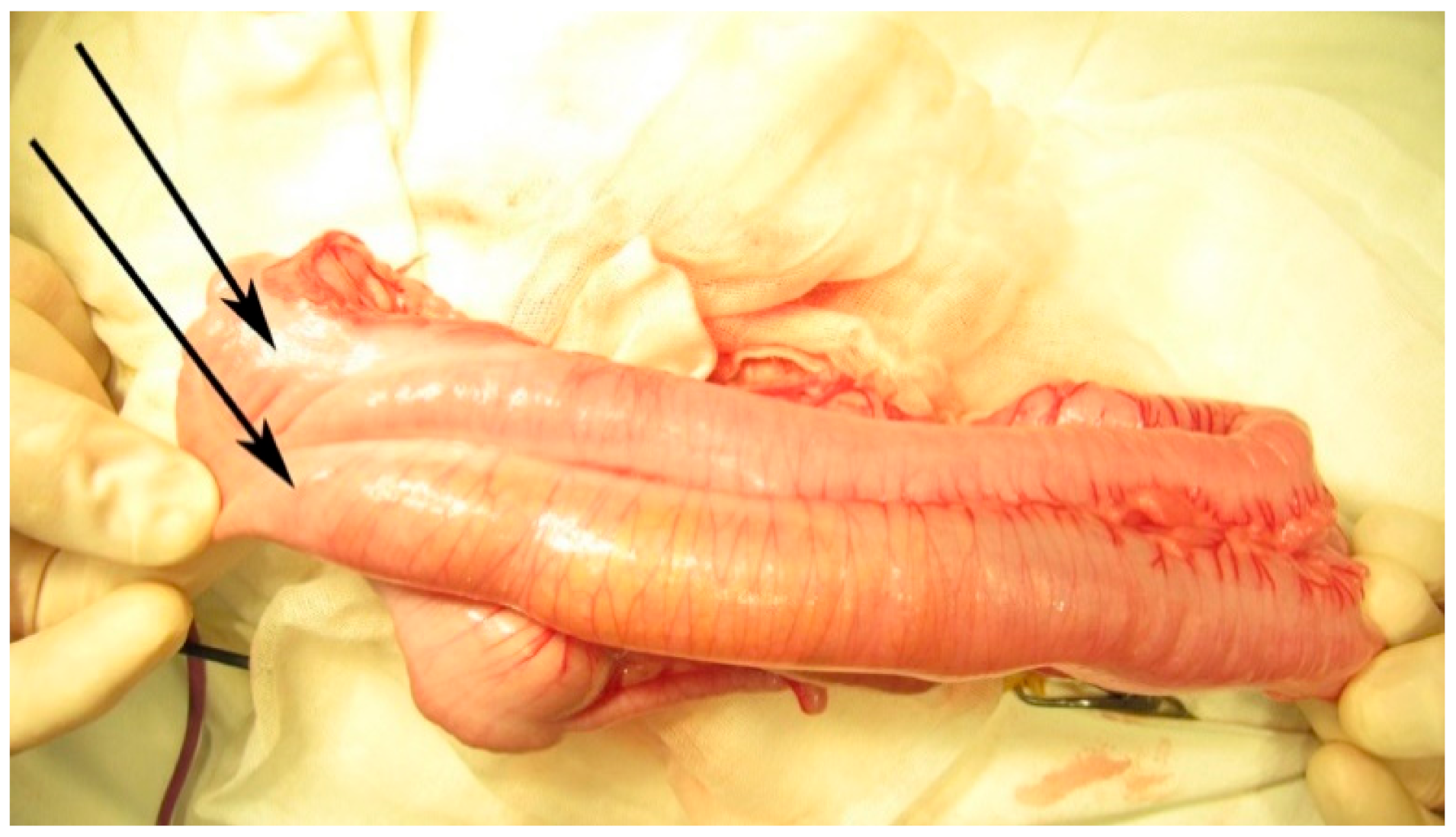
The second surgery was performed at 7 months of age, due to recurrent frequent episodes of intestinal obstruction with associating abdominal distension. During an exploratory laparotomy, malrotation with the complete duplication of the 10 cm terminal ileum and the appendix was noted, continued with partial (common wall and shared mesentery) duplication of the colon up to the proximal descending colon (
Figure 5,
Figure 6 and
Figure 7). It continued with the complete duplication of the rest of descending and sigmoid colon accompanied by the duplication of the distal mesentery. The left hypoplastic rectum opened on the inferior-posterior wall of the left vagina. An exam of the internal genital organs showed a complete duplication of the vagina and uterus with hypoplasia on the left side, with each uterus continuing with a fallopian tube and normal ovary. The two bladder halves had a separate muscular wall connected by the peritoneum, with each ureter draining in the ipsilateral bladder (
Figure 8).
Resection of the left anovestibular fistula and distal hypoplastic rectum was performed at this time by latero-oblique colo-colic anastomosis.
At 2 years old, reconstruction of the urinary bladder was done by the suturing of the two halves into a new bladder and the excision of the left hypoplastic bladder neck together with part of the proximal urethra. The decision to leave the hypoplastic bladder was made by taking into consideration the desire to avoid an antireflux procedure since the preoperative cystogram showed no signs of VUR and each ureter opening in the ipsilateral bladder. At the same time, excision of the left hypoplastic vagina and uterus was performed.
Six months later, plastic reconstruction of the perineum with left major labia plasty and excision of the left urethra and vaginal vestibule were performed (
Figure 9).
The patient was followed for 9 years up to present day and is scheduled for yearly multidisciplinary follow-ups. Bladder function was evaluated once she became potty trained, and she is fully continent for both urine and feces. No further work-up regarding urine or stool continence was done since clinical evaluation of the patient and her history show normal bowel movements, no soiling, wetting, or UTIs, though the work-up elements that were periodically performed were ultrasound and urine samples. At this moment, she is under orthopedic care for the left leg hypoplasia.
3. Discussion
Digestive duplications may occur anywhere along the digestive tube and may or may not be associated with other congenital anomalies [
4]. In addition to the duplication of the gastrointestinal, genitourinary, and spinal systems at different levels of severity, CDS cases have been reported to be associated with a series of other malformations not included into the definition of Dominguez et al. [
1]. From 31 articles and 90 cases reviewed by Acer et al. [
5] and Alfadhel et al. [
6], an imperforated anus is the most commonly associated gastrointestinal malformation; kidney agenesis is the most common urogenital malformation, and partial/total agenesis of the sacrum and coccyx is the most commonly encountered spinal anomaly. All of these are features of the caudal regression syndrome. Very few abnormalities outside of the caudal region have been described (e.g., cardiovascular anomalies).
Many theories trying to explain the pathogenesis of these two complex syndromes have been postulated, both involving developmental anomalies of the organs and systems derived from the caudal eminence caused by fetal insult at the early stages of embryogenesis. Dominguez [
1] mentioned that “complex anomalies of the distal caudal end of the trunk have a wide spectrum ranging from organ agenesis and fusion to splitting and duplication”, a theory supported previously by Duhamel [
3] when writing about caudal regression syndrome.
We reviewed four similar reports of CDS [
2,
7,
8,
9] with vertebral and lower limb malformations as components of caudal regression syndrome, but we believe that this association might be more frequent than is reported in the published literature. The reported cases involve older children (aged between 2and 10 years old) presenting with complications resulting from a duplicated gastrointestinal tract (fecal incontinence [
2], constipation, and abdominal distention [
7]) or complications resulting from a duplicated urinary system (urinary frequency with severe vesico-ureteric reflux (VUR) and renal hypoplasia [
7] or stress incontinence with recurrent episodes of UTIs [
8]). Myelomeningocele was present in two cases [
2,
9], but none of the patients had any neurological deficits in the lower limbs.
High incidence of type I and II sacral agenesis sometimes associated with theprogressive deterioration in the upper urinary tract due to long-standing untreated bladder dysfunction have been reported before [
10], the main reason for this being failure to recognize the sacral defect [
11]. When sacral agenesis is accompanied by myelomeningocele, it is difficult to attribute the neurogenic deficit to one condition or the other [
12].Moreover, our experience shows that besides debilitating motor disorders, children with spinal anomalies present a surprising spectrum of conditions associated with urinary or fecal incontinence [
13]. The case becomes more complicated when, in addition to these, we have a duplication of the urinary tract as was described in our report. In this case a detailed MRI exam can help the diagnosis because the neurologic manifestations correspond to the level of vertebral agenesis: a wedge-shaped conus terminalis without tethering of the cord corresponds to a worse prognosis, and a tethered spinal cord, which is often not associated with agenesis of the cord terminalis, [
14] can cause progressive neurologic symptoms but is surgically treatable.
In our case the MRI exam demonstrated conus terminalis and cauda equina abnormalities and the relation between the lypomatous tissues and the neural structures. Surgical treatment for type III lypomielomeningocele had the goal of reducing the lipoma and reestablishing the continuity of the meningeal sheets. This will dramatically diminish the risk of the tethered cord syndrome, which is responsible for progressive neurological deficits. In the reviewed cases, including ours, CDS was associated with type I, partial sacral agenesis and a tethered spinal cord image on MRI [
2,
8] but without spinal cord malformations.
The urologic complications in the reviewed cases [
8,
9] are more likely related to the late age of bladder duplication treatment and not by the spinal anomalies, proven by improvement after surgical reconstruction. In our case with type I sacral agenesis without spinal cord anomalies, the early treatment of both lipomyelomeningocele and bladder duplication led to excellent results regarding continence without urinary infection during follow-up.
The surgical treatment plan was carried out to give the patient fecal and urine continence, maintain potential fertility, and a normal cosmetic appearance. The surgical treatment of the duplicated colon depends on the presenting symptoms and the complexity of the duplication. Most authors [
5,
6] chose resectioning with colocolic anastomosis of the duplicated colon and posterior sagittal anorectoplasty in cases associated with imperforated anus [
15]. If no dysfunction is present at the time of diagnosis, some authors support temporization of surgical intervention [
16]. Reconstructive surgery for genital malformations has been performed in a few patients for cosmetic reasons at such a young age and mostly have been completed in boys [
14,
15].




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}








