Glutathione Transferase (GST)-Activated Prodrugs

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
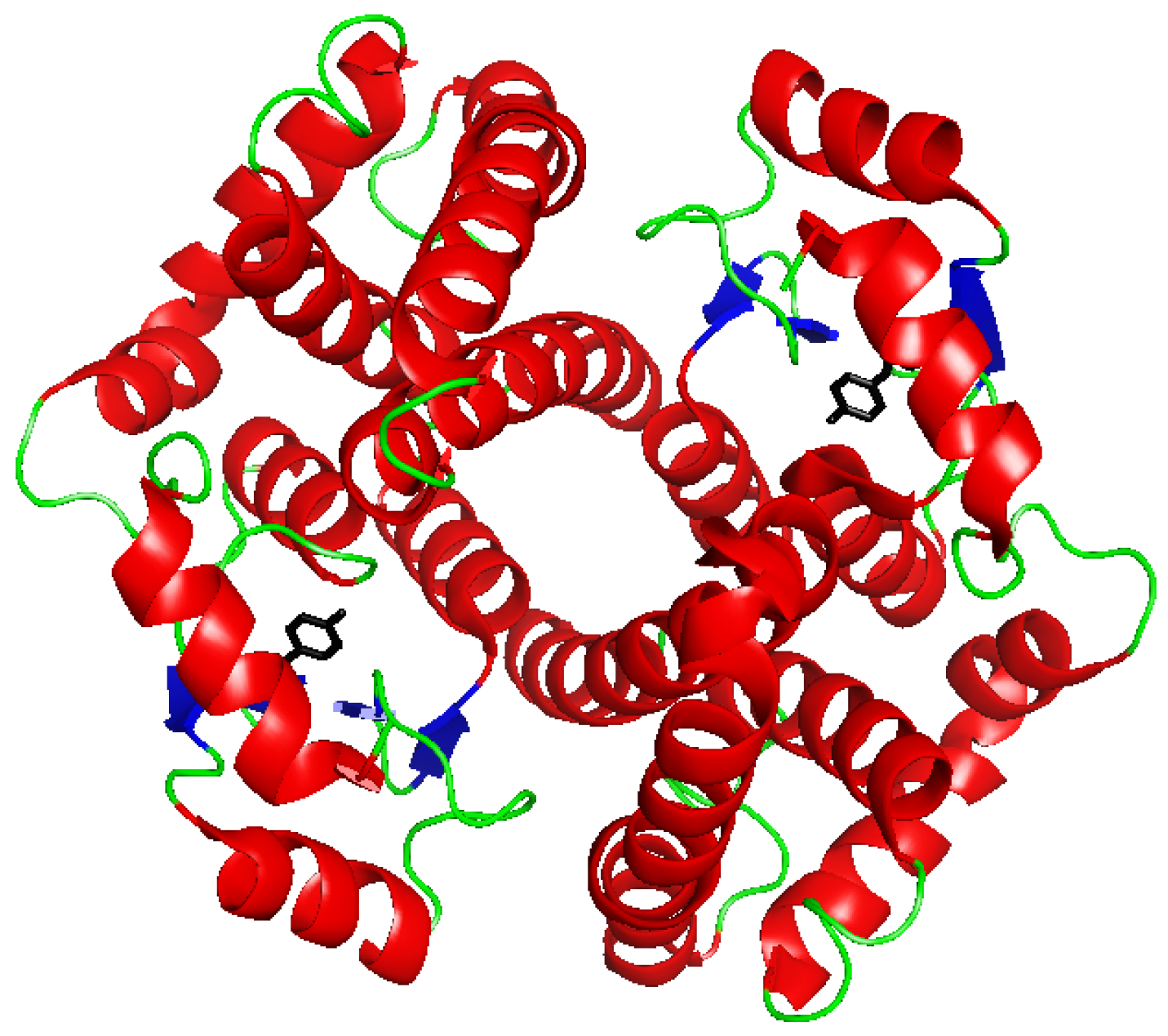
2. Glutathione and Glutathione Transferase

2.1. GSH-Dependent Prodrugs Activation
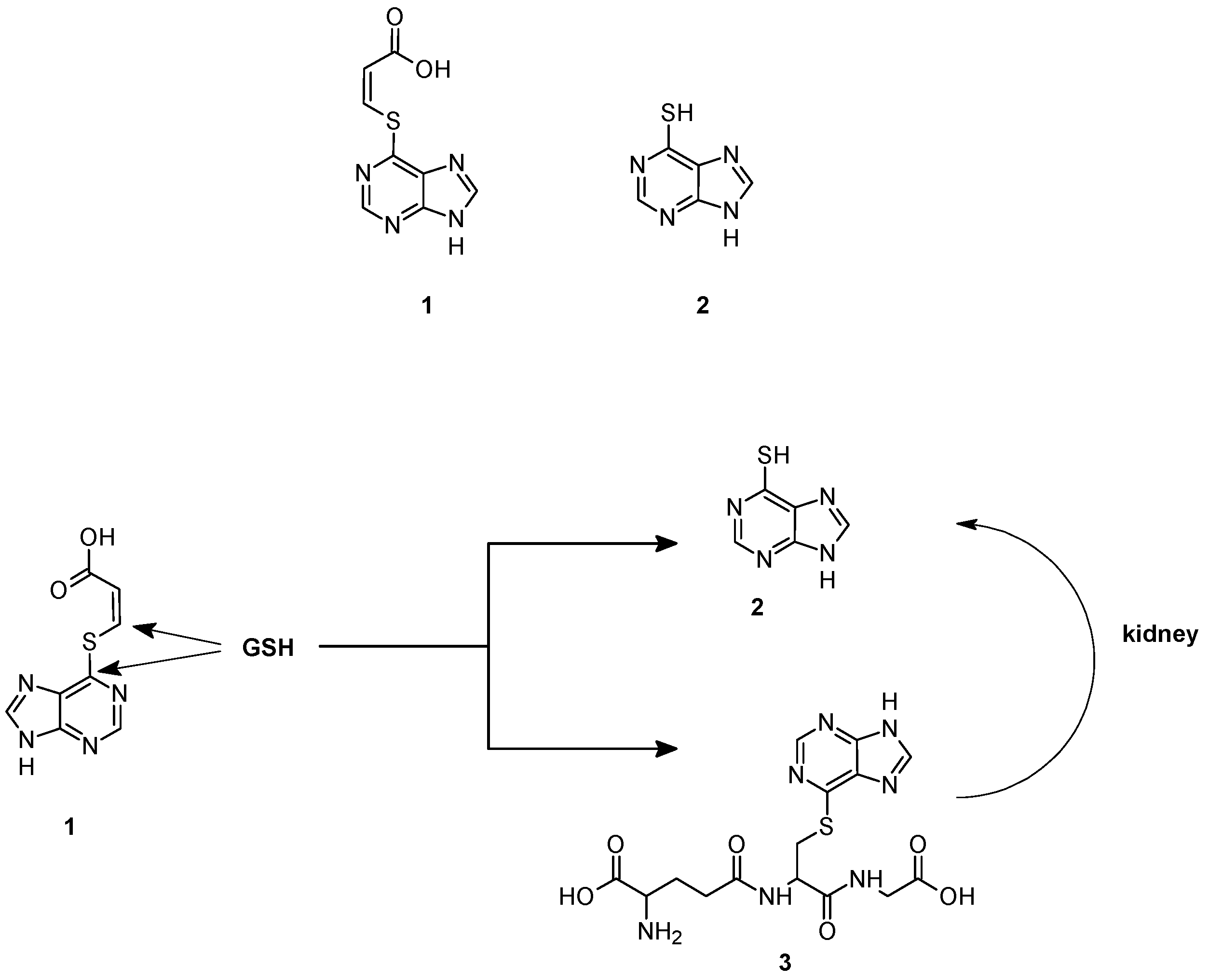
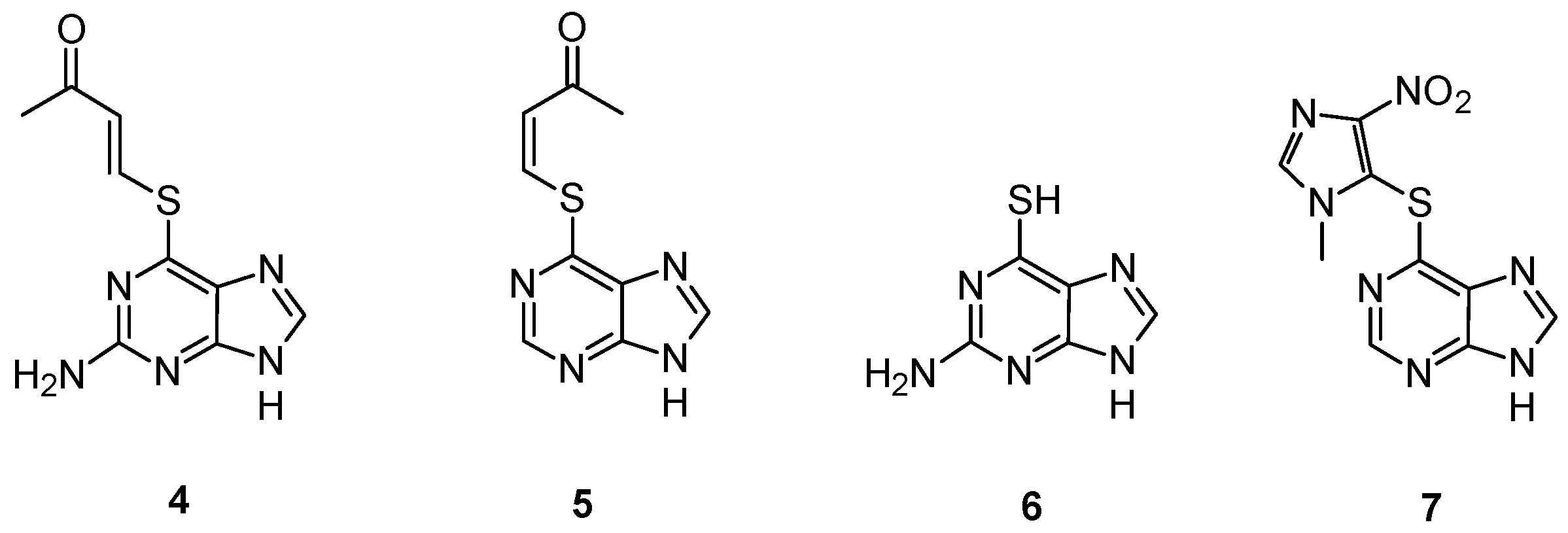
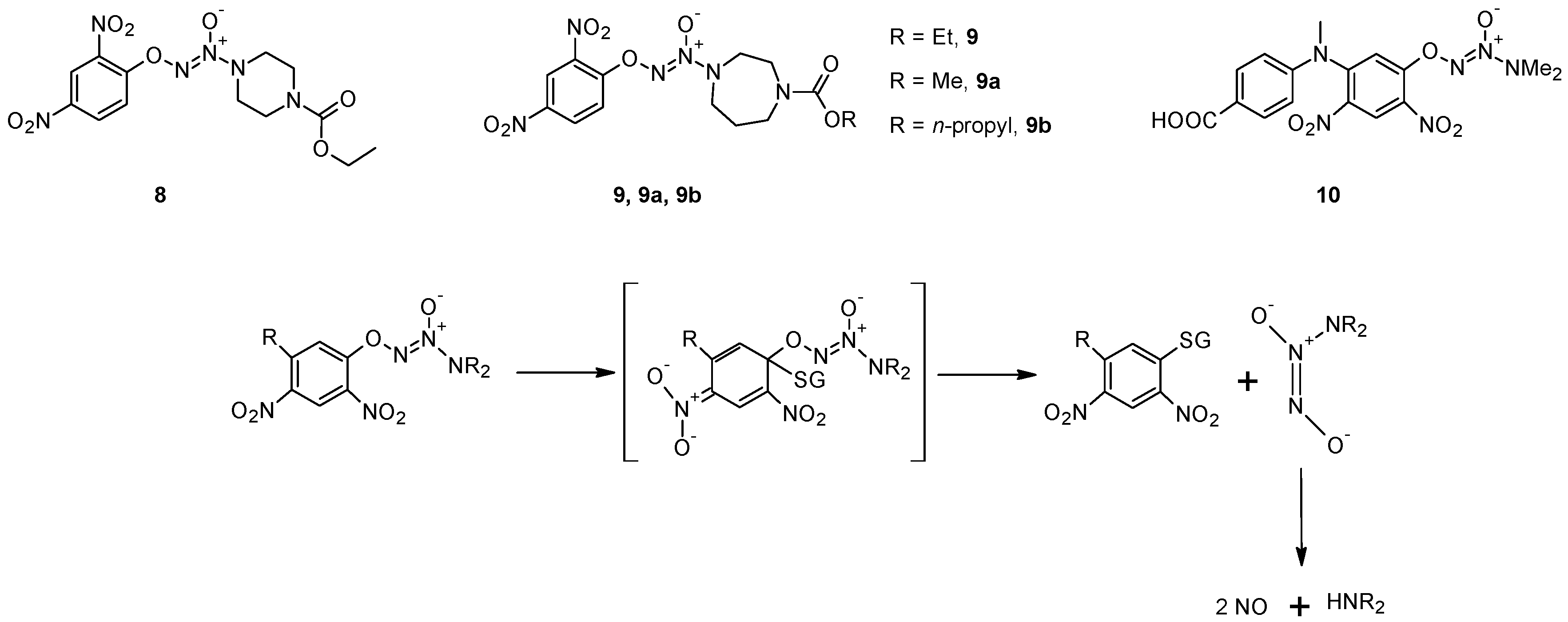
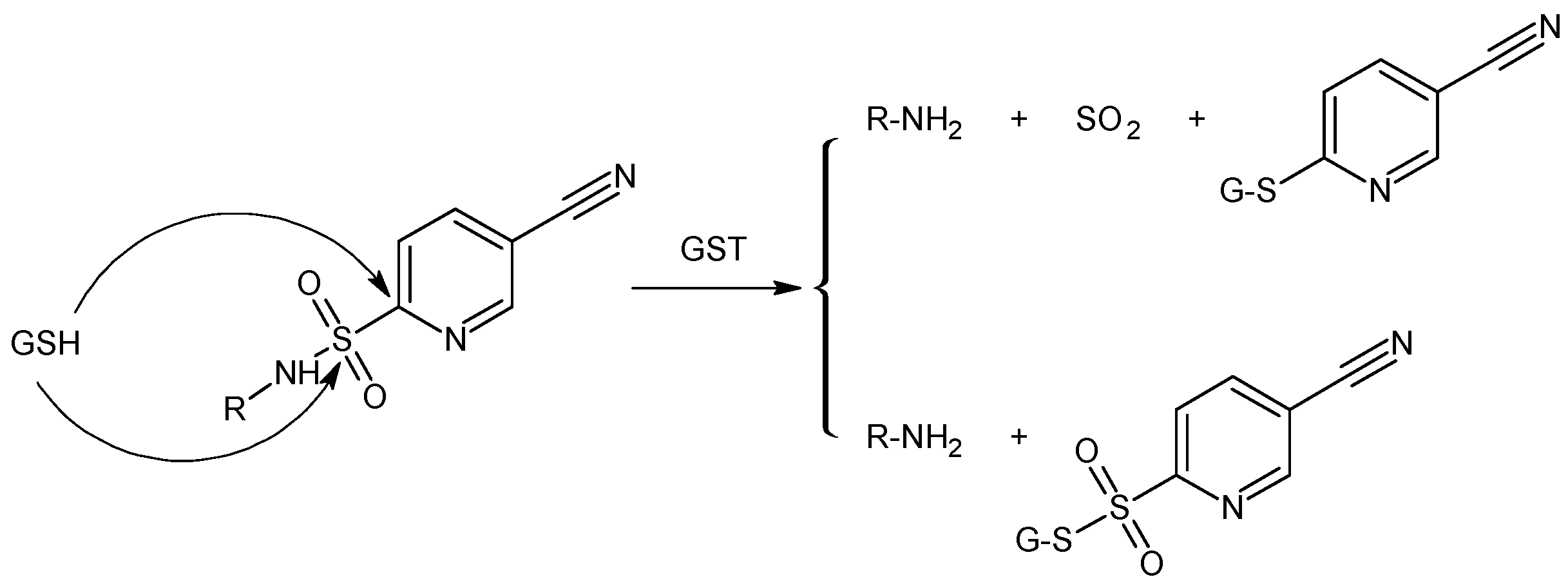
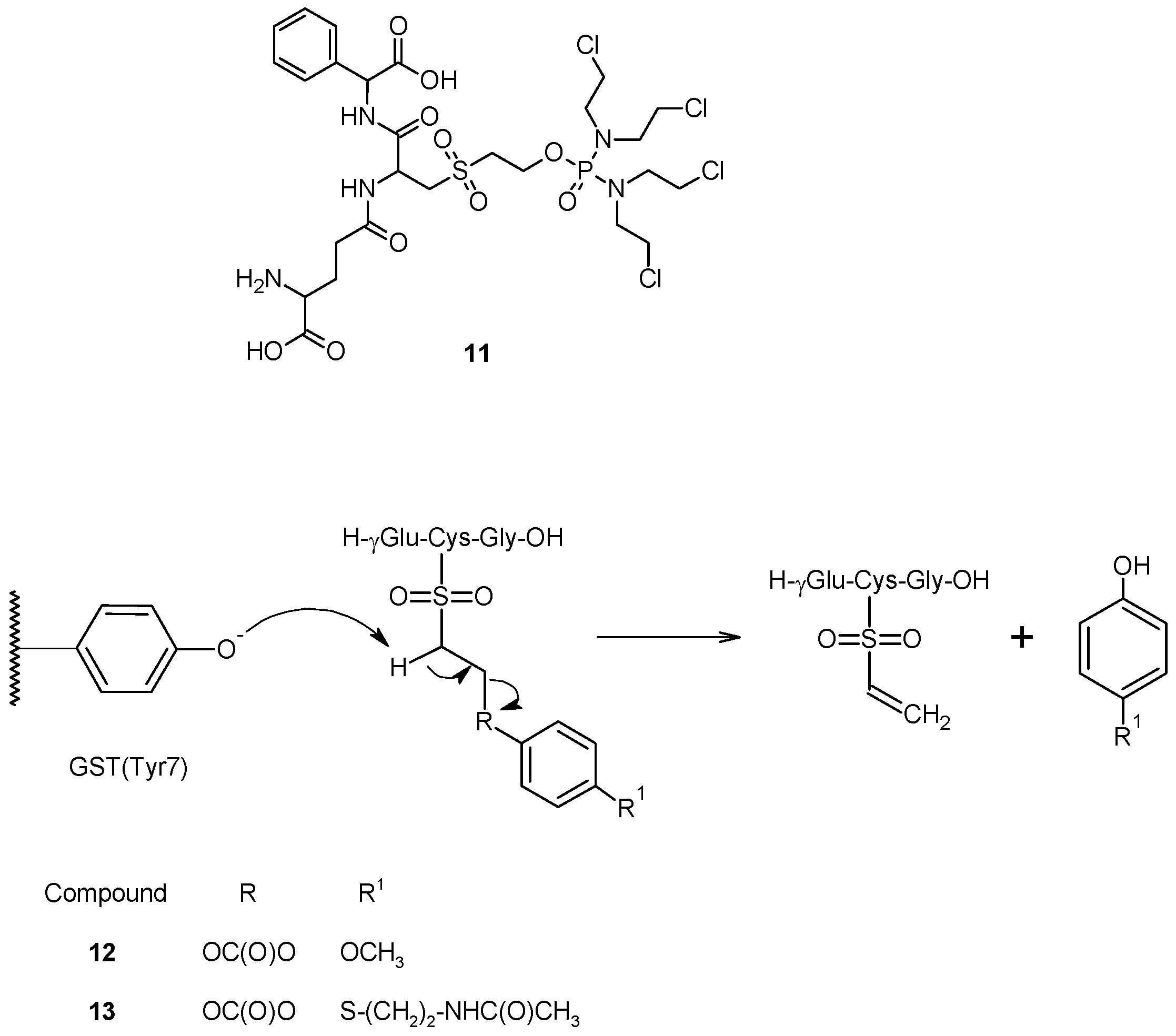
2.2. GSH-Independent Prodrugs Activation
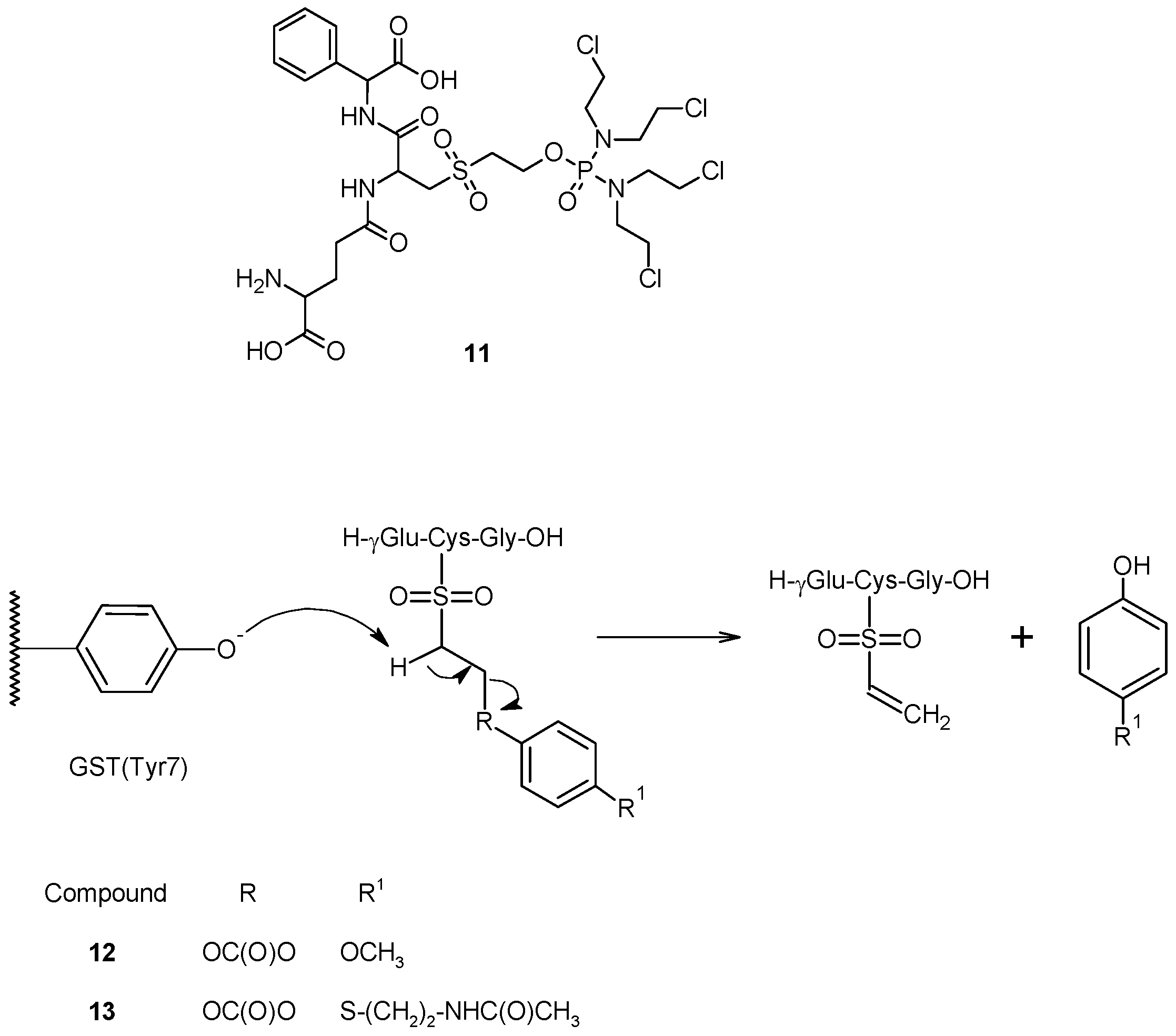
3. Conclusions
Conflict of Interest
References
- Lowenthal, R.M.; Eaton, K. Toxicity of chemotherapy. Hematol. Oncol. Clin. North Am. 1996, 10, 967–990. [Google Scholar] [CrossRef]
- Nielsen, D.; Maare, C.; Skovsgaard, T. Cellular resistance to anthracyclines. Gen. Pharmacol. 1996, 27, 251–255. [Google Scholar] [CrossRef]
- Stavrovskaya, A.A. Cellular mechanisms of multidrug resistance of tumor cells. Biochem. Moscow 2000, 65, 95–106. [Google Scholar]
- Arpicco, S.; Dosio, F.; Stella, B.; Cattel, L. Anticancer prodrugs: An overview of major strategies and recent developments. Curr. Top. Med. Chem. 2011, 11, 2346–2381. [Google Scholar] [CrossRef]
- Hayes, J.D.; Flanagan, J.U.; Jowsey, I.R. Glutathione transferases. Annu. Rev. Pharmacol. Toxicol. 2005, 45, 51–88. [Google Scholar] [CrossRef]
- Commandeur, J.N.M.; Stijntjes, G.J.; Vermeulen, N.P.E. Enzymes and transport-systems involved in the formation and disposition of glutathione S-conjugates—Role in bioactivation and detoxication mechanisms of xenobiotics. Pharmacol. Rev. 1995, 47, 271–330. [Google Scholar]
- Van Bladeren, P.J. Glutathione conjugation as a bioactivation reaction. Chem. Biol. Interact. 2000, 129, 61–76. [Google Scholar] [CrossRef]
- Mannervik, B.; Board, P.G.; Hayes, J.D.; Listowsky, I.; Pearson, W.R. Nomenclature for mammalian soluble glutathione transferases. Methods Enzymol. 2005, 401, 1–8. [Google Scholar] [CrossRef]
- Ranson, H.; Rossiter, L.; Ortelli, F.; Jensen, B.; Wang, X.L.; Roth, C.W.; Collins, F.H.; Hemingway, J. Identification of a novel class of insect glutathione S-transferases involved in resistance to DDT in the malaria vector Anopheles gambiae. Biochem. J. 2001, 359, 295–304. [Google Scholar] [CrossRef]
- Dixon, D.P.; Lapthorn, A.; Edwards, R. Plant glutathione transferases. Genome Biol. 2002, 3, 1–10. [Google Scholar]
- Rossjohn, J.; Polekhina, G.; Feil, S.C.; Allocati, N.; Masulli, M.; Di Ilio, C.; Parker, M.W. A mixed disulfide bond in bacterial glutathione transferase: Functional and evolutionary implications. Struct. Fold. Des. 1998, 6, 721–734. [Google Scholar] [CrossRef]
- DePierre, J.; Morgenstern, R. Comparison of the distribution of microsomal and cytosolic glutathione S-transferase activities in different organs of the rat. Biochem. Pharmacol. 1983, 32, 721–723. [Google Scholar] [CrossRef]
- Pacifici, G.; Franchi, M.; Bencini, C.; Repetti, F.; Di Lascio, N.; Muraro, G. Tissue distribution of drug-metabolizing enzymes in humans. Xenobiotica 1988, 18, 849–856. [Google Scholar] [CrossRef]
- Armstrong, R.N. Structure, catalytic mechanism, and evolution of the glutathione transferases. Chem. Res. Toxicol. 1997, 10, 2–18. [Google Scholar] [CrossRef]
- Grahn, E.; Novotny, M.; Jakobsson, E.; Gustafsson, A.; Grehn, L.; Olin, B.; Madsen, D.; Wahlberg, M.; Mannervik, B.; Kleywegt, G.J. New crystal structures of human glutathione transferase A1-1 shed light on glutathione binding and the conformation of the C-terminal helix. Acta Crystallogr. Sect. D Biol. Crystallogr. 2006, 62, 197–207. [Google Scholar]
- Tan, K.L.; Chelvanayagam, G.; Parker, M.W.; Board, P.G. Mutagenesis of the active site of the human Theta-class glutathione transferase GSTT2–2: Catalysis with different substrates involves different residues. Biochem. J. 1996, 319, 315–321. [Google Scholar]
- Stenberg, G.; Board, P.G.; Mannervik, B. Mutation of an evolutionarily conserved tyrosine residue in the active-site of a human class alpha-glutathione transferase. FEBS Lett. 1991, 293, 153–155. [Google Scholar] [CrossRef]
- Kong, K.H.; Takasu, K.; Inoue, H.; Takahashi, K. Tyrosine-7 in human class-Pi glutathione-S-transferase is important for lowering the pka of the thiol-group of glutathione in the enzyme-glutathione complex. Biochem. Biophys. Res. Commun. 1992, 184, 194–197. [Google Scholar] [CrossRef]
- Graminski, G.F.; Kubo, Y.; Armstrong, R.N. Spectroscopic and kinetic evidence for the thiolate anion of glutathione at the active site of glutathione S-transferase. Biochemistry 1989, 28, 3562–3568. [Google Scholar] [CrossRef]
- Widersten, M.; Bjornestedt, R.; Mannervik, B. Involvement of the carboxyl groups of glutathione in the catalytic mechanism of human glutathione transferase A1-1. Biochemistry 1996, 35, 7731–7742. [Google Scholar] [CrossRef]
- Parraga, A.; Garcia-Saez, I.; Walsh, S.B.; Mantle, T.J.; Coll, M. The three-dimensional structure of a class-Pi glutathione S-transferase complexed with glutathione: The active-site hydration provides insights into the reaction mechanism. Biochem. J. 1998, 333, 811–816. [Google Scholar]
- Dourado, D.; Fernandes, P.A.; Mannervik, B.; Ramos, M.J. Glutathione transferase: New model for glutathione activation. Chem. Eur. J. 2008, 14, 9591–9598. [Google Scholar] [CrossRef]
- Board, P.G.; Coggan, M.; Chelvanayagam, G.; Easteal, S.; Jermiin, L.S.; Schulte, G.K.; Danley, D.E.; Hoth, L.R.; Griffor, M.C.; Kamath, A.V.; et al. Identification, characterization, and crystal structure of the omega class glutathione transferases. J. Biol. Chem. 2000, 275, 24798–24806. [Google Scholar] [CrossRef]
- Dixon, D.P.; Davis, B.G.; Edwards, R. Functional divergence in the glutathione transferase superfamily in plants. Identification of two classes with putative functions in redox homeostasis in Arabidopsis thaliana. J. Biol. Chem. 2002, 277, 30859–30869. [Google Scholar] [CrossRef]
- Mannervik, B.; Castro, V.M.; Danielson, U.H.; Tahir, M.K.; Hansson, J.; Ringborg, U. Expression of class Pi-glutathione transferase in human-malignant melanoma-cells. Carcinogenesis 1987, 8, 1929–1932. [Google Scholar] [CrossRef]
- Hayes, J.D.; Pulford, D.J. The glutathione S-transferase supergene family: Regulation of GST and the contribution of the isoenzymes to cancer chemoprotection and drug resistance. Crit. Rev. Biochem. Mol. Biol. 1995, 30, 445–600. [Google Scholar] [CrossRef]
- O’Brien, M.L.; Tew, K.D. Glutathione and related enzymes in multidrug resistance. Eur. J. Cancer 1996, 32A, 967–978. [Google Scholar]
- Batist, G.; Tulpule, A.; Sinha, B.K.; Katki, A.G.; Myers, C.E.; Cowan, K.H. Overexpression of a novel anionic glutathione transferase in multidrug-resistant human-breast cancer-cells. J. Biol. Chem. 1986, 261, 5544–5549. [Google Scholar]
- Sargent, J.M.; Williamson, C.; Hall, A.G.; Elgie, A.W.; Taylor, C.G. Evidence for the Involvement of the Glutathione Pathway in Drug Resistance in AML. In Drug Resistance in Leukemia and Lymphoma III; Kaspers, G.J.L., Pieters, R., Veerman, A.J.P., Eds.; Kluwer Academic/Plenum Publishers: New York, NY, USA, 1999; pp. 205–209. [Google Scholar]
- Kodera, Y.; Isobe, K.; Yamauchi, M.; Kondo, K.; Akiyama, S.; Ito, K.; Nakashima, I.; Takagi, H. Expression of glutathione-S-transferase-Alpha and glutathione-S-transferase-Pi in gastric-cancer—A correlation with cisplatin resistance. Cancer Chemother. Pharmacol. 1994, 34, 203–208. [Google Scholar] [CrossRef]
- Adler, V.; Yin, Z.M.; Fuchs, S.Y.; Benezra, M.; Rosario, L.; Tew, K.D.; Pincus, M.R.; Sardana, M.; Henderson, C.J.; Wolf, C.R.; et al. Regulation of JNK signaling by GSTp. EMBO J. 1999, 18, 1321–1334. [Google Scholar] [CrossRef]
- Sweeney, C.; Coles, B.F.; Nowell, S.; Lang, N.P.; Kadlubar, F.F. Novel markers of susceptibility to carcinogens in diet: Associations with colorectal cancer. Toxicology 2002, 181, 83–87. [Google Scholar] [CrossRef]
- Ruzza, P.; Rosato, A.; Rossi, C.R.; Floreani, M.; Quintieri, L. Glutathione transferases as targets for cancer therapy. Anti-Cancer Agents Med. Chem. 2009, 9, 763–777. [Google Scholar]
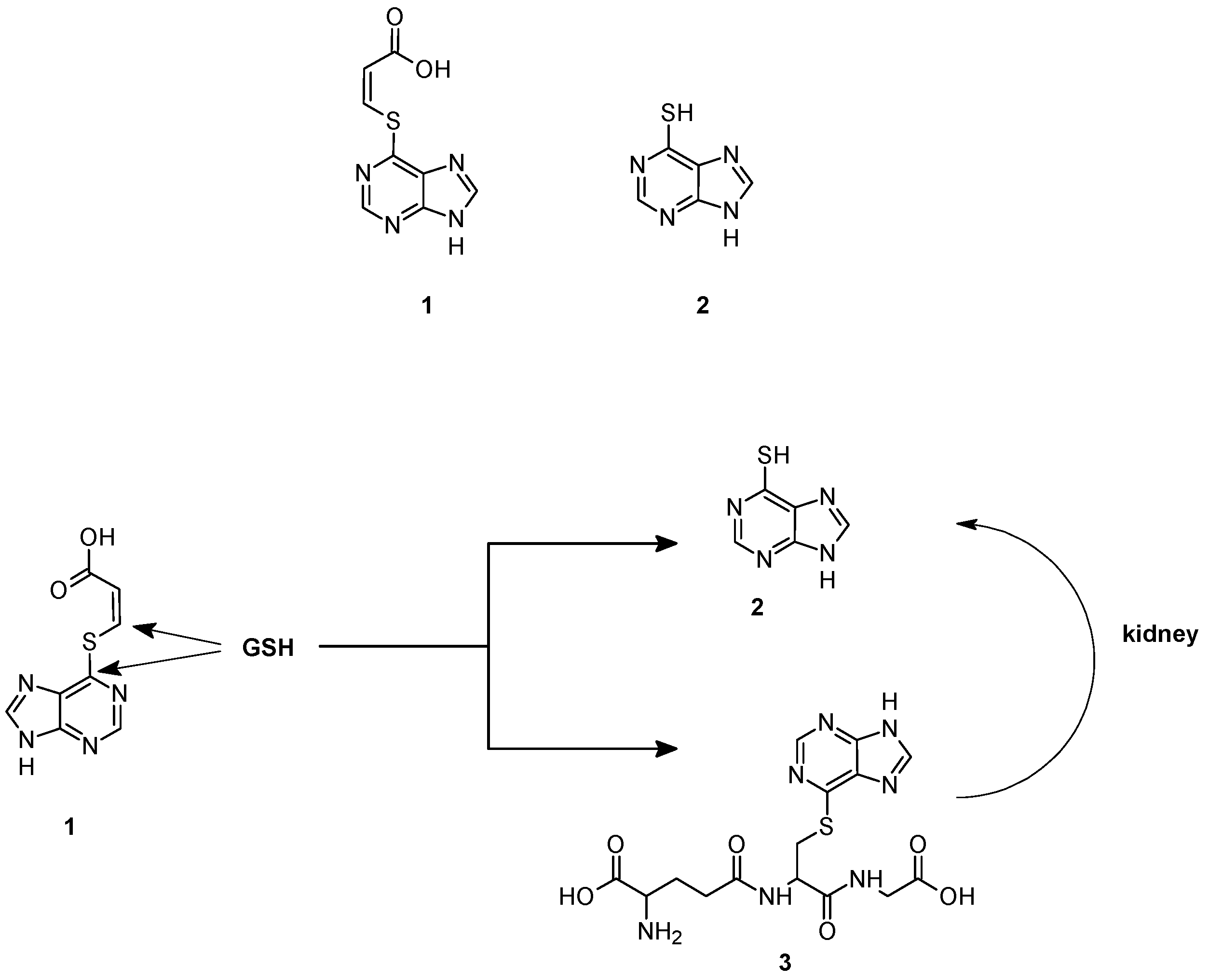
- Gunnarsdottir, S.; Elfarra, A.A. Glutathione-Dependent metabolism of cis-3-(9H-purin-6-ylthio)acrylic acid to yield the chemotherapeutic drug 6-mercaptopurine: Evidence for two distinct mechanisms in rats. J. Pharmacol. Exp. Ther. 1999, 290, 950–957. [Google Scholar]
- Elfarra, A.; Hwang, I. Targeting of 6-mercaptopurine to the kidneys. Metabolism and kidney-selectivity of S-(6-purinyl)-l-cysteine analogs in rats. Drug Metab. Dispos. 1993, 21, 841–845. [Google Scholar]
- Maellaro, E.; Dominaci, S.; del Bello, B.; Valentini, M.A.; Pieri, L.; Perego, P.; Supino, R.; Zumino, F.; Lorenzini, E.; Paolicchi, A.; et al. Membrane gamma-glutamyl transpeptidase activity of melanoma cells: Effects on cellular H2O2 production, cell surface protein thiol oxidation and NF-kappa B activation status. J. Cell Sci. 2000, 113, 2671–2678. [Google Scholar]
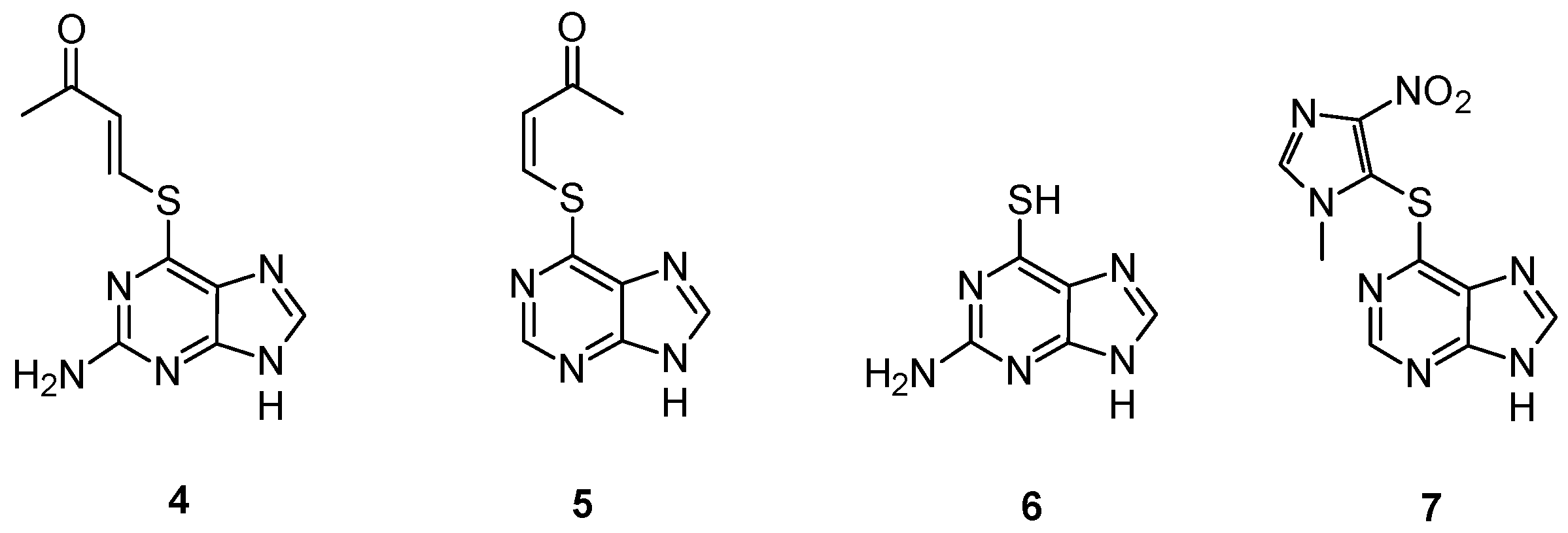
- Gunnarsdottir, S.; Rucki, M.; Elfarra, A.A. Novel glutathione-dependent thiopurine prodrugs: Evidence for enhanced cytotoxicity in tumor cells and for decreased bone marrow toxicity in mice. J. Pharmacol. Exp. Ther. 2002, 301, 77–86. [Google Scholar] [CrossRef]
- Gunnarsdottir, S.; Elfarra, A.A. Cytotoxicity of the novel glutathione-activated thiopurine prodrugs cis-avtp [cis-6-(2-acetylvinylthio)purine] and trans-avtg [trans-6-(2-acetylvinylthio)guanine] results from the National Cancer Institute’s anticancer drug screen. Drug Metab. Dispos. 2004, 32, 321–327. [Google Scholar]
- Eklund, B.I.; Gunnarsdottir, S.; Elfarra, A.A.; Mannervik, B. Human glutathione transferases catalyzing the bioactivation of anticancer thiopurine prodrugs. Biochem. Pharmacol. 2007, 73, 1829–1841. [Google Scholar] [CrossRef]
- Tew, K.D. Glutathione-associated enzymes in anticancer drug-resistance. Cancer Res. 1994, 54, 4313–4320. [Google Scholar]
- Eklund, B.I.; Moberg, M.; Bergquist, J.; Mannervik, B. Divergent activities of human glutathione transferases in the bioactivation of azathioprine. Mol. Pharmacol. 2006, 70, 747–754. [Google Scholar] [CrossRef]
- Cara, C.J.; Pena, A.S.; Sans, M.; Rodrigo, L.; Guerrero-Esteo, M.; Hinojosa, J.; Garcia-Paredes, J.; Guijarro, L.G. Reviewing the mechanism of action of thiopurine drugs: Towards a new paradigm in clinical practice. Med. Sci. Monit. 2004, 10, 247–254. [Google Scholar]
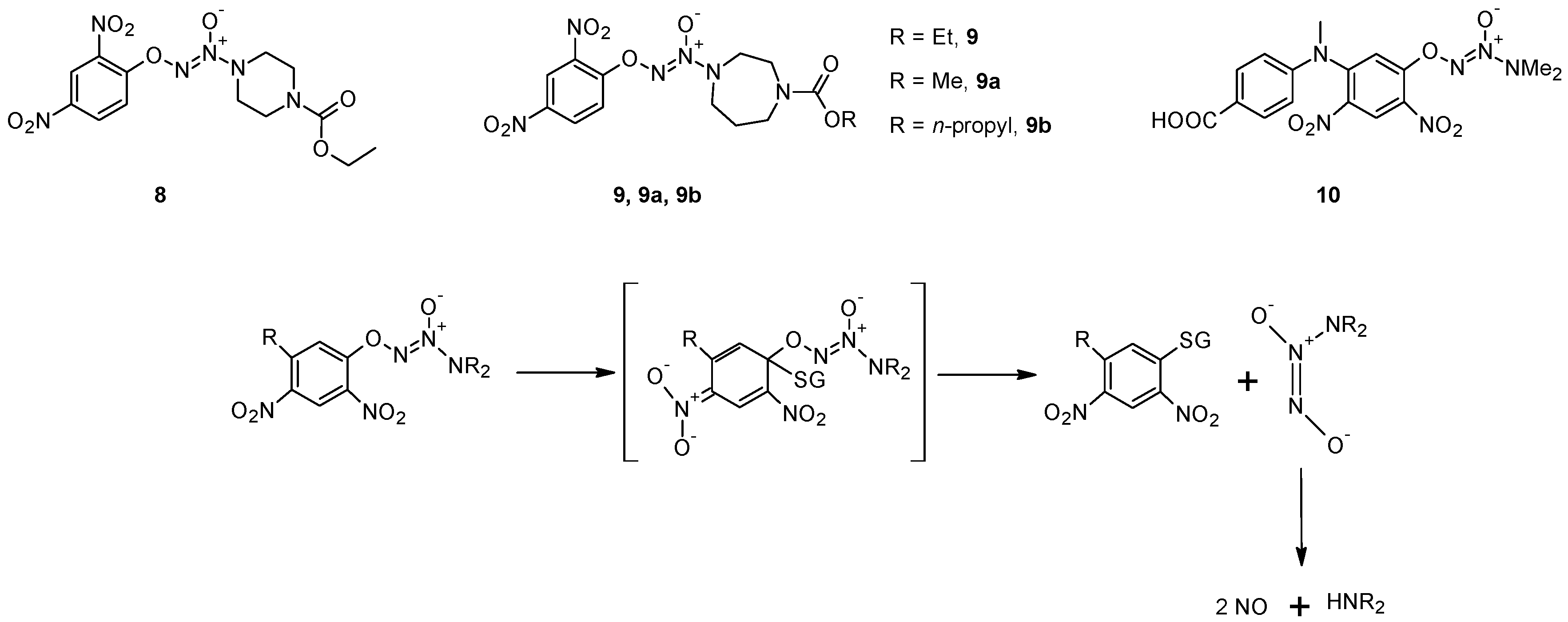
- Shami, P.J.; Saavedra, J.E.; Wang, L.Y.; Bonifant, C.L.; Diwan, B.A.; Singh, S.V.; Gu, Y.J.; Fox, S.D.; Buzard, G.S.; Citro, M.L.; et al. JS-K, a glutathione/glutathione S-transferase-activated nitric oxide donor of the diazeniumdiolate class with potent antineoplastic activity. Mol. Cancer Ther. 2003, 2, 409–417. [Google Scholar]
- Kiziltepe, T.; Hideshima, T.; Ishitsuka, K.; Ocio, E.M.; Raje, N.; Catley, L.; Li, C.Q.; Trudel, L.J.; Yasui, H.; Vallet, S.; et al. JS-K, a GST-activated nitric oxide generator, induces DNA double-strand breaks, activates DNA damage response pathways, and induces apoptosis in vitro and in vivo in human multiple myeloma cells. Blood 2007, 110, 709–718. [Google Scholar] [CrossRef]
- Laschak, M.; Spindler, K.D.; Schrader, A.J.; Hessenauer, A.; Streicher, W.; Schrader, M.; Cronauer, M.V. JS-K, a glutathione/glutathione S-transferase-activated nitric oxide releasing prodrug inhibits androgen receptor and WNT-signaling in prostate cancer cells. BMC Cancer 2012, 12, 130:1–130:10. [Google Scholar]
- Chakrapani, H.; Kalathur, R.C.; Maciag, A.E.; Citro, M.L.; Ji, X.H.; Keefer, L.K.; Saavedra, J.E. Synthesis, mechanistic studies, and anti-proliferative activity of glutathione/glutathione S-transferase-activated nitric oxide prodrugs. Bioorg. Med. Chem. 2008, 16, 9764–9771. [Google Scholar] [CrossRef]
- Findlay, V.J.; Townsend, D.M.; Saavedra, J.E.; Buzard, G.S.; Citro, M.L.; Keefer, L.K.; Ji, X.H.; Tew, K.D. Tumor cell responses to a novel glutathione S-transferase-activated nitric oxide-releasing prodrug. Mol. Pharmacol. 2004, 65, 1070–1079. [Google Scholar] [CrossRef]
- Zhao, Z.; Koeplinger, K.A.; Peterson, T.; Conradi, R.A.; Burton, P.S.; Suarato, A.; Heinrikson, R.L.; Tomasselli, A.G. Mechanism, structure-activity studies, and potential applications of glutathione S-transferase-catalyzed cleavage of sulfonamides. Drug Metab. Dispos. 1999, 27, 992–998. [Google Scholar]
- Koeplinger, K.A.; Zhao, Z.Y.; Peterson, T.; Leone, J.W.; Schwende, F.S.; Heinrikson, R.L.; Tomasselli, A.G. Activated sulfonamides are cleaved by glutathione-S-transferases. Drug Metab. Dispos. 1999, 27, 986–991. [Google Scholar]
- Axarli, I.; Labrou, N.E.; Petrou, C.; Rassias, N.; Cordopatis, P.; Clonis, Y.D. Sulphonamide-based bombesin prodrug analogues for glutathione transferase, useful in targeted cancer chemotherapy. Eur. J. Med. Chem. 2009, 44, 2009–2016. [Google Scholar] [CrossRef]
- Lyttle, M.H.; Satyam, A.; Hocker, M.D.; Bauer, K.E.; Caldwell, C.G.; Hui, H.C.; Morgan, A.S.; Mergia, A.; Kauvar, L.M. Glutathione-S-Transferase activates novel alkylating agents. J. Med. Chem. 1994, 37, 1501–1507. [Google Scholar] [CrossRef]
- Satyam, A.; Hocker, M.D.; Kane-Maguire, K.A.; Morgan, A.S.; Villar, H.O.; Lyttle, M.H. Design, synthesis, and evaluation of latent alkylating agents activated by glutathione S-transferase. J. Med. Chem. 1996, 39, 1736–1747. [Google Scholar] [CrossRef]
- Townsend, D.M.; Shen, H.X.; Staros, A.L.; Gate, L.; Tew, K.D. Efficacy of a glutathione S-transferase pi-activated prodrug in platinum-resistant ovarian cancer. Mol. Cancer Ther. 2002, 1, 1089–1095. [Google Scholar]
- Morgan, A.S.; Sanderson, P.E.; Borch, R.F.; Tew, K.D.; Niitsu, Y.; Takayama, T.; von Hoff, D.D.; Izbicka, E.; Mangold, G.; Paul, C.; et al. Tumor efficacy and bone marrow-sparing properties of ter286, a cytotoxin activated by glutathione S-transferase. Cancer Res. 1998, 58, 2568–2575. [Google Scholar]
- Rosario, L.A.; O’Brien, M.L.; Henderson, C.J.; Wolf, C.R.; Tew, K.D. Cellular response to a glutathione S-transferase P1-1 Activated Prodrug. Mol. Pharmacol. 2000, 58, 167–174. [Google Scholar]
- Kavanagh, J.J.; Gershenson, D.M.; Choi, H.; Lewis, L.; Patel, K.; Brown, G.L.; Garcia, A.; Spriggs, D.R. Multi-Institutional phase 2 study of TLK286 (TELCYTA, a glutathione S-transferase P1-1 activated glutathione analog prodrug) in patients with platinum and paclitaxel refractory or resistant ovarian cancer. Int. J. Gynecol. Cancer 2005, 15, 593–600. [Google Scholar] [CrossRef]
- Ruzza, P.; Calderan, A.; Nassi, A.; Quintieri, L. Synthesis of GSH-linked tyrosinase-activated melanoma prodrugs. J. Pept. Sci. 2012, 18, S157. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Ruzza, P.; Calderan, A. Glutathione Transferase (GST)-Activated Prodrugs. Pharmaceutics 2013, 5, 220-231. https://doi.org/10.3390/pharmaceutics5020220
Ruzza P, Calderan A. Glutathione Transferase (GST)-Activated Prodrugs. Pharmaceutics. 2013; 5(2):220-231. https://doi.org/10.3390/pharmaceutics5020220
Chicago/Turabian StyleRuzza, Paolo, and Andrea Calderan. 2013. "Glutathione Transferase (GST)-Activated Prodrugs" Pharmaceutics 5, no. 2: 220-231. https://doi.org/10.3390/pharmaceutics5020220
APA StyleRuzza, P., & Calderan, A. (2013). Glutathione Transferase (GST)-Activated Prodrugs. Pharmaceutics, 5(2), 220-231. https://doi.org/10.3390/pharmaceutics5020220