Enhanced Dissolution and Oral Bioavailability of Piroxicam Formulations: Modulating Effect of Phospholipids

Abstract
:1. Introduction
2. Experimental Section
2.1. Materials
2.2. Preparation of Phospholipid-Based Solid Dispersions and Physical Mixture
2.3. Dissolution Studies
2.4. Differential Scanning Calorimetry (DSC)
2.5. Powder X-ray Diffraction (PXRD)
2.6. Bioavailability Studies
2.7. HPLC Analysis
3. Results and Discussion
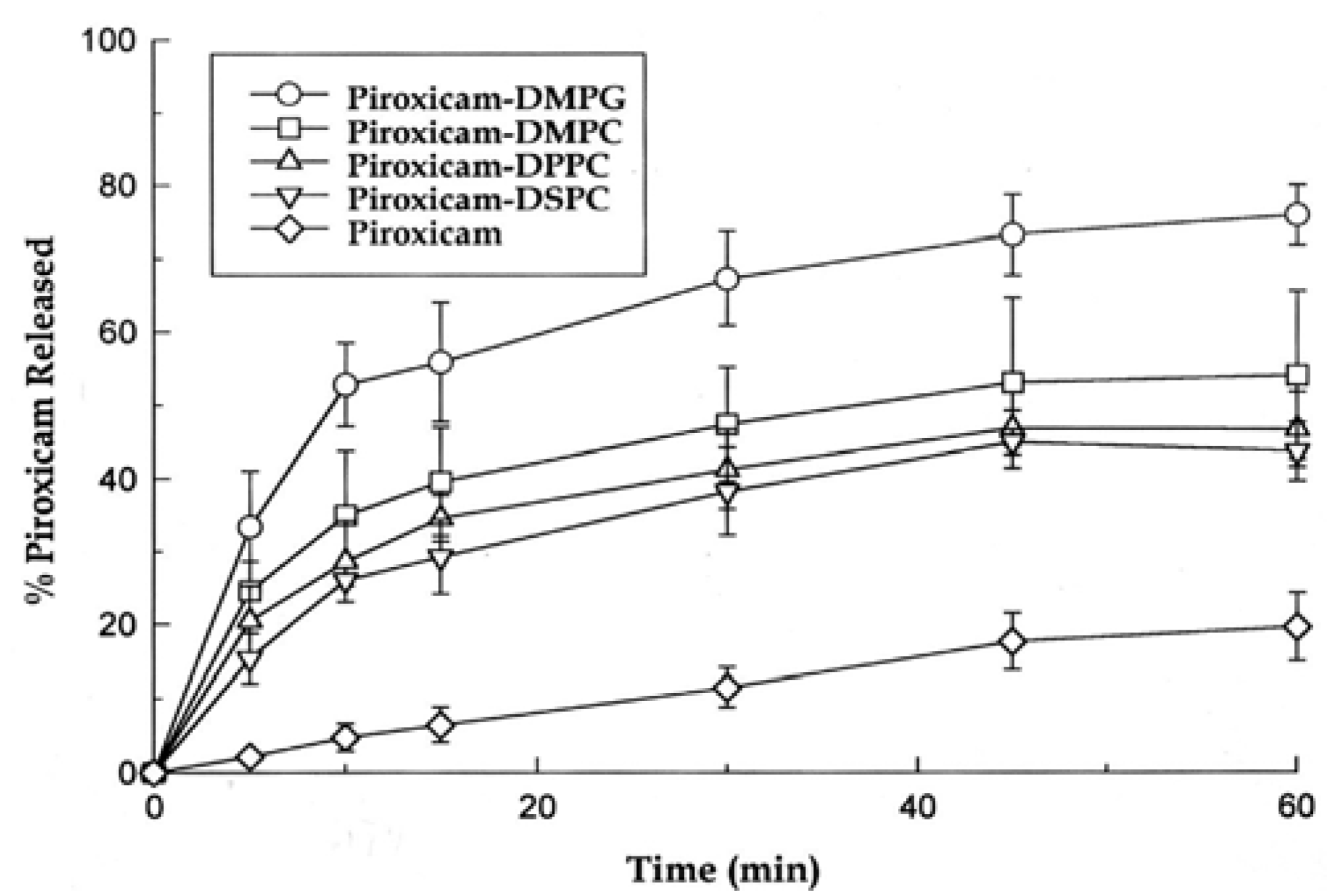
3.1. Effect of the Carrier Phospholipid on Dissolution Rate of Piroxicam

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Composition | Initial Dissolution Rate (% released/min) | % Released after 60 min |
|---|---|---|
| Control | 0.4 ± 0.2 | 19.7 ± 4.6 |
| Piroxicam-DMPG | 6.7 ± 1.5 | 76.1 ± 4.1 |
| Piroxicam-DMPC | 4.9 ± 0.8 | 54.2 ± 11.5 |
| Piroxicam-DPPC | 4.1 ± 1.0 | 46.8 ± 5.1 |
| Piroxicam-DSPC | 3.1 ± 0.7 | 43.8 ± 4.0 |
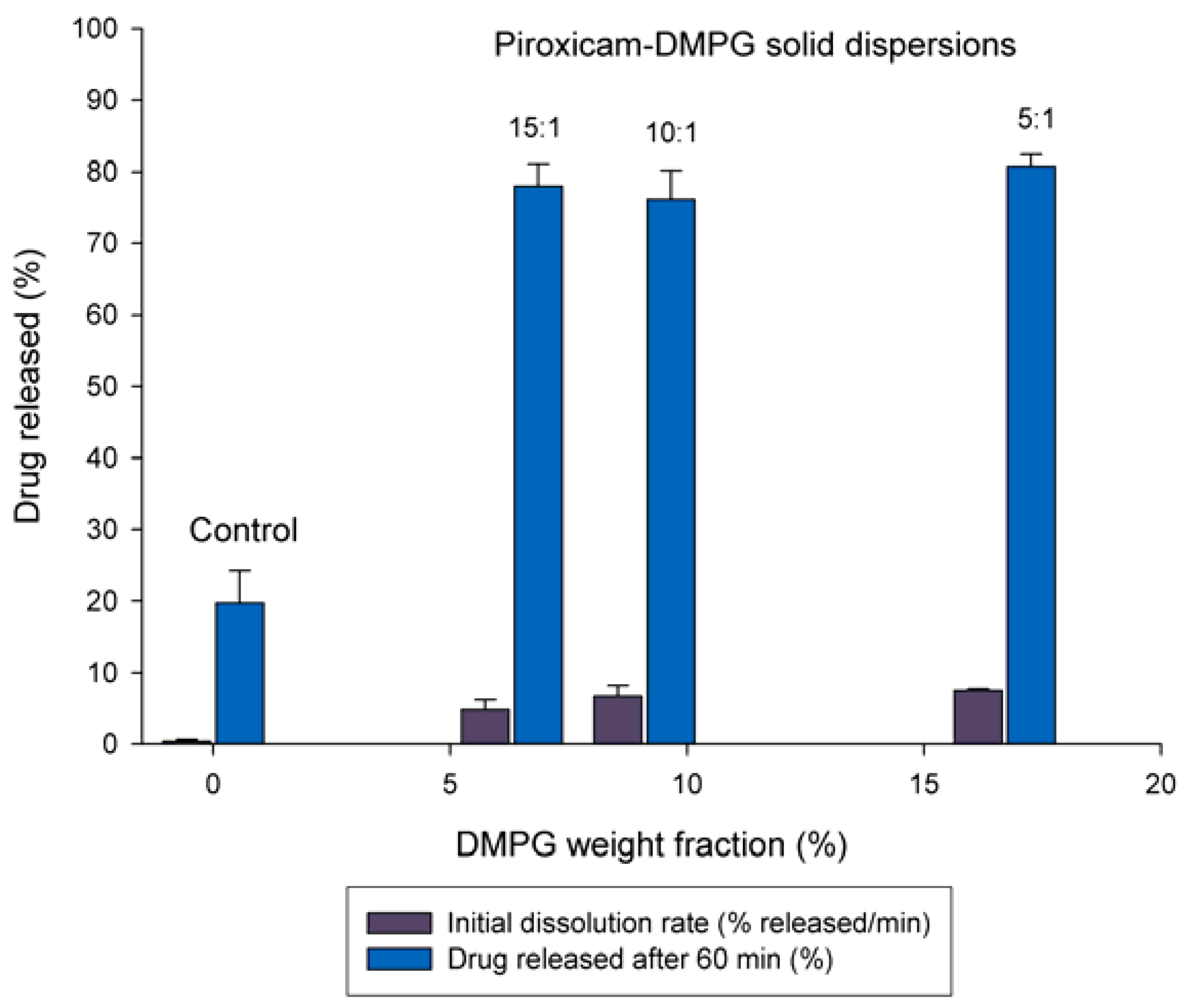
3.2. Effect of DMPG Weight Fraction on the Dissolution Rate of Piroxicam
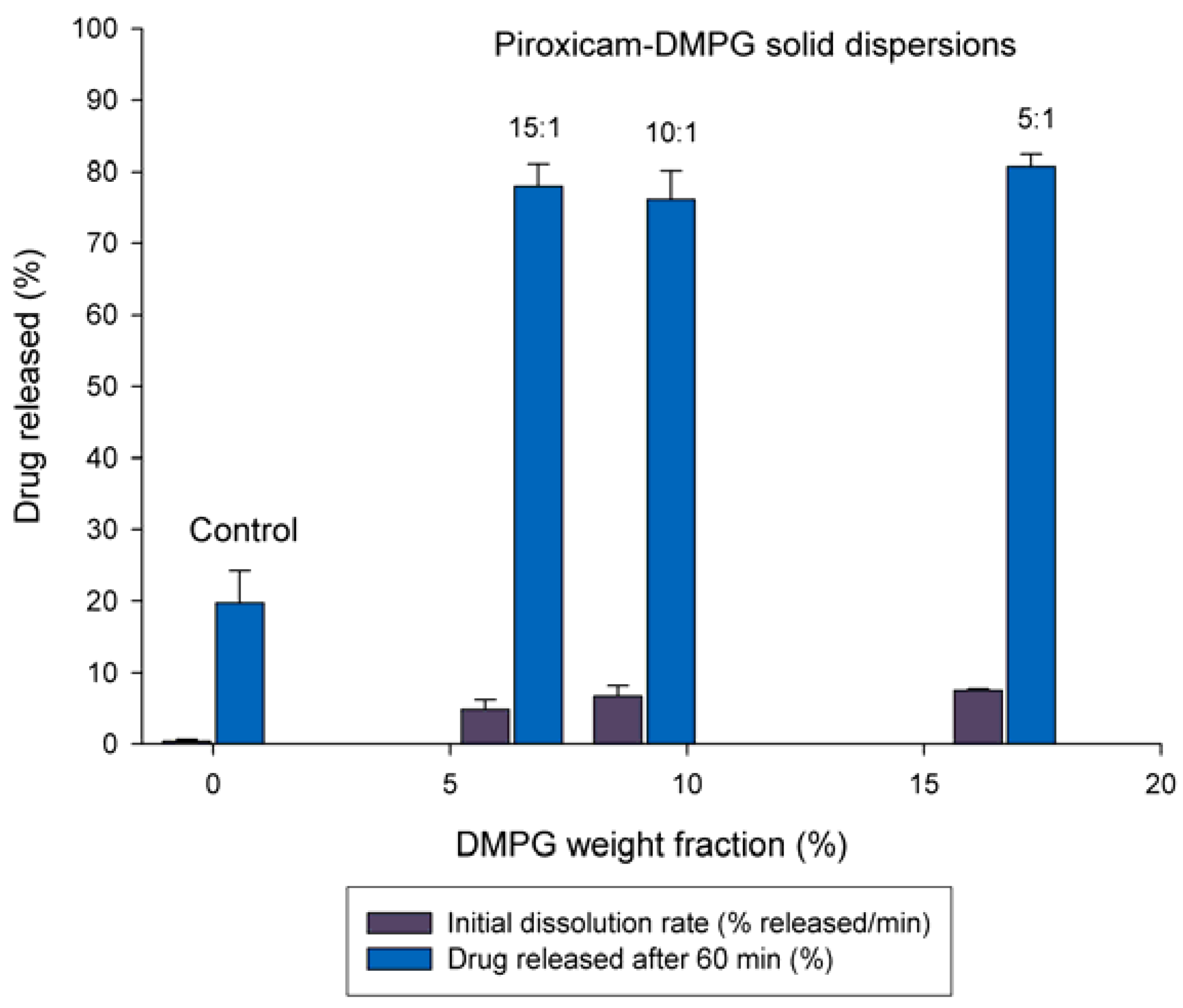
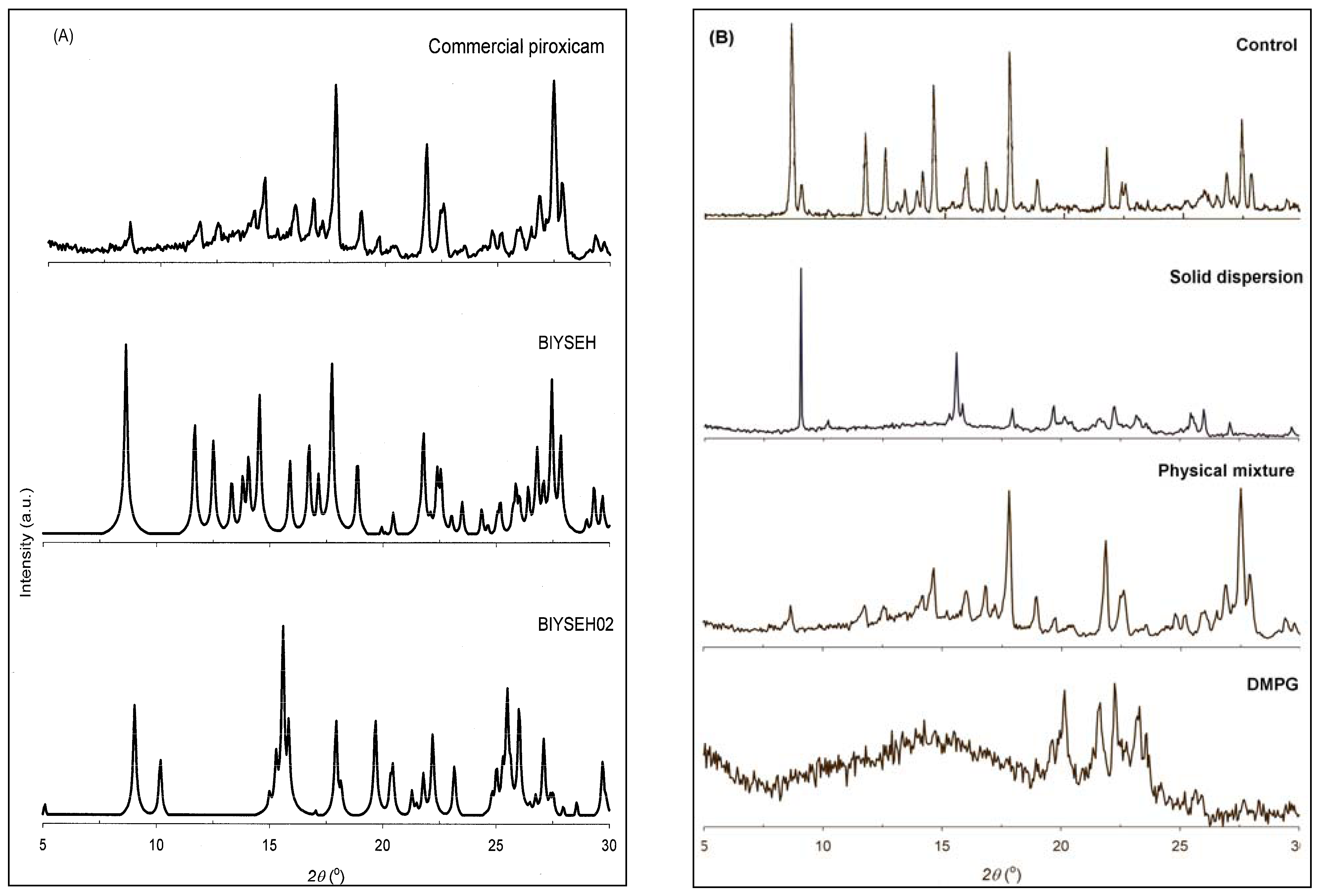
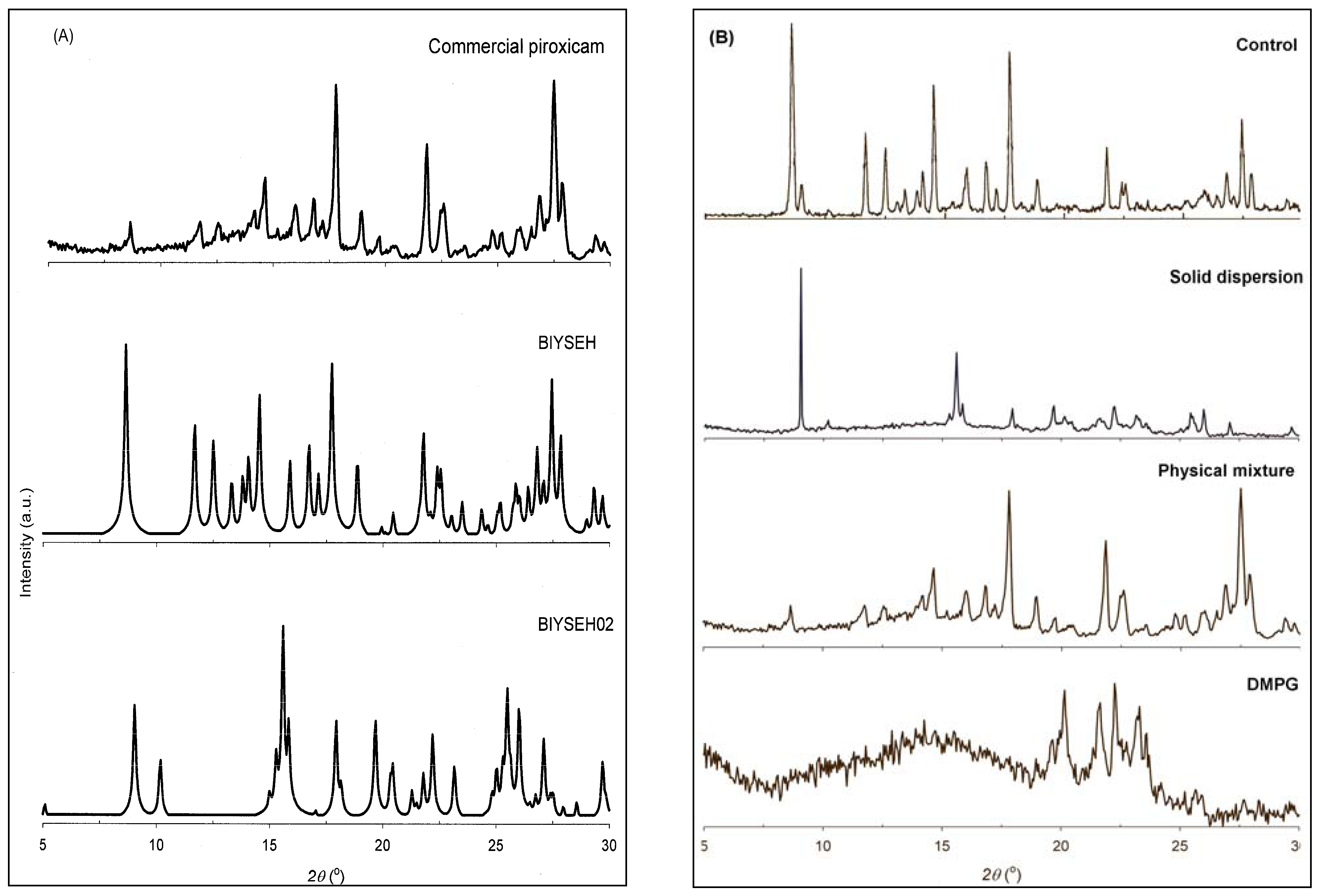
3.3. Solid-state Characterization of DMPG-based Solid Dispersions
| Sample | Melting temperature (onset, ºC) | Heat of melting (∆H, J/g) |
|---|---|---|
| Commercial Piroxicam | 201.1 ± 0.6 | 103.3 ± 1.0 |
| Control Piroxicam | 199.3 ± 1.1 | 98.5 ± 1.8 |
| Piroxicam:DMPG physical mixture (15:1, w/w) | 198.9 ± 1.6 | 93.7 ± 0.9 |
| Piroxicam:DMPG solid dispersions | ||
| 15:1 (w/w) | 198.4 ± 2.3 | 91.1 ± 1.9 |
| 10:1 (w/w) | 196.8 ± 1.7 | 75.6 ± 2.5 |
| 5:1 (w/w) | 195.2 ± 2.1 | 74.4 ± 3.2 |
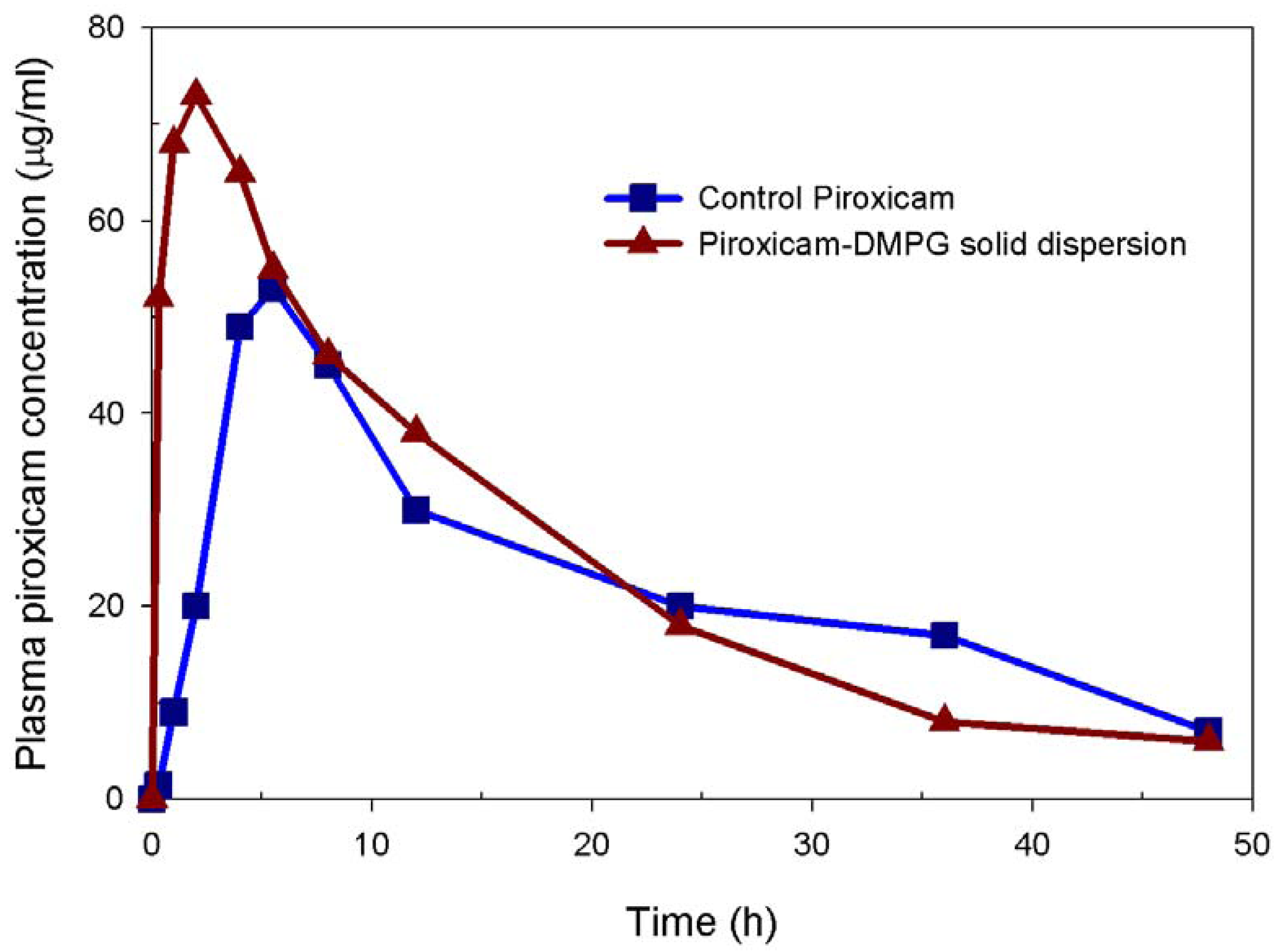
3.4. In vivo Performance of 15:1 Piroxicam-DMPG solid Dispersion
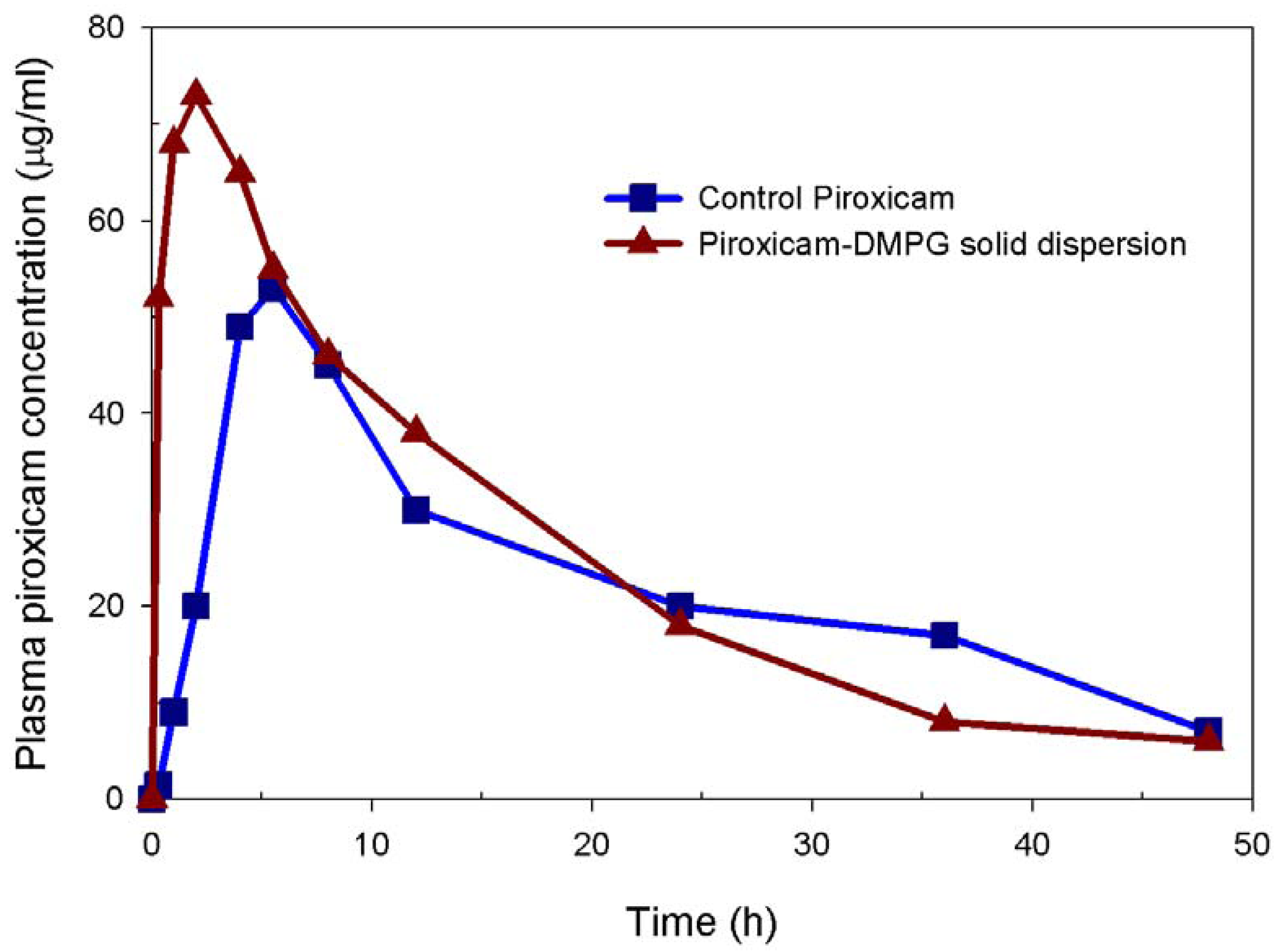
| Parameters | Formulation | |
|---|---|---|
| Control | DMPG-based solid dispersion (15:1, w/w) | |
| Peak plasma concentration (cmax, μg/mL) | 38.9 ±14.3 | 53.3 ± 15.1 |
| Time to peak concentration (Tmax, h) | 5.5 ± 2.1 | 2.0 ± 0.0* |
| AUC0-48 (μg/mL . h) | 921 ± 207 | 1210 ± 254 |
| Elimination half life (T1/2, h) | 13.0 ± 2.7 | 14.5 ± 5.6 |
| Mean residence time (MRT, h) | 25.9 ± 5.5 | 27.7 ± 8.2 |
4. Conclusions
Acknowledgements
References and Notes
- Amidon, G.L.; Lennernas, H.; Shah, V.P.; Crison, J.R. A theoretical basis for a biopharmaceutic drug classification: the correlation of in vitro drug product dissolution and in vivo bioavailability. Pharm. Res. 1995, 12, 413–420. [Google Scholar] [CrossRef]
- Serajuddin, A.T.M. Salt formation to improve solubility. Advan. Drug Delivery Rev. 2007, 59, 603–616. [Google Scholar] [CrossRef]
- Merisko-Liversidge, E.; Liversidge, G.G.; Cooper, E.R. Nanosizing: a formulation approach for poorly-water-soluble compounds. Eur. J. Pharm. Sci. 2003, 18, 113–120. [Google Scholar] [CrossRef]
- Chiou, W.; Riegelman, S. Pharmaceutical applications of solid dispersion system. J. Pharm. Sci. 1971, 60, 1281–1302. [Google Scholar] [CrossRef]
- Serajuddin, A.T.M. Solid dispersion of poorly water soluble drugs, early promises, subsequent problems and recent breakthroughs. J. Pharm. Sci. 1999, 88, 1058–1066. [Google Scholar] [CrossRef]
- Leuner, C.; Dressman, J. Improving drug solubility for oral delivery using solid dispersions. Eur. J. Pharm. Biopharm. 2000, 50, 47–60. [Google Scholar] [CrossRef]
- Copland, M.J.; Rades, T.; Davies, N.M.; Baird, M.A. Lipid based particulate formulations for the delivery of antigen. Immunol. Cell Biol. 2005, 83, 97–105. [Google Scholar] [CrossRef]
- Marsac, P.J.; Li, T.; Taylor, L.S. Estimation of drug-polymer miscibility and solubility in amorphous solid dispersions using experimentally determined interaction parameters. Pharm. Res. 2009, 26, 139–151. [Google Scholar] [CrossRef]
- Shikov, A.N.; Pozharitskaya, O.N.; Miroshnyk, I.; Mirza, S.; Urakova, I.N.; Hirsjärvi, S.; Makarov, V.G.; Heinämäki, J.; Yliruusi, J.; Hiltunen, R. Nanodispersions of taxifolin: impact of solid-state properties on dissolution behavior. Int. J. Pharm. 2009, 377, 148–152. [Google Scholar] [CrossRef]
- Fini, A.; Cavallari, C.; Ceschel, G.; Rabasco, A.M. Bimodal release of olanzapine from lipid microspheres. J. Pharm. Sci. 2010, 99, 4251–4260. [Google Scholar] [CrossRef]
- Craig, D.Q.M. The mechanisms of drug release from solid dispersions in water soluble polymers. Int. J. Pharm. 2002, 231, 131–144. [Google Scholar] [CrossRef]
- Forster, A.; Rades, T.; Hempenstall, J. Selection of suitable drug and excipient candidates to prepare glass solutions by melt extrusion for immediate release oral formulations. Pharm. Tech. Eur. 2002, 14, 27–37. [Google Scholar]
- Biswas, M.; Akogyeram, C.O.; Scott, K.R.; Potti, G.K.; Gallelli, J.F.; Habib, M.J. Development of carbamazepine:phospholipid solid dispersion formulations. J. Control. Release 1993, 23, 239–245. [Google Scholar] [CrossRef]
- Potti, G.; Gallelli, J.; Akoggeram, C.; Ahmadi, B.; Habib, M. Improved dissolution of indomethacin in coprecipitates with phospholipids - II. Drug Dev. Ind. Pharm. 1993, 19, 1221–1229. [Google Scholar] [CrossRef]
- Yamamura, S.; Rogers, J.A. Characterization and dissolution behavior of nifedipine and phosphatidylcholine binary systems. Int. J. Pharm. 1996, 130, 65–73. [Google Scholar] [CrossRef]
- Venkataram, S.; Rogers, J. Characteristics of drug-phospholipid coprecipitates I: Physical properties and dissolution behavior of griseofulvin-dimyristoylphosphatidylcholine systems. J. Pharm. Sci. 1984, 73, 757–761. [Google Scholar] [CrossRef]
- Lichenberger, L.; Wang, Z.-M.; Romero, J.; Ulloa, C.; Perez, J.; Giraud, M.N.; Barreto, J. Nonsteroidal anti-inflammatory drugs (NSAIDs) associate with zwitterionic phospholipids: Insight into the mechanism and reversal of NSAID-induced gastrointestinal injury. Nat. Med. 1995, 1, 154–158. [Google Scholar] [CrossRef]
- Budavari, S. The Merck Index, 12th ed.; Merck & Co., Inc.: Whitehouse Station, NJ, USA, 1996; p. 839. [Google Scholar]
- Tantishaiyakul, V.; Kaewnopparat, N.; Ingkatawornwong, S. Properties of solid dispersions of piroxicam in polyvinylpyrrolidone K-30. Int. J. Pharm. 1996, 143, 59–66. [Google Scholar] [CrossRef]
- Pan, R.N.; Chen, J.H.; Chen, R.R. Enhancement of dissolution and bioavailability of piroxicam in solid dispersion systems. Drug Dev. Ind. Pharm. 2000, 26, 989–994. [Google Scholar] [CrossRef]
- Wu, K.; Li, J.; Wang, W.; Winstead, D.A. Formation and characterization of solid dispersions of piroxicam and polyvinylpyrrolidone using spray drying and precipitation with compressed antisolvent. J. Pharm. Sci. 2009, 98, 2422–2431. [Google Scholar] [CrossRef]
- Prabhu, S.; Ortega, M.; Ma, C. Novel lipid-based formulations enhancing the in vitro dissolution and permeability characteristics of a poorly water –soluble model drug, piroxicam. Int. J. Pharm. 2005, 301, 209–216. [Google Scholar] [CrossRef]
- Rocci, M.L.; Jusko, W.J. Lagran program for area and moments in pharmacokinetic analysis. Comput. Progr. Biomed. 1983, 16, 203–216. [Google Scholar] [CrossRef]
- Boudinot, F.D.; Ibrahim, S.S. High-performance liquid chromatographic assay for piroxicam in human plasma. J. Chromatogr. 1998, 430, 424–428. [Google Scholar]
- Lingyun, Z.; Feng, S.S. Effects of lipid chain length on molecular interactions between paclitaxel and phospholipid within model biomembranes. J. Colloid Interface Sci. 2004, 274, 55–68. [Google Scholar] [CrossRef]
- Riske, K.A.; Döbereiner, H.-G.; Lamy-Freund, M.T. Gel-Fluid Transition in Dilute versus Concentrated DMPG Aqueous Dispersions. J. Phys. Chem. B 2002, 106, 239–246. [Google Scholar] [CrossRef]
- Vrecer, F.; Vrbink, M.; Meden, A. Characterization of piroxicam crystal modifications. Int. J. Pharm. 2003, 256, 3–15. [Google Scholar] [CrossRef]
- Sheth, A.R.; Bates, S.; Muller, F.X.; Grant, D.J.W. Local structure in amorphous phases of piroxicam from powder x-ray diffractometry. Cryst. Growth Des. 2005, 5, 571–578. [Google Scholar] [CrossRef]
- Kojic-Prodic, B.; Ruzic-Toros, Z. Structure of the anti-inflammatory drug 4-hydroxy-2-methyl-N-2-pyridyl-2H-1λ6,2-benzothiazine-3-carboxamide 1,1-dioxide (piroxicam). Acta Crystallogr. Sect. B 1982, 38, 2948–2951. [Google Scholar] [CrossRef]
- Reck, G.; Dietz, G.; Laban, G.; Gunther, W.; Bannier, G.; Hohne, E. X-ray studies on piroxicam modifications. Pharmazie 1988, 43, 477–481. [Google Scholar]
- Lee, C.R.; Balfour, J.A. Piroxicam-β-cyclodextrin: A review of its pharmacodynamic and pharmacokinetic properties, and therapeutic potential in rheumatic diseases and pain states. Drugs 1994, 48, 907–929. [Google Scholar] [CrossRef]
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Mirza, S.; Miroshnyk, I.; Habib, M.J.; Brausch, J.F.; Hussain, M.D. Enhanced Dissolution and Oral Bioavailability of Piroxicam Formulations: Modulating Effect of Phospholipids. Pharmaceutics 2010, 2, 339-350. https://doi.org/10.3390/pharmaceutics2040339
Mirza S, Miroshnyk I, Habib MJ, Brausch JF, Hussain MD. Enhanced Dissolution and Oral Bioavailability of Piroxicam Formulations: Modulating Effect of Phospholipids. Pharmaceutics. 2010; 2(4):339-350. https://doi.org/10.3390/pharmaceutics2040339
Chicago/Turabian StyleMirza, Sabiruddin, Inna Miroshnyk, Muhammad J. Habib, James F. Brausch, and Muhammad D. Hussain. 2010. "Enhanced Dissolution and Oral Bioavailability of Piroxicam Formulations: Modulating Effect of Phospholipids" Pharmaceutics 2, no. 4: 339-350. https://doi.org/10.3390/pharmaceutics2040339
APA StyleMirza, S., Miroshnyk, I., Habib, M. J., Brausch, J. F., & Hussain, M. D. (2010). Enhanced Dissolution and Oral Bioavailability of Piroxicam Formulations: Modulating Effect of Phospholipids. Pharmaceutics, 2(4), 339-350. https://doi.org/10.3390/pharmaceutics2040339