
Perspectives on the pH-Influenced Design of Chitosan–Genipin Nanogels for Cell-Targeted Delivery

 , , , , , and
, , , , , and
Abstract

1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Generation of CS-GNP NG
2.3. NG Formation Monitoring
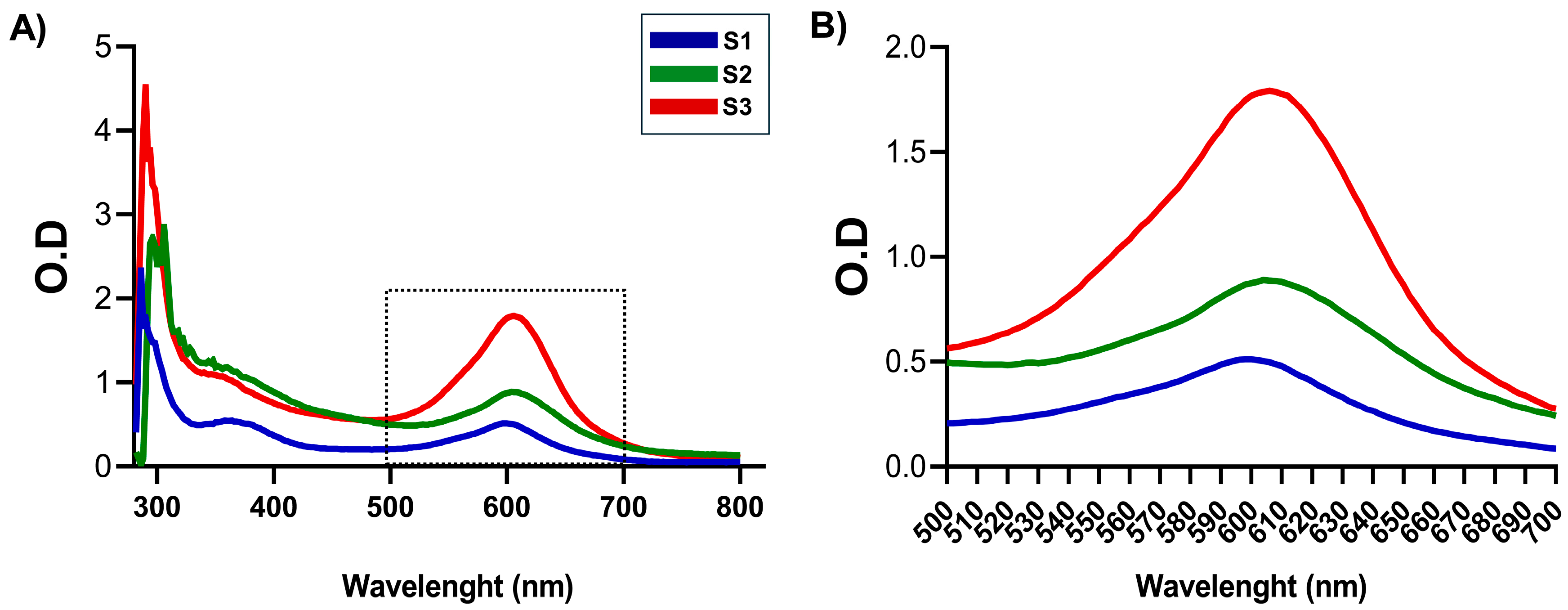
2.3.1. Spectral Analysis
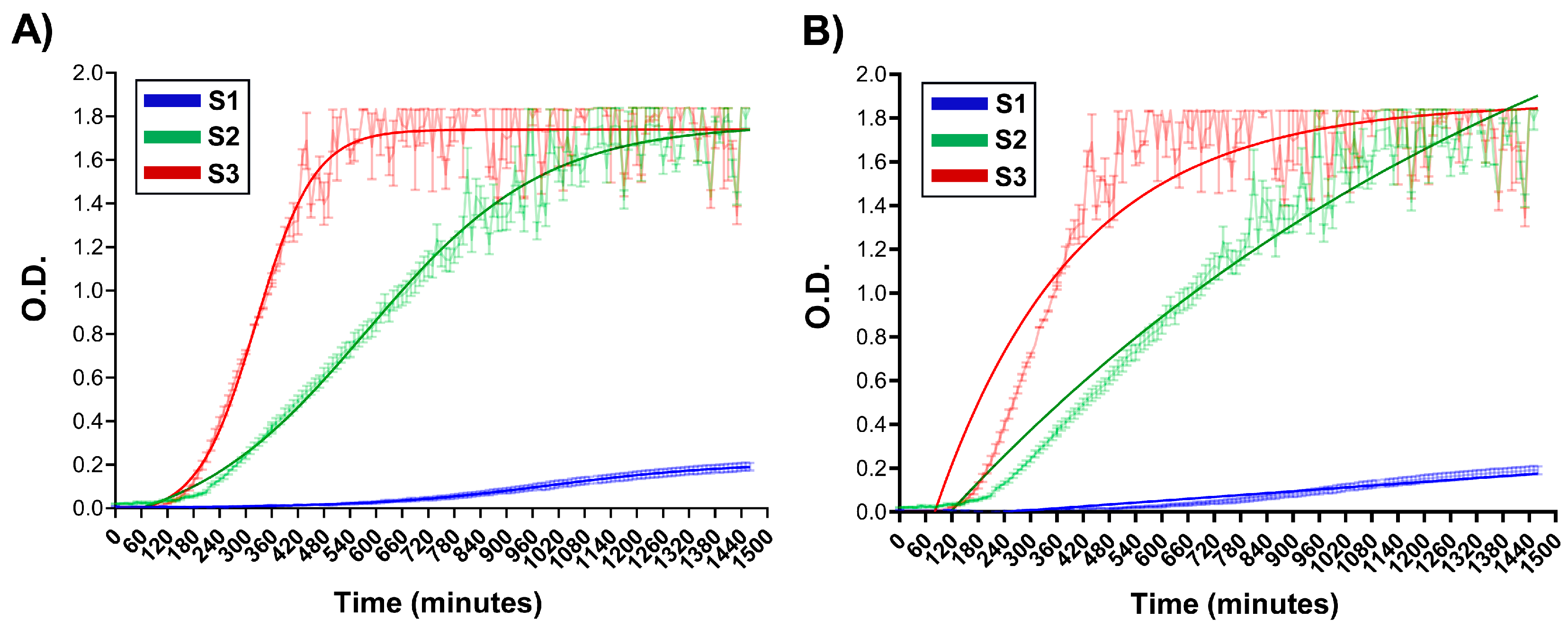
2.3.2. Kinetics Approach
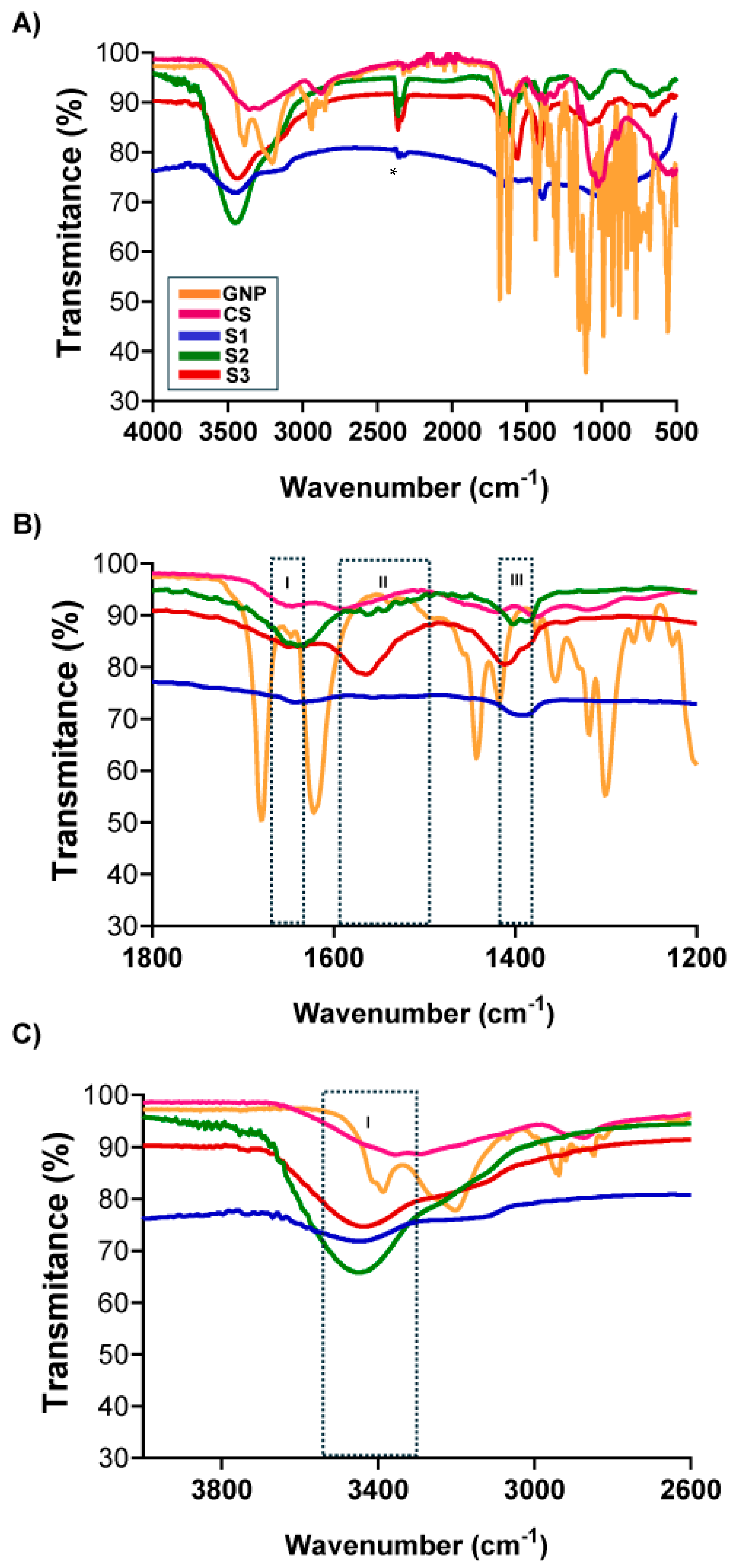
2.3.3. Fourier-Transform Infrared (FTIR)
2.4. NG Characterisation
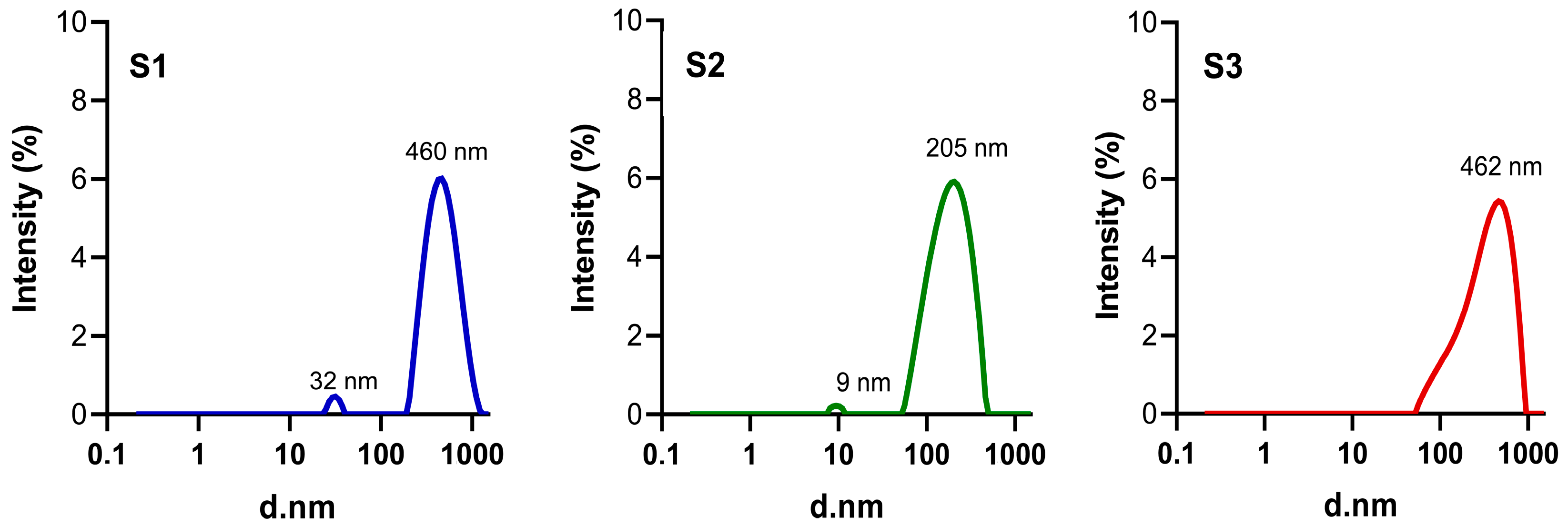
2.4.1. Particle Size Distribution and ζ-Potential
2.4.2. Stability Measurement of NG
2.4.3. Scanning Electron Microscopy (SEM) Analysis
2.4.4. Transmission Electron Microscopy (TEM) Analysis
2.4.5. Rheological Analysis
2.4.6. Fluorescence Spectra
2.4.7. Small-Angle X-Ray Scattering (SAXS)
2.4.8. Swelling Test
2.5. NGs in Biological Systems
2.5.1. Cell Culture Conditions
2.5.2. Cell Metabolic Activity Determination
2.5.3. Cellular Internalisation
2.6. Statistical Analysis
3. Results and Discussion
3.1. NG Construction and Kinetics of Formation
3.2. Chemical Groups Interaction
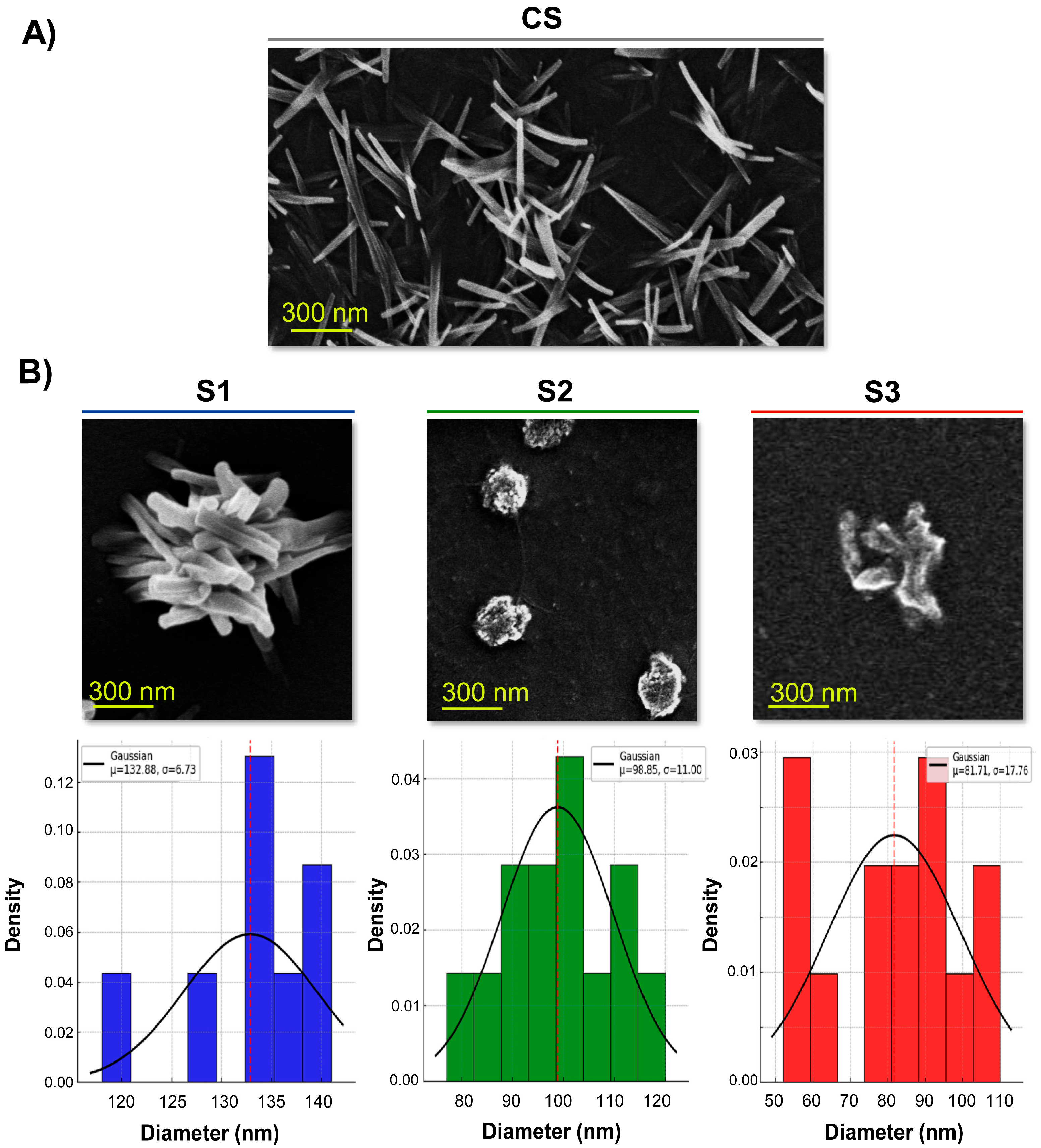
3.3. Particle Size Distribution, PDI and ζ-Potential
3.4. Stability of NG Formulation Under Physiological Temperature Conditions
3.5. Ultrastructural Analysis of NG
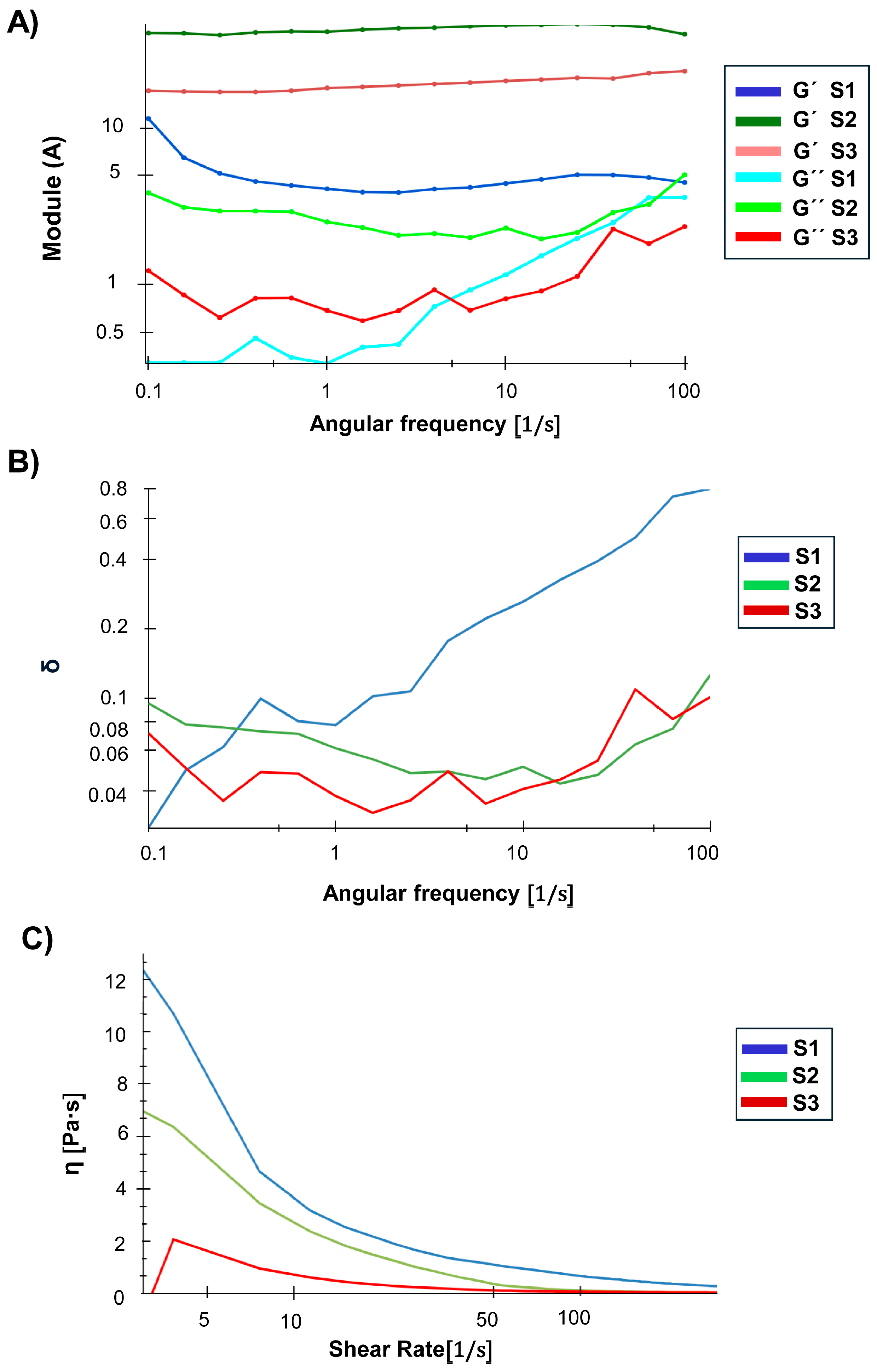
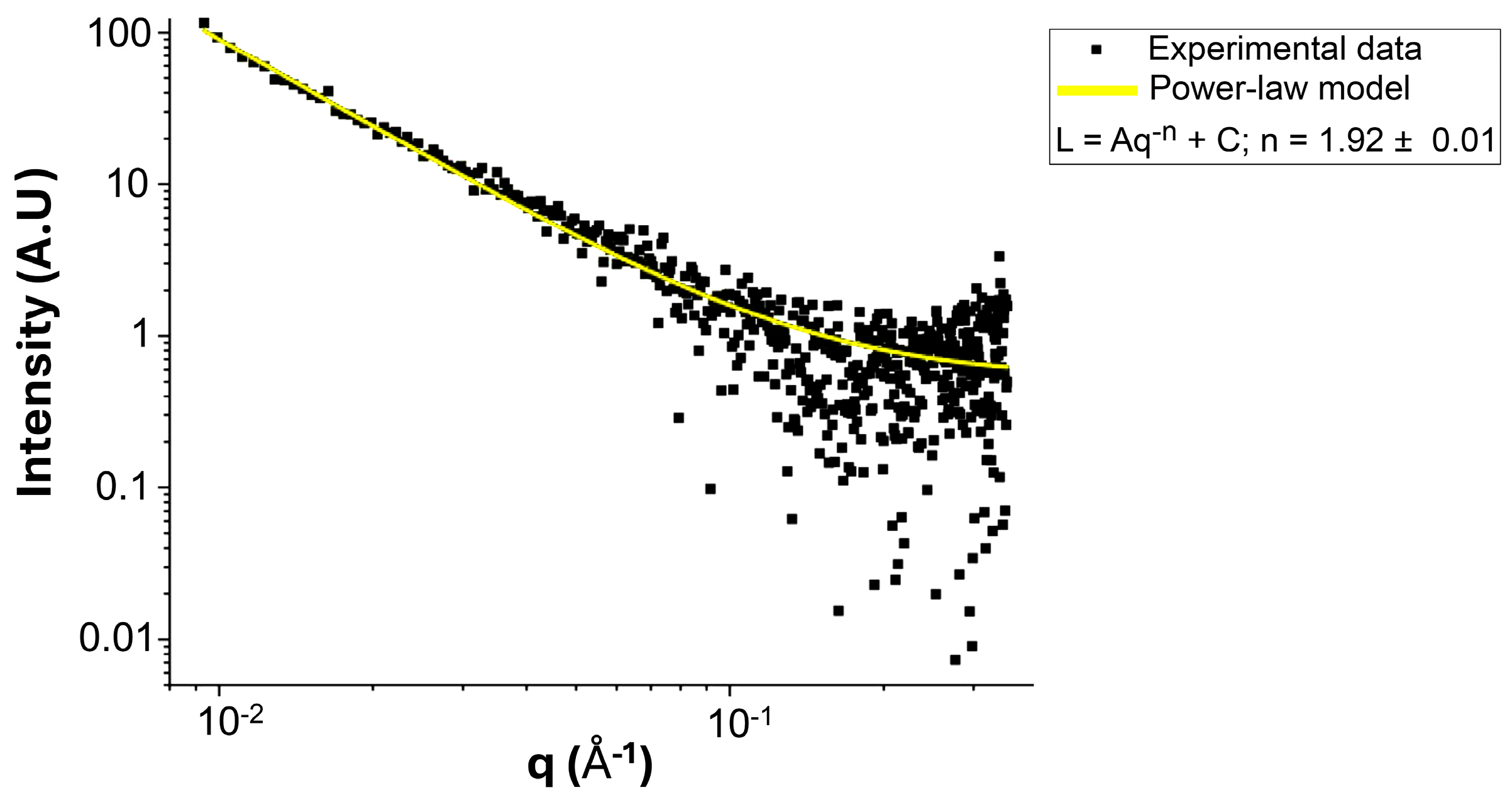
3.6. NG Rheological and SAXS Insights
3.7. Swelling Behaviour of NGs
3.8. Biocompatibility Assays
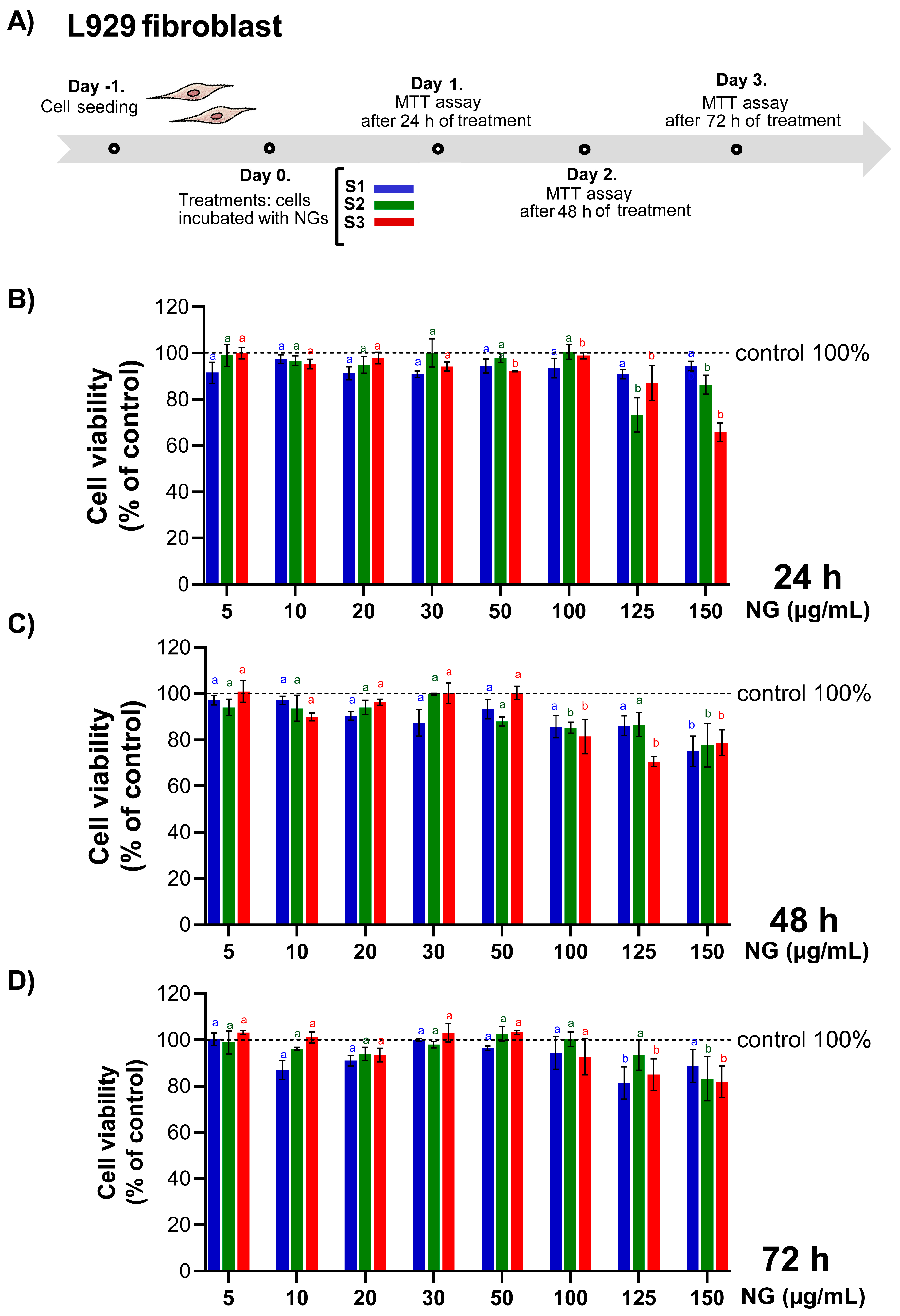
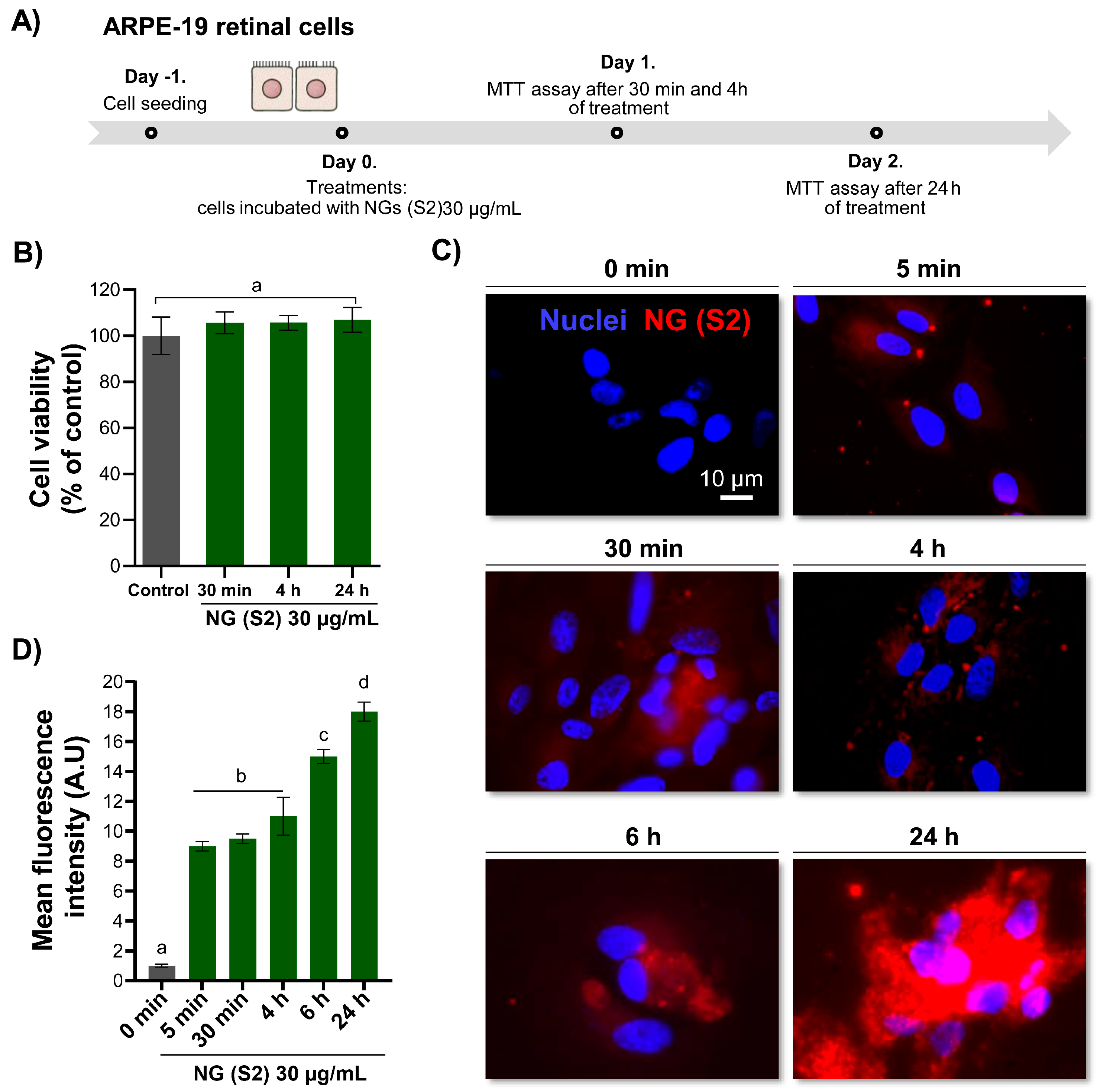
3.8.1. Impact of CS-GNP NG on Cell Viability
3.8.2. Cellular Internalisation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ξ | correlation length |
| η | viscosity |
| δ | phase angle |
| γ | shear rate |
| ARPE-19 | human retinal pigment epithelial cells |
| CS | chitosan |
| CS-GNP NG | chitosan–genipin nanogel |
| d.nm | diameter |
| DAPI | 4’,6-diamidino-2-fenilindol fluorescent probe |
| DMEM | Dulbecco’s modified Eagle’s medium |
| DLS | dynamic light scattering |
| ESR | equilibrium swelling ratio |
| FBS | foetal bovine serum |
| FOE | first-order exponential |
| FTIR | Fourier-transform infrared spectroscopy |
| G′ | viscoelastic storage |
| G″ | loss module |
| GNP | genipin |
| HIUS | High-intensity ultrasound |
| m | power-law exponent |
| MTT | colorimetric assay used to assess cytotoxicity |
| NG | nanogel |
| NP | nanoparticle |
| L929 | mouse fibroblast cell line |
| PDI | polydispersity index |
| SAXS | small-angle X-ray scattering |
| SEM | scanning electron microscopy |
| SK | sigmoidal kinetics |
| TEM | transmission electron microscopy |
References
- Cao, Y.; Tan, Y.F.; Wong, Y.S.; Liew, M.W.J.; Venkatraman, S. Recent Advances in Chitosan-Based Carriers for Gene Delivery. Mar. Drugs 2019, 17, 381. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Álvarez, L.; Ruiz-Rubio, L.; Vilas-Vilela, J.L. Handbook of Materials for Nanomedicine, 1st ed.; Torchilin, V., Ed.; Jenny Stanford Publishing: New York, NY, USA, 2020; ISBN 9781003045113. [Google Scholar]
- Biswas, R.; Mondal, S.; Ansari, M.A.; Sarkar, T.; Condiuc, I.P.; Trifas, G.; Atanase, L.I. Chitosan and Its Derivatives as Nanocarriers for Drug Delivery. Molecules 2025, 30, 1297. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Qian, J.; Ding, F. Recent Advances in Engineered Chitosan-Based Nanogels for Biomedical Applications. J. Mater. Chem. B 2017, 5, 6986–7007. [Google Scholar] [CrossRef]
- Rinaudo, M. Chitin and Chitosan: Properties and Applications. Prog. Polym. Sci. 2006, 31, 603–632. [Google Scholar] [CrossRef]
- Lai, J.Y. Biocompatibility of Genipin and Glutaraldehyde Cross-Linked Chitosan Materials in the Anterior Chamber of the Eye. Int. J. Mol. Sci. 2012, 13, 10970–10985. [Google Scholar] [CrossRef] [PubMed]
- Muhamad, I.I.; Fen, L.S.; Hui, N.H.; Mustapha, N.A. Genipin-Cross-Linked Kappa-Carrageenan/Carboxymethyl Cellulose Beads and Effects on Beta-Carotene Release. Carbohydr. Polym. 2011, 83, 1207–1212. [Google Scholar] [CrossRef]
- Sharma, K.; Kaur, M.; Rattan, G.; Kaushik, A. Effective Biocatalyst Developed via Genipin Mediated Acetylcholinesterase Immobilization on Rice Straw Derived Cellulose Nanofibers for Detection and Bioremediation of Organophosphorus Pesticide. Colloids Surf. A Physicochem. Eng. Asp. 2022, 640, 128484. [Google Scholar] [CrossRef]
- Hackenhaar, C.R.; Rosa, C.F.; Flores, E.E.E.; Santagapita, P.R.; Klein, M.P.; Hertz, P.F. Development of a Biocomposite Based on Alginate/Gelatin Crosslinked with Genipin for β-Galactosidase Immobilization: Performance and Characteristics. Carbohydr. Polym. 2022, 291, 119483. [Google Scholar] [CrossRef]
- Gonçalves, I.; Hernández, D.; Cruz, C.; Lopes, J.; Barra, A.; Nunes, C.; da Silva, J.A.L.; Ferreira, P.; Coimbra, M.A. Relevance of Genipin Networking on Rheological, Physical, and Mechanical Properties of Starch-Based Formulations. Carbohydr. Polym. 2021, 254, 117236. [Google Scholar] [CrossRef]
- Zawawi, N.A.; Maarof, M.; Fadilah, N.I.M.; Hao, D.L.Q.; Tabata, Y.; Fauzi, M.B. Hybrid Adhesive Hydrogel Patch Containing Genipin-Crosslinked Gelatin–Hyaluronic Acid for Future Use in Atopic Dermatitis. J. Funct. Biomater. 2025, 16, 195. [Google Scholar] [CrossRef]
- Roether, J.; Oelschlaeger, C.; Willenbacher, N. Hyaluronic Acid Cryogels with Non-Cytotoxic Crosslinker Genipin. Mater. Lett. X 2019, 4, 100027. [Google Scholar] [CrossRef]
- Priddy-Arrington, T.R.; Edwards, R.E.; Colley, C.E.; Nguyen, M.M.; Hamilton-Adaire, T.; Caldorera-Moore, M.E. Characterization and Optimization of Injectable In Situ Crosslinked Chitosan-Genipin Hydrogels. Macromol. Biosci. 2023, 23, e2200505. [Google Scholar] [CrossRef]
- Butler, M.F.; Ng, Y.-F.; Pudney, P.D.A. Mechanism and Kinetics of the Crosslinking Reaction between Biopolymers Containing Primary Amine Groups and Genipin. J. Polym. Sci. Part A Polym. Chem. 2003, 41, 3941–3953. [Google Scholar] [CrossRef]
- Muzzarelli, R.A.A.; El Mehtedi, M.; Bottegoni, C.; Aquili, A.; Gigante, A. Genipin-Crosslinked Chitosan Gels and Scaffolds for Tissue Engineering and Regeneration of Cartilage and Bone. Mar. Drugs 2015, 13, 7314–7338. [Google Scholar] [CrossRef]
- Delmar, K.; Bianco-Peled, H. The Dramatic Effect of Small PH Changes on the Properties of Chitosan Hydrogels Crosslinked with Genipin. Carbohydr. Polym. 2015, 127, 28–37. [Google Scholar] [CrossRef]
- Mi, F.L.; Tan, Y.C.; Liang, H.F.; Sung, H.W. In Vivo Biocompatibility and Degradability of a Novel Injectable-Chitosan-Based Implant. Biomaterials 2002, 23, 181–191. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Tang, J.; Liu, Y.; Hu, B.; Chen, Q.; Liu, Y. Reaction Kinetics of Chitosan Nanogels Crosslinked by Genipin. J. Chromatogr. A 2023, 1710, 464427. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Xu, S.; Li, S.; Pan, H. Genipin-Cross-Linked Hydrogels Based on Biomaterials for Drug Delivery: A Review. Biomater. Sci. 2021, 9, 1583–1597. [Google Scholar] [CrossRef]
- Buosi, F.S.; Alaimo, A.; Di Santo, M.C.; Elías, F.; García Liñares, G.; Acebedo, S.L.; Castañeda Cataña, M.A.; Spagnuolo, C.C.; Lizarraga, L.; Martínez, K.D.; et al. Resveratrol Encapsulation in High Molecular Weight Chitosan-Based Nanogels for Applications in Ocular Treatments: Impact on Human ARPE-19 Culture Cells. Int. J. Biol. Macromol. 2020, 165, 804–821. [Google Scholar] [CrossRef]
- Rivera López, E.; Samaniego López, C.; Spagnuolo, C.C.; Berardino, B.G.; Alaimo, A.; Pérez, O.E. Chitosan-Tricarbocyanine-Based Nanogels Were Able to Cross the Blood–Brain Barrier Showing Its Potential as a Targeted Site Delivery Agent. Pharmaceutics 2024, 16, 964. [Google Scholar] [CrossRef]
- De Matteo, R.; Rey, J.M.; Corfield, R.; Gómez Andrade, V.A.; Santagapita, P.R.; Di Salvo, F.; Pérez, O.E. Chitosan-Inspired Matrices for Folic Acid. Insightful Structural Characterization and Ensured Bioaccessibility. Food Biophys. 2024, 19, 412–424. [Google Scholar] [CrossRef]
- Miras, J.; Liu, C.; Blomberg, E.; Thormann, E.; Vílchez, S.; Esquena, J. PH-Responsive Chitosan Nanofilms Crosslinked with Genipin. Colloids Surf. A Physicochem. Eng. Asp. 2021, 616, 126229. [Google Scholar] [CrossRef]
- Flores, E.E.E.; Cardoso, F.D.; Siqueira, L.B.; Ricardi, N.C.; Costa, T.H.; Rodrigues, R.C.; Klein, M.P.; Hertz, P.F. Influence of Reaction Parameters in the Polymerization between Genipin and Chitosan for Enzyme Immobilization. Process Biochem. 2019, 84, 73–80. [Google Scholar] [CrossRef]
- Kildeeva, N.; Chalykh, A.; Belokon, M.; Petrova, T.; Matveev, V.; Svidchenko, E.; Surin, N.; Sazhnev, N. Influence of Genipin Crosslinking on the Properties of Chitosan-Based Films. Polymers 2020, 12, 1086. [Google Scholar] [CrossRef]
- Arteche Pujana, M.; Pérez-Álvarez, L.; Cesteros Iturbe, L.C.; Katime, I. Biodegradable Chitosan Nanogels Crosslinked with Genipin. Carbohydr. Polym. 2013, 94, 836–842. [Google Scholar] [CrossRef]
- Moura, M.J.; Martins, S.P.; Duarte, B.P.M. Production of Chitosan Microparticles Cross-Linked with Genipin–Identification of Factors Influencing Size and Shape Properties. Biochem. Eng. J. 2015, 104, 82–90. [Google Scholar] [CrossRef]
- Silva, C.P.; Martínez, J.H.; Martínez, K.D.; Farías, M.E.; Leskow, F.C.; Pérez, O.E. Proposed Molecular Model for Electrostatic Interactions between Insulin and Chitosan. Nano-Complexation and Activity in Cultured Cells. Colloids Surf. A Physicochem. Eng. Asp. 2018, 537, 425–434. [Google Scholar] [CrossRef]
- Mi, F.-L.; Sung, H.-W.; Shyu, S.-S. Synthesis and Characterization of a Novel Chitosan-Based Network Prepared Using Naturally Occurring Crosslinker. J. Appl. Polym. Sci. 2000, 28, 2804–2814. [Google Scholar] [CrossRef]
- Pérez, O.E.; David-Birman, T.; Kesselman, E.; Levi-Tal, S.; Lesmes, U. Milk Protein–Vitamin Interactions: Formation of Beta-Lactoglobulin/Folic Acid Nano-Complexes and Their Impact on in Vitro Gastro-Duodenal Proteolysis. Food Hydrocoll. 2014, 38, 40–47. [Google Scholar] [CrossRef]
- Moore, J.; Cerasoli, E. Particle Light Scattering Methods and Applications. Available online: https://nanoqam.ca/wiki/lib/exe/fetch.php?media=particle_light_scattering_methods_and_applications.pdf (accessed on 2 June 2025).
- Blees, M.H. Foundations of Colloid Science. Colloids Surf. A Physicochem. Eng. Asp. 2002, 210, 125. [Google Scholar] [CrossRef]
- Bruno, S.G.; Alaimo, A.; Samaniego López, C.; Rubinstein, A.J.; Nemirovsky, S.I.; Moretton, M.A.; Chiappetta, D.A.; Pérez, O.E. Progress in the Design of Chitosan-Based Nanogels for Folic Acid Delivery for Ocular Applications. Int. J. Biol. Macromol. 2025, 318, 145053. [Google Scholar] [CrossRef] [PubMed]
- Vo, N.T.N.; Huang, L.; Lemos, H.; Mellor, A.L.; Novakovic, K. Genipin-Crosslinked Chitosan Hydrogels: Preliminary Evaluation of the in Vitro Biocompatibility and Biodegradation. J. Appl. Polym. Sci. 2021, 138, 50848. [Google Scholar] [CrossRef]
- Gruber, S.; Nickel, A. Toxic or Not Toxic? The Specifications of the Standard ISO 10993-5 Are Not Explicit Enough to Yield Comparable Results in the Cytotoxicity Assessment of an Identical Medical Device. Front. Med. Technol. 2023, 5, 1195529. [Google Scholar] [CrossRef] [PubMed]
- Pizzolitto, C.; Cok, M.; Asaro, F.; Scognamiglio, F.; Marsich, E.; Lopez, F.; Donati, I.; Sacco, P. On the Mechanism of Genipin Binding to Primary Amines in Lactose-Modified Chitosan at Neutral PH. Int. J. Mol. Sci. 2020, 21, 6831. [Google Scholar] [CrossRef]
- Touyama, R.; Takeda, Y.; Inoue, K.; Kawamura, I.; Yatsuzuka, M.; Ikumoto, T.; Shingu, T.; Yokoi, T.; Inouye, H. Studies on blue pigments produced from genipin and methylamine. I. Structures of reddish-brown pigments, intermediates leading to blue pigments. Chem. Pharm. Bull. 1994, 42, 668–673. [Google Scholar] [CrossRef]
- Dadashi, H.; Vandghanooni, S.; Karamnejad-Faragheh, S.; Karimian-Shaddel, A.; Eskandani, M.; Jahanban-Esfahlan, R. A Rapid Protocol for Synthesis of Chitosan Nanoparticles with Ideal Physicochemical Features. Heliyon 2024, 10, e32228. [Google Scholar] [CrossRef]
- Lárez Velásquez, C.; Rivas, A.; Santos Ocanto, I. Obtención de Genipina a Partir de Frutos de Caruto (Genipa americana L.) Del Llano Venezolano. Av. Química 2014, 9, 75–86. [Google Scholar] [CrossRef]
- Murugan, K.; Choonara, Y.E.; Kumar, P.; Bijukumar, D.; du Toit, L.C.; Pillay, V. Parameters and Characteristics Governing Cellular Internalization and Trans-Barrier Trafficking of Nanostructures. Int. J. Nanomed. 2015, 10, 2191–2206. [Google Scholar] [CrossRef]
- Reay, S.L.; Jackson, E.L.; Ferreira, A.M.; Hilkens, C.M.U.; Novakovic, K. In Vitro Evaluation of the Biodegradability of Chitosan-Genipin Hydrogels. Mater. Adv. 2022, 3, 7946–7959. [Google Scholar] [CrossRef]
- Malvern Panalytical. Dynamic Light Scattering (DLS). Available online: https://www.malvernpanalytical.com/en/products/technology/light-scattering/dynamic-light-scattering (accessed on 30 June 2025).
- Williams, P.M. Encyclopedia of Membranes; Springer: Berlin/Heidelberg, Germany, 2016. [Google Scholar]
- Rasmussen, M.K.; Pedersen, J.N.; Marie, R. Size and Surface Charge Characterization of Nanoparticles with a Salt Gradient. Nat. Commun. 2020, 11, 2337. [Google Scholar] [CrossRef]
- Chunhachaichana, C.; Sawatdee, S.; Rugmai, S.; Srichana, T. Development and Characterization of Nanodispersion-Based Sildenafil Pressurized Metered-Dose Inhaler Using Combined Small-Angle X-ray Scattering, Dynamic Light Scattering, and Impactors. J. Drug Deliv. Sci. Technol. 2022, 76, 103749. [Google Scholar] [CrossRef]
- Dimida, S.; Demitri, C.; De Benedictis, V.M.; Scalera, F.; Gervaso, F.; Sannino, A. Genipin-Cross-Linked Chitosan-Based Hydrogels: Reaction Kinetics and Structure-Related Characteristics. J. Appl. Polym. Sci. 2015, 132, 42256. [Google Scholar] [CrossRef]
- Della Sala, F.; Barretta, M.; di Gennaro, M.; Paradiso, R.; Borriello, G.; Borzacchiello, A. Bio-Composite Nanogels Based on Chitosan and Hyaluronic Acid for the Treatment of Lung Infections. Gels 2024, 10, 709. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Meng, X.; Liu, S.; Poisson, J.; Vana, P.; Zhang, K. Dehydration Regulates Structural Reorganization of Dynamic Hydrogels. Nat. Commun. 2024, 15, 6886. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Sande, S.A. A Dynamic Light Scattering Study of Hydrogels with the Addition of Surfactant: A Discussion of Mesh Size and Correlation Length. Polym. J. 2015, 47, 302–310. [Google Scholar] [CrossRef]
- John, D.F. Viscoelastic Properties of Polymers; Wiley: Hoboken, NJ, USA, 1980. [Google Scholar]
- Parès, L.; Naasri, S.; Delattre, L.; Therriault, H.; Liberelle, B.; De Crescenzo, G.; Lauzon, M.A.; Faucheux, N.; Paquette, B.; Virgilio, N. Macroporous Chitosan/Alginate Hydrogels Crosslinked with Genipin Accumulate and Retain Glioblastoma Cancer Cells. RSC Adv. 2024, 14, 35286–35304. [Google Scholar] [CrossRef]
- Rhim, J.W.; Park, H.M.; Ha, C.S. Bio-Nanocomposites for Food Packaging Applications. Prog. Polym. Sci. 2013, 38, 1629–1652. [Google Scholar] [CrossRef]
- Matos Ruiz, M.E.; Muller-Karger, C.; Sabino, M.A. Caracterización de Hidrogeles de Quitosano Entrecruzados Covalentemente Con Genipina Hazem Nicola. Rev. Iberoam. Polim. 2008, 9, 326–330. [Google Scholar]
- Ahmed, E.M. Hydrogel: Preparation, Characterization, and Applications: A Review. J. Adv. Res. 2015, 6, 105–121. [Google Scholar] [CrossRef]
- Teixema, J. Small-Angle Scattering by Fractal Systems. J. Appl. Crystallogr. 1988, 21, 781–785. [Google Scholar] [CrossRef]
- Beaucage, G. Small-Angle Scattering. J. Appl. Cryst 1996, 28, 134–146. [Google Scholar] [CrossRef]
- Karchoubi, F.; Afshar Ghotli, R.; Pahlevani, H.; Baghban Salehi, M. New Insights into Nanocomposite Hydrogels; a Review on Recent Advances in Characteristics and Applications. Adv. Ind. Eng. Polym. Res. 2024, 7, 54–78. [Google Scholar] [CrossRef]
- Butler, M.F.; Clark, A.H.; Adams, S. Swelling and Mechanical Properties of Biopolymer Hydrogels Containing Chitosan and Bovine Serum Albumin. Biomacromolecules 2006, 7, 2961–2970. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zheng, Y.; Wang, W.; Wang, A. Preparation and Swelling Properties of Semi-IPN Hydrogels Based on Chitosan-g-Poly(Acrylic Acid) and Phosphorylated Polyvinyl Alcohol. J. Appl. Polym. Sci. 2009, 114, 643–652. [Google Scholar] [CrossRef]
- Chen, S.C.; Wu, Y.C.; Mi, F.L.; Lin, Y.H.; Yu, L.C.; Sung, H.W. A Novel PH-Sensitive Hydrogel Composed of N,O-Carboxymethyl Chitosan and Alginate Cross-Linked by Genipin for Protein Drug Delivery. J. Control. Release 2004, 96, 285–300. [Google Scholar] [CrossRef]
- Torabi, S.; Oleimani, S.; Mahravani, H.; Ebrahimi, M.M.; Shahsavandi, S. Mouse Fibroblast L929 Cell Line as a Useful Tool for Replication and Adaptation of Infectious Bursal Disease Virus. Arch. Razi Inst. 2023, 78, 863–871. [Google Scholar] [CrossRef] [PubMed]
- Del Prado-Audelo, M.L.; Magaña, J.J.; Mejía-Contreras, B.A.; Borbolla-Jiménez, F.V.; Giraldo-Gomez, D.M.; Piña-Barba, M.C.; Quintanar-Guerrero, D.; Leyva-Gómez, G. In Vitro Cell Uptake Evaluation of Curcumin-Loaded PCL/F68 Nanoparticles for Potential Application in Neuronal Diseases. J. Drug Deliv. Sci. Technol. 2019, 52, 905–914. [Google Scholar] [CrossRef]
- Di Santo, M.C.; Alaimo, A.; Domínguez Rubio, A.P.; De Matteo, R.; Pérez, O.E. Biocompatibility Analysis of High Molecular Weight Chitosan Obtained from Pleoticus Muelleri Shrimps. Evaluation in Prokaryotic and Eukaryotic Cells. Biochem. Biophys. Rep. 2020, 24, 100842. [Google Scholar] [CrossRef]
- Bhattamisra, S.K.; Shak, A.T.; Xi, L.W.; Safian, N.H.; Choudhury, H.; Lim, W.M.; Shahzad, N.; Alhakamy, N.A.; Anwer, M.K.; Radhakrishnan, A.K.; et al. Nose to Brain Delivery of Rotigotine Loaded Chitosan Nanoparticles in Human SH-SY5Y Neuroblastoma Cells and Animal Model of Parkinson’s Disease. Int. J. Pharm. 2020, 579, 119148. [Google Scholar] [CrossRef]
- Kanimozhi, K.; Khaleel Basha, S.; Sugantha Kumari, V.; Kaviyarasu, K.; Maaza, M. In Vitro Cytocompatibility of Chitosan/PVA/Methylcellulose—Nanocellulose Nanocomposites Scaffolds Using L929 Fibroblast Cells. Appl. Surf. Sci. 2018, 449, 574–583. [Google Scholar] [CrossRef]
- Foroozandeh, P.; Aziz, A.A. Insight into Cellular Uptake and Intracellular Trafficking of Nanoparticles. Nanoscale Res. Lett. 2018, 13, 339. [Google Scholar] [CrossRef] [PubMed]
- Budai, L.; Budai, M.; Fülöpné Pápay, Z.E.; Vilimi, Z.; Antal, I. Rheological Considerations of Pharmaceutical Formulations: Focus on Viscoelasticity. Gels 2023, 9, 469. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | pH (CS) | %CS | %GNP |
|---|---|---|---|
| S1 | 3.6 | 0.3 | 0.1 |
| S2 | 4.5 | 0.3 | 0.1 |
| S3 | 5.5 | 0.3 | 0.1 |
| Sample | Phase | (1/t) | R2 |
|---|---|---|---|
| S1 | Initial Nucleation | 1.01 × 10−6 ± 4.76 × 10−8 | 0.9784 |
| S2 | Initial Nucleation | 9.25 × 10−6 ± 3.45 × 10−7 | 0.9775 |
| S3 | Initial Nucleation | 2.65 × 10−5 ± 1.34 × 10−6 | 0.9682 |
| S1 | Exponential phase | 0.00025 ± 1.56 × 10−6 | 0.9784 |
| S2 | Exponential Phase | 0.0023 ± 9.96 × 10−5 | 0.9775 |
| S3 | Exponential Phase | 0.0066 ± 1.21 × 10−5 | 0.9682 |
| Sample | PDI | ζ-Potential (mV) | G′ (1 Hz) | μap |
|---|---|---|---|---|
| S1 | 0.229 ± 0.035 a,* | 31.64 ± 1.43 a | 4.09 a | 0.331 ± 0.045 a |
| S2 | 0.252 ± 0.024 a | 29.35 ± 1.17 a | 41.2 b | 0.0418 ± 0.0052 b |
| S3 | 0.274 ± 0.033 a | 20.42 ± 0.75 c | 18.0 c | 0.0336 ± 0.0336 c |
| Time (h) | Hydrated NG Mass (g) | ESR (%) |
|---|---|---|
| 0 | 0.010 ± 0.000 | 0 |
| 4 | 0.0167 ± 0.006 | 66.7 |
| 6 | 0.1567 ± 0.015 | 1466.7 |
| 24 | 0.3033 ± 0.010 | 2933.3 |
| 48 | 0.3100 ± 0.012 | 3000.0 |
| 72 | 0.3200 ± 0.011 | 3100.0 |
| 96 | 0.3200 ± 0.012 | 3100.0 |
| 120 | 0.3200 ± 0.012 | 3100.0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Glasman, J.D.; Alaimo, A.; Samaniego López, C.; Farías, M.E.; Currá, R.B.; Lamas, D.G.; Pérez, O.E. Perspectives on the pH-Influenced Design of Chitosan–Genipin Nanogels for Cell-Targeted Delivery. Pharmaceutics 2025, 17, 876. https://doi.org/10.3390/pharmaceutics17070876
Glasman JD, Alaimo A, Samaniego López C, Farías ME, Currá RB, Lamas DG, Pérez OE. Perspectives on the pH-Influenced Design of Chitosan–Genipin Nanogels for Cell-Targeted Delivery. Pharmaceutics. 2025; 17(7):876. https://doi.org/10.3390/pharmaceutics17070876
Chicago/Turabian StyleGlasman, Julieta D., Agustina Alaimo, Cecilia Samaniego López, María Edith Farías, Romina B. Currá, Diego G. Lamas, and Oscar E. Pérez. 2025. "Perspectives on the pH-Influenced Design of Chitosan–Genipin Nanogels for Cell-Targeted Delivery" Pharmaceutics 17, no. 7: 876. https://doi.org/10.3390/pharmaceutics17070876
APA StyleGlasman, J. D., Alaimo, A., Samaniego López, C., Farías, M. E., Currá, R. B., Lamas, D. G., & Pérez, O. E. (2025). Perspectives on the pH-Influenced Design of Chitosan–Genipin Nanogels for Cell-Targeted Delivery. Pharmaceutics, 17(7), 876. https://doi.org/10.3390/pharmaceutics17070876