To enhance the pharmacotechnical and pharmacological properties of hydroxybenzamide derivatives, these compounds were incorporated into β-cyclodextrin, a naturally occurring and cost-effective cyclodextrin, whose hydrophobic cavity is well-suited for the encapsulation of such molecules.
3.1. Molecular Modeling
The HOMO–LUMO gap, the energy difference between the frontier molecular orbitals, was employed for the evaluation of the stability of the compounds.
A previous study by our research group [
38] dealt with the investigation of the frontier molecular energies of the ester by using a different functional (BLYP, a local density approximation functional). For the present investigation, which involves the comparison among the three investigated compounds, a more accurate prediction of the molecular structures and properties is needed. Therefore, the more accurate hybrid functional B3LYP [
39] was chosen over the one previously employed.
The calculated results, together with the energies of the HOMO and LUMO orbitals, are given in
Table 1:
The results suggest a higher stability for both the ester and hydrazide, compared to the results calculated for hydrazone. For the latter compound, a decrease in the HOMO energy and an increase in the LUMO energy were obtained.
A graphic representation of the frontier molecular orbitals is depicted in
Figure 3:
The results depicted in
Figure 3 outline the similarities between the ester and hydrazide; for both compounds, the HOMO orbitals are located on the 2-bromo-phenyl moiety, while the LUMO orbitals are delocalized over the two aromatic cycles. Concerning the hydrazone, the main difference appears at the HOMO orbitals; they are present over the benzaldehyde moiety, not on the 2-bromo-phenyl residue. Also, the LUMO orbitals of the hydrazone are located on the two aromatic rings of the salicylanilide structure.
The results presented in
Table 2 outline the similarities of properties like ovality, molecular area and volume for the ester and the hydrazide. Regarding the calculated values of the polar surface area, larger values were obtained for hydrazide and hydrazone, compounds with free amine and hydroxyl groups. According to the results in
Table 3, all the investigated compounds are hydrophobic (a larger logP value being obtained for hydrazone, a compound with three phenyl rings).
The results of the molecular docking study are included in
Table 4:
The size of β-cyclodextrin cavity allows the partial inclusion of the
N-(2-bromo-phenyl)-2-hydroxy-benzamide derivatives (
Figure 4). As shown in
Table 4, the best binding affinities were obtained for hydrazone (−5.59 kcal/mol).
The interactions of the
N-(2-bromo-phenyl)-2-hydroxy-benzamide derivatives with β-cyclodextrin are shown in
Table 5.
Regarding the interaction between the
N-(2-bromo-phenyl)-2-hydroxy-benzamide derivatives and β-cyclodextrin (
Table 5), in all three cases, atoms in close contact were present, and only in case of hydrazone (HN), one hydrogen bond is formed. These results suggest the partial inclusion of the compounds within the β-CD cavity.
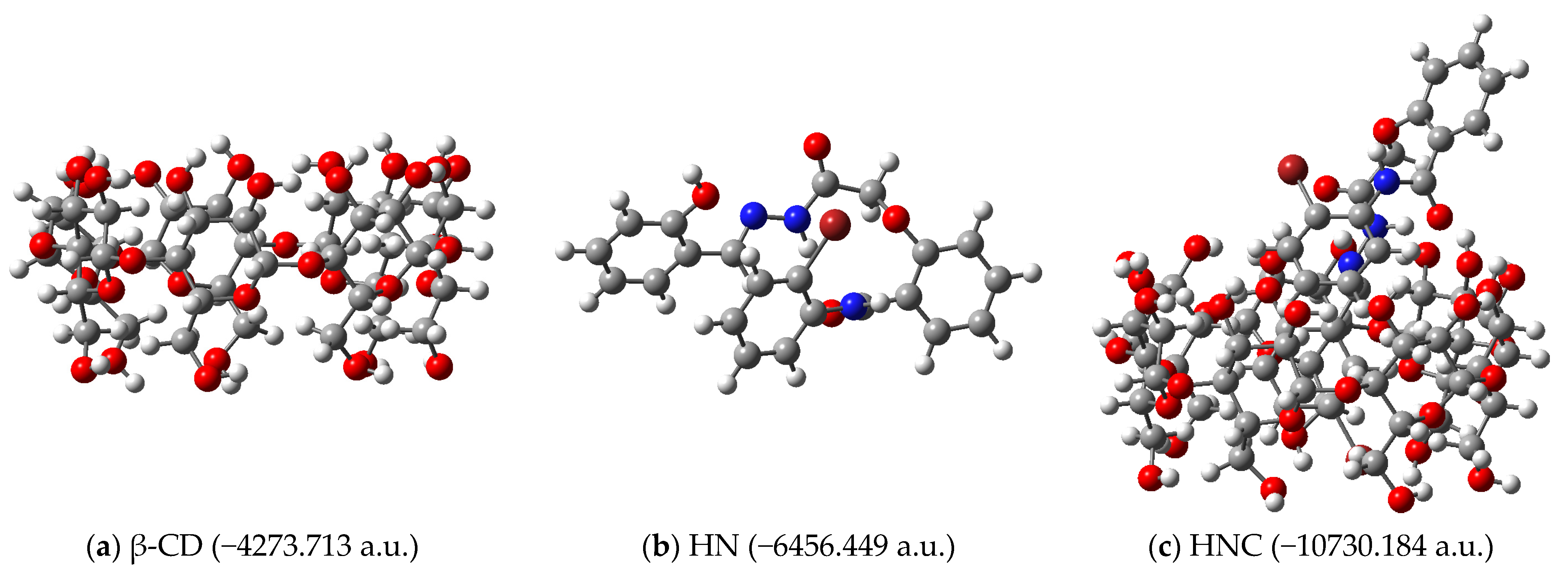
The complexation energies associated with obtaining the inclusion complexes depicted in
Figure 4 were calculated. The structures of the receptors, ligands and complexes (together with their calculated energies) are depicted in
Figure 5 for ethyl ester EE as ligand,
Figure 6 for hydrazide HD as ligand and
Figure 7 for hydrazine HN as ligand, respectively.
The results presented in
Table 6 suggest the endothermic process for the obtaining of the inclusion complexes of ethyl ester EE and hydrazide HD, and an exothermic one for the hydrazine HN complex with β-cyclodextrin.
Molecular docking computations also suggested that hydrazine HN, characterized by the largest area, volume and polarizability within the series, has the highest binding affinity towards β-cyclodextrin.
3.2. Spectral and Thermal Characterization
In order to compare the results obtained for the inclusion complexes, the physical mixtures containing the two components of the complex were also prepared and analyzed. Characterization measurements were performed using the following modern analytical methods: X-ray diffraction, SEM, FTIR, 1H-NMR, TGA and DSC.
The following abbreviations were used to express the obtained results: EE—ethyl ester; CD—β-cyclodextrin; EEPM—physical mixture of CD and EE; EEC—inclusion complex of CD and EE; HD—hydrazide; HDPM—physical mixture of CD and HD; HDC—inclusion complex of CD and HD; HN—hydrazone; HNPM—physical mixture of CD and HN; HNC—inclusion complex of CD and HN.
Figure 8 presents the X-ray diffraction spectra of the three analyzed sets corresponding to the ethyl ester, hydrazide and hydrazone series, along with the β-cyclodextrin, their physical mixtures and inclusion complexes.
The XRD patterns of the physical mixture slightly differ from the pure mathematical sum of its components. This could be due to the microstructural changes, partial amorphization and weak interactions during mixing, even if a true inclusion complex has not yet formed [
40,
41,
42].
The analysis of the compounds by X-ray diffraction indicates, in the case of the binary compounds, the decrease in the crystallinity degree in comparison with the guest substances (EE, HD, HN) due to the amorphization phenomenon, and also changes in the position and intensity of the diffraction peaks. These alterations prove the formation of new solid states and demonstrate the presence of interactions between the hydroxybenzamide compounds and the cyclodextrin.
In order to evaluate the surface morphology and the dimension of the crystals, the SEM images of the
N-(2-bromo-phenyl)-2-hydroxy-benzamide derivatives, β-cyclodextrin, their physical mixtures and inclusion complexes were recorded (
Figure 9).
Regarding the physical mixtures (
Figure 9c), there are no major differences between these and the cyclodextrin sample (
Figure 9b), only in the case of the HNPM, hydrazone crystals can be better distinguished. However, the crystals are more agglomerated in the PMs compared to the inclusion complexes. For the inclusion complexes (
Figure 9d), amorphous aggregates with asymmetrical shapes were observed, with a certain uniformity of the obtained precipitated particles. The dimensions of the particles are in the range of hundreds of nanometers, which is better observed at higher magnification.
FTIR is an important analysis to demonstrate the existence of host–guest interaction in the molecule of inclusion complexes. FTIR spectra of the guest, β-cyclodextrin and their mixtures are presented in
Figure 10.
Some characteristics of the IR spectra of the inclusion complexes (EEC, HDC, HNC), also observed by other researchers and important to prove the complex’s formation [
43,
44,
45], are the similarity between spectra of the complexes and the CD spectra, and also the narrowing of the absorption band (3400.36 cm
−1) corresponding to the hydroxyl groups of pure CD in the spectra of complexes. The IR spectra of the physical mixtures are a combination of the spectra of the neat compounds, where the characteristic bands of the guest and the cyclodextrin stand out.
Table 7 and
Table 8 show minor differences between the spectra of the inclusion complexes and the spectra of the components (guests and host). Even small, the changes in the characteristic vibrations of the guests and host after complexation may suggest the existence of interactions between the components of the complexes and the partial entrapment of the guest within the hydrophobic cavity of β-cyclodextrin. However, the IR shifts must be correlated with other data sets, such as
1H-NMR, as the other technique provides additional evidence to support the significance of those changes.
The observed changes in the IR spectra of the complexes consist in minor shifts in certain bands frequency to higher or lower values, and also the modification of certain bands intensity. For example, in the case of the ethyl ester and hydrazide, the frequency corresponding to N–H stretching vibrations (νNH) of secondary amide, slightly shifts to a higher value in the complexes (
Table 7), which can be related to the location of the amide group inside the cyclodextrin cavity (
Figure 4b). In addition, for hydrazone, the intensity of the carbonyl group (νC=O ester) increases in the complex compared with the physical mixture, probably due to the hydrogen bond [
46] formed with the hydroxyl group of the cyclodextrin (
Figure 10c), which is in agreement with the molecular modeling (
Figure 4c). The ether linkage (COC aromatic), present in all three guests, seems to be more affected by complexation phenomena in case of the ethyl ester, being in accordance with the molecular docking data, showing the presence of ethyl ester molecule inside the CD cavity, only the bromophenyl part of the molecule remaining outside (
Figure 4a).
The entrapment of the guests within the hydrophobic cavity of β-cyclodextrin is also suggested by the slight changes in the C–H stretching vibrations (ν[CH
2]) of β-cyclodextrin, which are more evident in the hydrazone inclusion complex (
Table 8). The insertion of the benzene core inside the cyclodextrin, consecutively with the increase in electron cloud density, results in the changes in frequency [
47]. On the other hand, the alteration of the microclimate with the development of hydrogen bonds and van der Waals interactions lead to the decrease in some band frequency [
48]. The absence of extra bands in the inclusion complexes’ spectra prove that the interactions between the complexes’ components are only of a physical nature.
1H-NMR, along with FTIR, represents a significant investigation to prove the formation of inclusion complexes.
1H-NMR data for the [2-(2-bromophenylcarbamoyl)phenoxy]acetic acid ethyl ester, β-cyclodextrin and their inclusion complex were presented in a previous paper [
38].
1H-NMR spectra of hydrazide, hydrazone, β-cyclodextrin and their complexes are presented in
Figures S1–S5 (Supplementary File).
Figure 11 shows the number of the host and guests protons used for the interpretation of NMR spectra.
The chemical shift changes (Δδ) between the free and complexed state of CD are presented in
Table 9. Changes in the chemical shifts in the host and guest molecules, visible in the NMR spectra, are due to the insertion of the more hydrophobic part of the guest molecule in the hydrophobic cavity of the cyclodextrin. Thus, the most affected cyclodextrin protons should be the ones positioned inside the cavity of the cyclodextrin (H-3 and H-5) [
49].
Table 9 reveals that the chemical shift changes corresponding to the protons located inside the β-cyclodextrin cavity are more evident than those manifested by the protons located outside the cavity, especially for the inclusion complex of hydrazone, demonstrating the formation of the inclusion complexes [
49]. These findings are also supported by IR results, where the inclusion of the guests into the cavity of β-cyclodextrin was shown by the changes in the C-H stretching vibrations of β-cyclodextrin, and also more evident in the hydrazone inclusion complex (
Table 8). Moreover, all computed supramolecular architectures of the studied complexes illustrate the partial inclusion of the guests into the β-cyclodextrin cavity and the presence of hydrophobic interactions inside the cyclodextrin cavity.
Table 10 and
Table 11 present the chemical shift changes between the free and complexed hydrazide, for aromatic and non-aromatic protons, respectively.
The
1H-NMR results shown in
Table 10 indicate slight modification of the chemical shifts corresponding to the aliphatic part of the hydrazide (∆δ
H ≤ ±0.005), the most affected by complexation seems to be the protons of OC
H2CO group. Encapsulation at this level was also suggested by modification of the ether linkage frequency in the IR spectra of the hydrazide (
Table 7) and supported by the molecular docking (
Figure 4b). Regarding the chemical shift changes corresponding to non-aromatic protons, both the salicylic and benzanilide part of the hydrazide molecule seems to be influenced by complexation (∆δ
H-3 = −0.004, ∆δ
H-6 = −0.010, ∆δ
H-10 = −0.007, ∆δ
H-12 = −0.008), as shown in
Table 11. The frequency shift in the N–H stretching vibrations observed in the IR spectra of the hydrazide complex (
Table 7), corroborated NMR results and molecular modeling data (
Figure 4b), attesting to the inclusion of salicylic nucleus inside the cyclodextrin cavity.
Table 12 and
Table 13 present the chemical shift changes between the free and complexed hydrazone, for aromatic and non-aromatic protons, respectively.
Thus, regarding the hydrazone, the hydroxyl group (∆δ
OH = +0.009) attached on the benzaldehyde moiety and the -NH- group (∆δ
CONHN = +0.006) bonded to imine group seem to be affected by the inclusion into the cyclodextrin cavity (
Table 12). These findings support the results obtained from IR spectra, where also slight modification of the N–H frequency was observed (
Table 7).
Among the aromatic protons (
Table 13), the most affected by complexation appear to be the H-10 (∆δ
H-10 = −0.004), which is enclosed by the cyclodextrin OH groups, and H-17 (∆δ
H-17 = −0.004), contained in the benzaldehyde moiety, that is included in the hydrophobic environment of the cyclodextrin cavity, based on the computational data regarding the ligand–receptor interactions (
Figure 4c).
The NMR data for free and complexed hydrazide and hydrazone prove the hypothesis resulted from molecular modeling, namely, the partial encapsulation of the guest within the cyclodextrin cavity. This conclusion was also drawn in the case of ethyl ester [
38].
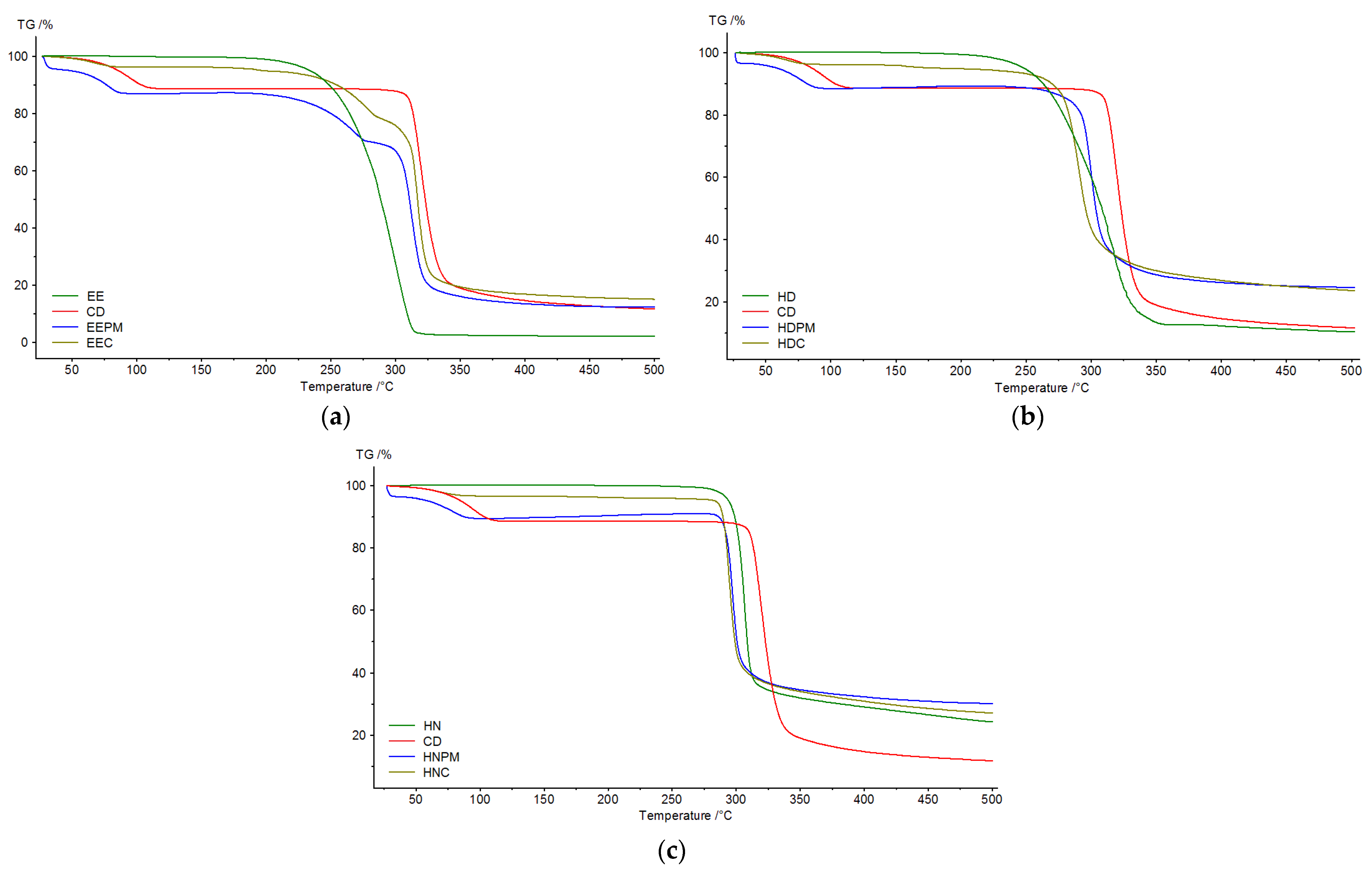
In
Figure 12, the TGA curves of the guest, β-cyclodextrin, physical mixture and inclusion complex for the three series, EE, HD and HN, are plotted.
Thermogravimetric analysis was accomplished to detect the mass loss in relation to the temperature change. Thermograms were recorded for pure guests (EE, HD, HN), β-cyclodextrin, physical mixtures and inclusion complexes. The TG data, in the temperature range 25–500 °C, are depicted in
Figure 12.
β-cyclodextrin displays two mass losses, one corresponds to the loss of water placed in the CD cavity (inflection point—95.7 °C) [
50,
51], and the other to the breakdown of the macrocycle, at 320.2 °C. The guests show a single mass loss in the temperature range 180–340 °C (EE), 200–360 °C (HD) and 260–500 °C (HN), explained by the degradation of benzene moiety from their structures.
The EE inclusion complex undergoes mass losses in three steps, the first step corresponds to the dehydration of the molecules, the second step to the decomposition of β-cyclodextrin and the last step can be associated with the decomposition of the ester. Regarding the behavior of the HD and HN complexes, the mass loss occurs in two stages. The first stage can also be related to the dehydration of the molecules, as for the second stage, this can be attributed to both, the decomposition of β-cyclodextrin, and the decomposition of the guests. In order to prove the formation of the inclusion complex, the physical mixtures of β-cyclodextrin and guests (EE, HD, HN) were subjected to thermal analysis and the result were compared with those obtained for inclusion complexes. Thus, the first mass loss in the case of the inclusion complexes occurs at 68.5 °C (EEC), 66.7 °C (HDC), and 68.4 °C (HNC), while in the case of the physical mixtures, it occurs at 78.8 °C (EEPM), 78.6 °C (HDPM), and 79.5 °C (HNPM). Moreover, the mass loss corresponding to the degradation of the CD macrocycle occurs at 320.2 °C, and that of the guest’s degradation at 294.5 (EE), 312.8 (HD), 306.7 (HN), as for the complexes, the values were lower (279.1 °C—EEC; 290.2 °C—HDC; 294.9 °C—HNC). These changes in the thermal stability of the host and guest suggest the formation of the inclusion complexes.
In addition, the results of the DSC analysis (
Figure 13) show changes in the enthalpy values corresponding to the thermal phenomena of the two components of the inclusion complexes, proved by the reduction in their peak area in the binary system, in comparison with the physical mixtures. Also, the peaks corresponding to the dehydration and melting/decomposition were shifted to different temperature values in cases of the complexes, proving that the formation of the inclusion complexes altered the thermal degradation property of the two components.
Thus, the ethyl ester (EE) showed a sharp endothermic peak at 92.7 °C, assignable to its melting point [
15] and one broad endothermic peak around 322.0 °C, assignable to its decomposition. β-CD has a broad endothermic peak around 143.7 °C, which corresponds to the loss of water molecules from its cavity, and two other endothermic peaks at 315.0 °C and 323.2 °C which can be assigned to the melting and decomposition of the cyclodextrin [
52]. The physical mixture (EEPM) and the inclusion complex (EEC) showed the peak of the ethyl ester (90.7 °C and 93.5 °C, respectively) and the endothermic water peak of cyclodextrin (120.4 °C and 105.8 °C, respectively), as well as the peaks around melting point (314.5 °C and 316.4 °C, respectively), and degradation (321.1 °C and 322.6 °C, respectively) of β-CD. Moreover, one broad endothermic peak appears in DSC of both the physical mixture and the complex (231.8 °C and 226.3 °C, respectively), without correspondence in the guest or host, which can likely represent the breaking of non-covalent interactions (hydrophobic interactions, hydrogen bonds) between the guest and β-cyclodextrin, and can be interpreted as a pre-decomposition of the binary compounds [
53]. However, when comparing the fusion enthalpy values (ΔH) of the EE melting process, 13.4 J/g (physical mixture) vs. 10.85 J/g (complex), it seems that the guest’s crystalline structure is disrupted more in the inclusion complex than in the physical mixture, suggesting molecular encapsulation or stronger host–guest interactions. An even bigger difference was observed for dehydration enthalpy values (ΔH) of CD, from 118.4 J/g (physical mixture) to 24.0 J/g (complex). Complex formation is driven by the release of high-enthalpy water molecules from the cyclodextrin cavity, which are poorly accommodated and energetically replaced by less polar drug molecules, leading to a more stable host–guest system [
54,
55]. Smaller changes were observed in case of the hydrazide inclusion complex (HDC), especially on the peak corresponding to the dehydration of the CD, and minor for the hydrazone complex (HNC), suggesting that the inclusion complexes are primarily stabilized by hydrophobic interactions. Also, it is worth noting that the endothermic decomposition of pure components changed to exothermic in the physical mixtures and complexes of EE and HD, maybe due to new molecular interactions that introduce more energetically favorable and heat-releasing degradation pathways, especially in the confined environment of an inclusion complex or through solid-state interactions in physical mixtures [
56].
Based on the differences in the X-ray spectra, SEM images, IR spectra, TG and DSC curves between the inclusion complexes and the pure cyclodextrin and guests, the formation of the inclusion complexes was proved.
3.3. Antimicrobial Effect
The antibacterial effect of the
N-(2-bromo-phenyl)-2-hydroxy-benzamide derivatives and their complexes is shown in
Table 14. As can be seen, only Gram-positive bacteria were inhibited by the tested compounds, and no inhibition on Gram-negative bacteria was observed at the tested concentrations. Such findings were also remarked in other studies, regarding the evaluation of the antimicrobial activity of some halogenated salicylanilide derivatives [
23,
57,
58,
59].
Esters of halogenated salicylanilide with amino acids were evaluated against
Staphylococcus aureus, methicillin-resistant
Staphylococcus aureus,
Staphylococcus epidermidis,
Enterococcus spp.,
Escherichia coli, and
Klebsiella pneumoniae. (
S)-2-(4-Bromophenylcarbamoyl)-5-chlorophenyl 2-acetamido-3-phenylpropanoate and (
S)-4-chloro-2-(4-(trifluoromethyl)phenyl carbamoyl)phenyl 2-acetamido-3-phenyl propanoate exhibited good antibacterial effect (MICs = 0.98–31.25 μmol/L) against Gram-positive bacteria. Gram-negative bacteria were less susceptible to the action of tested compounds (MICs = 15.62–500 μmol/L) [
59].
The antimicrobial activity of some salicylanilide 4-(trifluoromethyl) benzoates was evaluated. Gram-positive bacteria, including MRSA, were inhibited with MICs ≥ 0.49 μmol/L, while among Gram-negative strains, only
E. coli had a partial susceptibility (MICs ≥ 31.25 μmol/L) [
58].
Higher resistance up to 500 μmol/L of Gram-negative species was also observed for salicylanilide diethyl phosphates, meanwhile good activity against Gram-positive bacteria (MICs ≥ 1.95 µmol/L) was obtained [
57].
In a previous study conducted in our laboratory,
N-(2-chlorophenyl)-2-hydroxybenzamide and
N-(4-chlorophenyl)-2-hydroxybenzamide derivatives were tested against Gram-positive and Gram-negative bacteria. Only the 2-Cl-substituted derivatives showed good activity against Gram-positive bacteria strains (MICs = 0.125–0.5 mg/mL), with no inhibition on the Gram-negative ones. The ethyl/methyl esters were twice as active as their corresponding hydrazides. The activity of the inclusion complex of ethyl 2-(2-((2-chlorophenyl)carbamoyl)phenoxy)acetate in β-CD against the tested bacteria was similar with the activity of the neat ester, even if the amount of the ester in the complex was approximately 4 times smaller [
23].
The promising activity of salicylanilide derivatives was proven also by Pauk et al. (2013) [
13]. Thus, a series of salicylanilides and derivatives, namely esters of
N-phenylsalicylamides and 2-hydroxy-
N-[1-(2-hydroxyphenylamino)-1-oxoalkan-2-yl] benzamides, were synthesized, characterized and evaluated against bacterial and mycobacterial strains. Compared with ampicillin, ciprofloxacin or isoniazid standards, some compounds proved better or similar activity. For example,
N-(4-bromo-phenyl)-5-chloro-2-hydroxy-benzamide exhibited a 0.76 µmol/L CMI, and its ester, 5-chloro-2-[(4-bromophenyl)carbamoyl]phenyl(2S)-2{[(benzyloxy)carbonyl]amino}-3-methylbutanoate, showed 3.75 µmol/L CMI against
Staphylococcus aureus. The structure–activity relationships regarding antibacterial activity, proved the importance of R
1 substitution in the para-position to the carboxamide moiety, together with a lipophilic and electron-withdrawing R
2 and a bulky R
3 substituent [
13].
In the present research, the minimum inhibitory concentration values of the
N-(2-bromo-phenyl)-2-hydroxy-benzamide derivatives ranged between 2.5 and 5.0 mg/mL, being 10 times higher when compared with the MICs of the
N-(2-chlorophenyl)-2-hydroxybenzamide derivatives [
23]. It can be concluded that the presence of the bromine instead of chlorine on the benzanilide ring decreased the antimicrobial effect.
This time though, the activity of the ethyl ester (EE) and hydrazide (HD) were equivalent, and the inclusion complexes were similar or twice as active as their corresponding neat derivatives. It should be mentioned that salicylanilide derivatives are found in complex solutions in an amount 3–4 times smaller than in solutions of uncomplexed compounds, because the complexes were obtained using a 1:1 molar ratio.
Thus, the most active compounds were the complexes of the ethyl ester (EEC) and hydrazide (HDC), both of them exhibited an activity of MIC = 2.5 mg/mL against
Streptococcus pneumoniae. The advantage of complexation was also highlighted in the study of Inoue et al. (2020) where the CD complexes of hinokitiol showed identical or higher activity when compared with neat hinokitiol [
60]. The lack of activity for the hydrazone compound (HN) can be explained based on the low solubility of the compound in the culture medium due to its high lipophilicity, which can act as a barrier against the diffusion in the substrate.
3.4. In Vitro Anti-Inflammatory Activity
Due to the animal risk management, and ethical concerns, in vivo studies can be replaced, if possible, at least in the case of early studies, by the in vitro evaluation of the biological activity [
61].
In the present study, in vitro anti-inflammatory activity was evaluated using the protease inhibition assay, as protease enzyme activity is closely correlated with inflammatory processes. Trypsin is a serine protease enzyme secreted by the pancreas that specifically cleaves peptide bonds at the carboxyl side of lysine and arginine residues. Its activity has been associated with inflammatory responses that can result in tissue damage. As a proteolytic enzyme, trypsin activates immune cells such as eosinophils, which play a key role in the body’s first line of defense and immune response. However, excessive production and release of trypsin can trigger an uncontrolled cascade of events, ultimately contributing to the onset of various diseases [
62]. Thus, the evaluation of antiproteolytic effect of compounds represents a good way to predict their anti-inflammatory activity.
When using protease inhibitors, the inhibition of the enzyme activity can be explained based on the formation of an enzyme inhibitor complex, where van der Waals’ and hydrogen bonds [
63] or even covalent bonds [
18] are formed.
The antiproteolytic assay was performed using selected concentrations of hydroxybenzamide derivatives. Dimethyl sulfoxide (DMSO) was employed as the solvent for compound dissolution, while phosphate-buffered saline (PBS) was used for subsequent dilutions to minimize potential DMSO-induced protein denaturation. At low concentrations, DMSO allows proteins to remain preferentially hydrated in aqueous solutions, thereby preserving their native conformation. Protein denaturation typically occurs only at significantly higher DMSO concentrations [
64].
Salicylic acid, sodium diclofenac, acetylsalicylic acid are used as positive controls in different assays to compare the anti-inflammatory efficacy of tested compounds. For example, for trypsin inhibition assay, IC
50 value of salicylic acid (16.6 ± 0.41 µg/mL) was used to compare the anti-inflammatory potential of aconitine produced by endophytic fungus
Acremonium alternatum [
65], IC
50 of diclofenac sodium (2.61 mg/mL) was used to compare the anti-inflammatory activity of the extracts from
Gomphrena globosa L. [
66] and for the anti-inflammatory activity of the
Enteromorpha compressa extracts the reference compound, acetylsalicylic acid (IC
50 = 4.66 µg/mL) was used [
67].
In the present study, the antiproteinase action expressed as the percentage of trypsin inhibition, of both, hydroxybenzamide derivatives and acetylsalicylic acid, used as positive controls, was concentration-dependent.
The IC
50 value of the acetylsalicylic acid was established to be 0.4051 ± 0.0026 mg/mL and was further compared to the ICs
50 obtained for hydroxybenzamide derivatives (
Table 15).
The IC50 values of the title compounds were more than sixfold lower than that of the positive control, indicating their superior efficacy in inhibiting trypsin activity. For uncomplexed compounds, the best inhibition was obtained in the case of ethyl ester (IC50 = 0.0436 ± 0.0047 mg/mL), followed by the hydrazone (IC50 = 0.0560 ± 0.0109 mg/mL) and hydrazide (IC50 = 0.0622 ± 0.0070 mg/mL). The results proved the beneficial role of complexation on the efficiency of the title compounds. The most prominent example in this context is the hydrazide derivative, which demonstrated a fourfold enhancement in biological activity upon complexation (IC50 = 0.0592 ± 0.0057 mg/mL), taking into account the 1:1 molar ratio employed during complex synthesis.
Similar results were found for the ICs of fenofibric acid and cyclodextrins, which demonstrate superior anti-inflammatory activity, compared to fenofibric acid alone, and recommend the use of such drug release systems for enhanced pharmacological profile of the active substance [
25].


 ,
, 














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}