1. Introduction
Localized prostate cancer can be treated effectively with surgery, radiotherapy, or focal therapy [
1]. However, more than 10% of men diagnosed with PrC will develop metastatic disease. The combination of androgen deprivation therapy (ADT), androgen receptor pathway inhibitors, and chemotherapy, while creating a state of medical castration, has improved the overall survival of patients with metastatic hormone-sensitive prostate cancer (mHSPrC). Despite this improvement, the median survival is under 2 years [
2,
3,
4], and the 5-year survival rate is only 30% [
3,
5]. Therefore, mPrC still represents an important unmet medical need.
Recent advances in the treatment of mPrC include targeted radioligand therapy, particularly directed toward the prostate-specific membrane antigen (PSMA), whose level of expression reflects disease progression and recurrence [
6]. The FDA approved PSMA-labeled lutetium (
177Lu) (Vipivotide tetraxetan) for this purpose in 2022. Nonetheless, this therapy can lead to significant side effects [
7,
8], possibly because PSMA is expressed at low-to-moderate levels in several normal tissues [
9,
10]. In addition, 5–10% of PrC cells do not express surface PSMA, and long-term ADT can lead to a significant reduction in PSMA expression [
11]. PSMA, as well as other targeting ligand candidates (reviewed in Hu et al., 2024) [
12], were selected because of their overabundance of PrC cells rather than for their ability to initiate effective carrier-drug uptake and intracellular drug delivery, factors that ultimately limit the efficacy of targeted drug therapy for cancer. Another factor limiting therapies targeting only one cell surface component is that PrC cells (like many other cancers) are genetically and phenotypically heterogeneous, leading to the modulation of receptor expression. Prostate cancers are often multifocal, having topographically and morphologically distinct tumor foci [
13,
14]. Indeed, studies have shown marked heterogeneity in the PSMA labeling of multifocal disease [
15].
Several different technologies have been used to isolate cell-targeting peptides, although arguably, the most efficient of these is based on phage peptide display techniques [
16]. Regarding prostate cancer, various approaches have been used to derive a pool of peptide candidates for the detection of and targeted drug delivery (TDD) to mPrC cells, but these have yet to progress to clinical evaluation [
17].
Here, we describe a reverse strategy to overcome these limitations, which is based on the action of receptors with appropriate characteristics for TDD, but without a priori knowledge of their identity or the receptor’s natural ligand (unbiased). A phage display library devoid of normal tissue-binding clones was processed through a unique, biopanning protocol, which included wild-type and drug-resistant mPrC and normal prostate cells. This led to the discovery of a pool of mPrC-specific 7-mer cyclic peptides that were subsequently subjected to functional tests, including cell uptake kinetics, internalization pathways, selectivity for patient PrC cells, chemo- and biostability, efficient conjugation to a cytotoxin, and the ability of the resultant peptide–drug conjugates (PDCs) to kill target cells in vitro and in vivo selectively. The peptides were shown not to bind to PSMA. Detailed studies on one of the peptides (P10) demonstrated that it is endocytosed by mPrC cells via a clathrin and dynamin-independent, ARF6-dependent pathway and specifically targets PrC tumors in mice. In vitro, PDCs incorporating P10 selectively killed wild-type and drug-resistant PrC cell lines as well as patient PrC cells but not normal prostate tissue cells, while in vivo, the PDCs halted the growth of PrC tumors in a xenograft model. This unique pool of peptides fulfills the functional characteristics required for effective detection and TDD inside mPrC cells. The protocol described can be easily adapted to isolate peptides for targeting other types of cancer cells.
2. Materials and Methods
2.1. Cells, Kits, and Reagents
Cell lines were obtained from the ATCC (Biological Industries, Beit Haemek, Israel). The mPrC cell lines PC-3 (RRID:CVCL_0035), LNCaP (RRID:CVCL_0395), DU-145, (RRID:CVCL_0105), and 22RV1 (RRID:CVCL_1045), and human epithelial HEK 293T/17 cells (RRID:CVCL_1926), were grown in RPMI 1640 medium (Satorius, Beit Haemek, Israel) supplemented with 10% fetal bovine serum (FBS) (Thermo Fisher, Rhenium, Modiin, Israel), 3 mM l-glutamine, and penicillin–streptomycin solution (Satorius). The medium for LNCaP cells also contained 1 µM of insulin and 1 nM of testosterone (Sigma-Aldrich, Rehovot, Israel). RPWE-1 cells derived from normal human prostate epithelium were grown in KSFM supplemented with 0.05 mg/mL of bovine pituitary extract (BPE), 5 ng/mL of EGF, and penicillin–streptomycin solution. Peripheral blood mononuclear cells (PBMCs) were obtained with consent from a healthy donor and used immediately. Normal prostate and prostate cancer biopsy tissues were obtained from the Rabin Medical Center (Beilinson campus) after approval of the study by the Rabin Medical Center’s institutional review board (0176-20-RMC). Normal tissue was obtained from consenting donors undergoing transurethral resection of the prostate (TURP) for benign prostatic obstruction, while prostate tumor biopsies were obtained from tissue resections from patients undergoing radical prostatectomy. On resection, the tissues were immediately placed in KSFM supplemented with 2.5 µg/mL of EGF, 25 µg/mL of BPE, 5% FBS, 1% penicillin-streptomycin solution, 1% amphotericin B solution, 250 mg/mL, and 10 mM HEPES, transported to the laboratory where they were promptly centrifuged, resuspended in medium without FBS and cultured on collagen type I-coated 6-well plates (Greiner, Danyel Biotech, Rehovot, Israel). All cells were maintained at 37 °C in air containing 5% CO2. A sample of normal human blood plasma was obtained from the Assuta Medical Center, Ramat Hachayal, Tel Aviv, Israel, as a part of an experiment unrelated to the current study.
Mouse liver was obtained from a healthy male mouse sacrificed as part of an unrelated study approved by the Ariel University Ethics Committee. The organ was washed in cold 0.01 M PBS, pH 7.4, transferred to a test tube containing 10 mL of Tris-HCl solution, pH 7.4, and homogenized for 4 min on ice using a Potter–Elvehjem glass tissue grinder. The mixture was centrifuged at 28,500× g and 4 °C for 20 min, after which the supernatant was collected. The homogenate was used immediately for stability testing or aliquoted and stored at −80 °C until use.
The phage display peptide library (Ph.D.-C7C, New England Biolabs, Ipswich, MA, USA) contained approximately 50 copies each of 10
8 pfu/μL. Each phage clone displayed a unique 7-mer cyclic peptide. The kit was used according to the manufacturer’s instructions. The 7-mer cyclic library was chosen for several reasons. (a) Structural stability: Cyclic configuration improves resistance to enzymatic degradation compared to linear peptides [
18,
19]. (b) Selectivity: Short cyclic peptides exhibit enhanced selectivity as ligands due to constrained conformation reducing structural flexibility, promoting precise target binding [
20]. (c) Higher binding affinity/selectivity via optimized orientation of functional groups and increased interaction surface area [
21].
Chemicals, drugs, and reagents for peptide conjugation and other procedures were purchased from Sigma-Aldrich (Rehovot, Israel), Holland Moran (Yehud, Israel), S.L. Moran (Jerusalem, Israel), Hylabs (Rehovot, Israel) or Biolab (Jerusalem, Israel).
2.2. Development of Drug-Resistant Cell Lines
PrC cell lines resistant to Estramustine (ESM) were developed as the clinical use of this drug induces significant side effects and drug resistance [
13,
14]. The procedure of Mohr was used [
15] as described in the
Supplementary Information.
2.3. Isolation and Validation of mPrC Cell Internalizing Phage
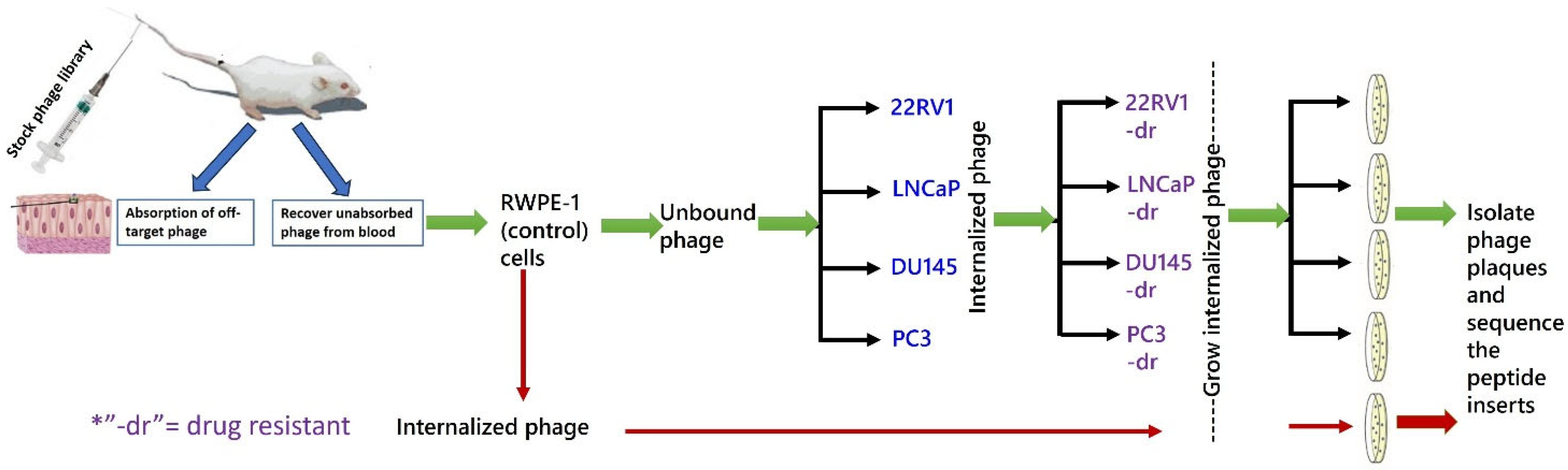
The protocol for phage selection is shown in
Figure 1. Briefly, the phage library was injected into a healthy male BALB/c mouse to remove phage clones displaying peptides with affinity for normal tissue or blood cells or soluble blood components [
16]. After 24 h, peripheral blood was collected, and the phages were recovered. This “absorbed” phage library was exposed to control RWPE-1 cells. Unbound and internalized phages were recovered separately, and the process was repeated. The non-binding phages were then exposed for 1 h to microplate wells seeded overnight with either PC-3, DU-145, 22Rv1, or LNCaP cells. The cells were washed in PBS, and membrane-bound phages were eluted by the addition of 400 μL of 0.2 M glycine-HCl, pH 2.2/1 mg/mL of BSA for 10 min. The solution was transferred to a sterile tube and neutralized by the addition of 65 μL 1 M Tris-HCl, pH 9.1. Four hundred microliters (400 µi) of PBS were added to the cells, which were then ruptured by freezing (−20 °C) and thawing (37 °C) (three cycles). The mixture was centrifuged at 3400×
g for 2 min, and the supernatants containing the internalized phage were exposed to their corresponding drug-resistant cell lines. Phage internalized by these cells were recovered and reprocessed through their respective drug-resistant cell lines two more times. Phages that were bound to or internalized by RWPE-1 cells were also collected and served as phage pool internalized by non-cancerous prostate cells. All recovered phages were titrated according to the library manufacturer’s protocol. From the bacterial lawn containing a phage dilution that produced approximately 200 blue (library phage) plaques, 15–20 plaques were individually collected into separate 15 mL tubes, amplified, recovered by PEG precipitation, and their DNA was isolated. Using kit primers, the region containing the peptide insert was Sanger-sequenced, and the peptide amino acid sequence was deduced.
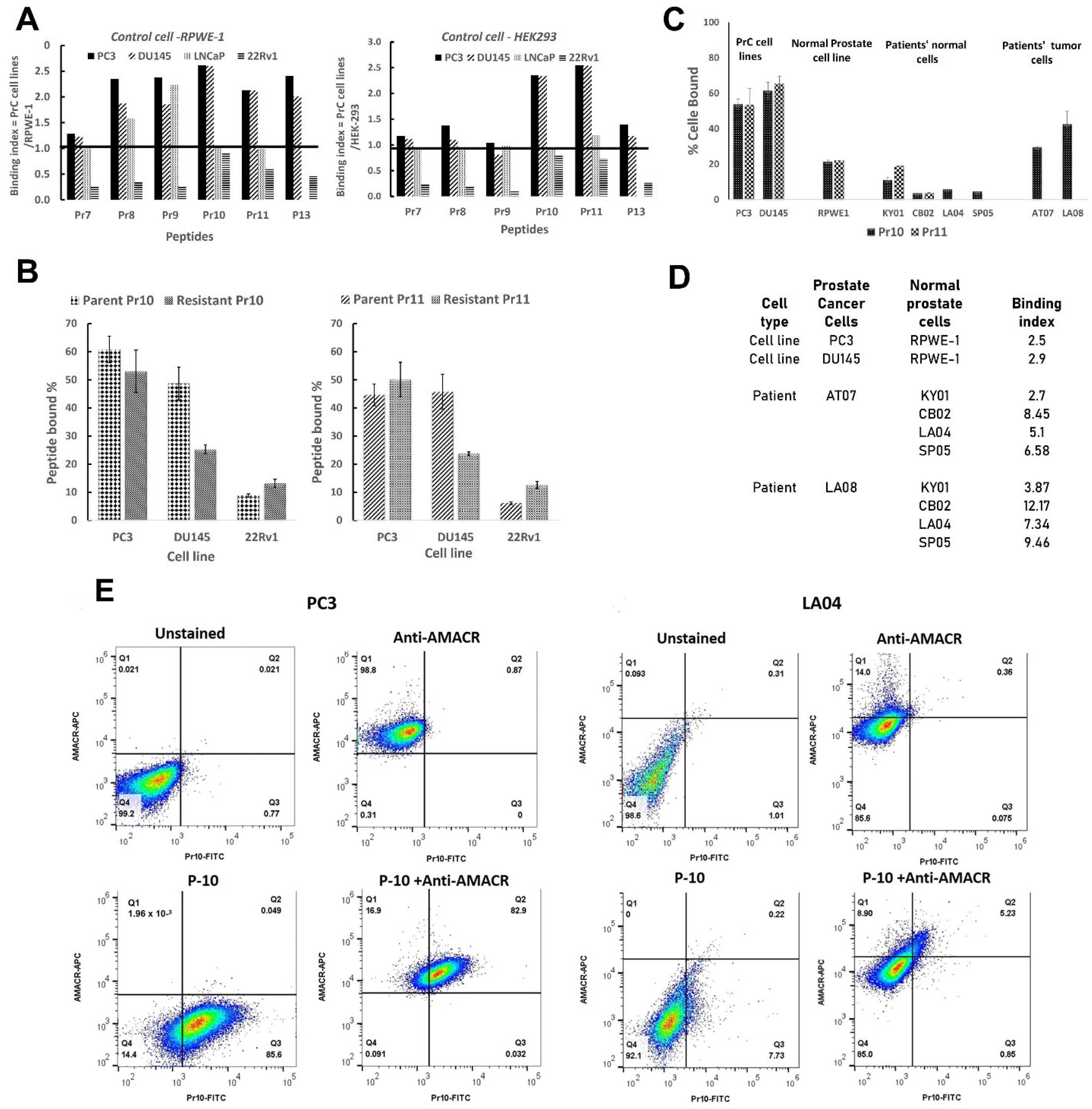
From the total list of phage-displayed peptide sequences, those also displayed by the RWPE-1-internalized phage were removed. The remaining mPrC clones were validated for PrC specificity by re-exposing them to RWPE-1, PC-3, DU-145, 22Rv1, or LNCaP cells for 1 h for 24 h. The internalized phages were recovered, and their titers were determined. The peptides displayed by the phage clones with the highest mPrC cell-to-RPWE-1 internalization titer ratio were synthesized and coupled to fluorescein isothiocyanate (FITC) (Pepmic Ltd., Suzhou, China) for flow cytometry analysis.
To validate the specificity of peptide binding, cells were seeded overnight, washed with PBS, and incubated for 15–60 min at 37 °C or 4 °C with 0–50 µM of peptide–FITC conjugates dissolved in RPMI/2% FBS. The cells were washed twice with PBS, trypsinized, and centrifuged at 340× g for 5 min. The pellet was resuspended in PBS and analyzed for FITC staining by flow cytometry, and the data were analyzed with FlowJo 10 software. Cells were first gated on a side scatter height (SSC-H) vs. side scatter area (SSC-A) plot to exclude doublets, as well as on a forward scatter area (FSC-A) and SSC-A plot to exclude small debris and cell aggregates. FITC-stained cells could then be compared to the unstained control. To negate the possibility of nonspecific FITC binding, competitive binding between FITC-labeled and unlabeled peptides was performed. PC3 and DU145 cells were seeded overnight, washed twice with PBS, and then exposed to various concentrations of unlabeled peptide in culture medium for 5 min. FITC–peptide conjugate was added at a constant concentration for 1 min, after which the cells were washed twice with PBS, trypsinized, and analyzed by flow cytometry.
Binding of Peptides to Normal Prostate and PrC Patient Cancer Cells
Biopsies of prostate tumor tissues and nonmalignant tissues were obtained from patients undergoing radical prostatectomy or TURP, as described above. Fresh tissue was transferred to Ariel University within 3 h, finely minced with a sterile blade, and then cultured on collagen-coated 6-well plates until the cells were attached to the well. The medium was changed twice a week. The cells used for the experiments were from passages 2–5. Passaged cells were seeded into 2 wells of 6-well plates for 24 h and then washed with PBS. One well received only medium, while the second well was exposed to 20 µM of FITC–peptide in full medium for 20 min. The cells were washed twice with PBS, trypsinized, centrifuged at 400×
g for 5 min at 4 °C, resuspended in PBS, and mixed with 1:1
v/
v fixation buffer (Invitrogen™, Rhenium, Modiin, Israel), prewarmed to 37 °C. After incubation for 10 min at 37 °C and washing in PBS, the cells were permeabilized in Perm buffer (BD™ PhoSFLOW, Biosciences, Ceasaria, Israel) for 15 min on ice, centrifuged, and resuspended in PBS. Alexa Fluor
® 647-conjugated anti-AMACR (alpha-methylacyl-CoA racemase) (P504S, Santa Cruz Biotechnology, Enco, Petah Tikva, Israel) was added to distinguish cancerous from normal cells. AMACR is a mitochondrial and peroxisomal enzyme that is overexpressed in prostate cancer [
17]. The cells were incubated on ice in the dark for 30 min, washed twice, resuspended in PBS, and analyzed by flow cytometry.
2.4. Analysis of Peptide Function
2.4.1. Influence of Cell Cycle on Peptide Binding
PrC cells were seeded for 24 h, washed with PBS, and exposed to 20 µM of FITC–peptide in complete medium for 60 min. The cells were washed, trypsinized, fixed in 66% ethanol on ice for 2 h, centrifuged at 500× g for 5 min, washed in cold PBS, and centrifuged again. The cells were resuspended and analyzed by flow cytometry for DNA content using the Propidium Iodide Flow Cytometry Kit (ab139418), (Abcam, Zotal, Tel Aviv, Israel).
2.4.2. Internalization of mPrC Peptides
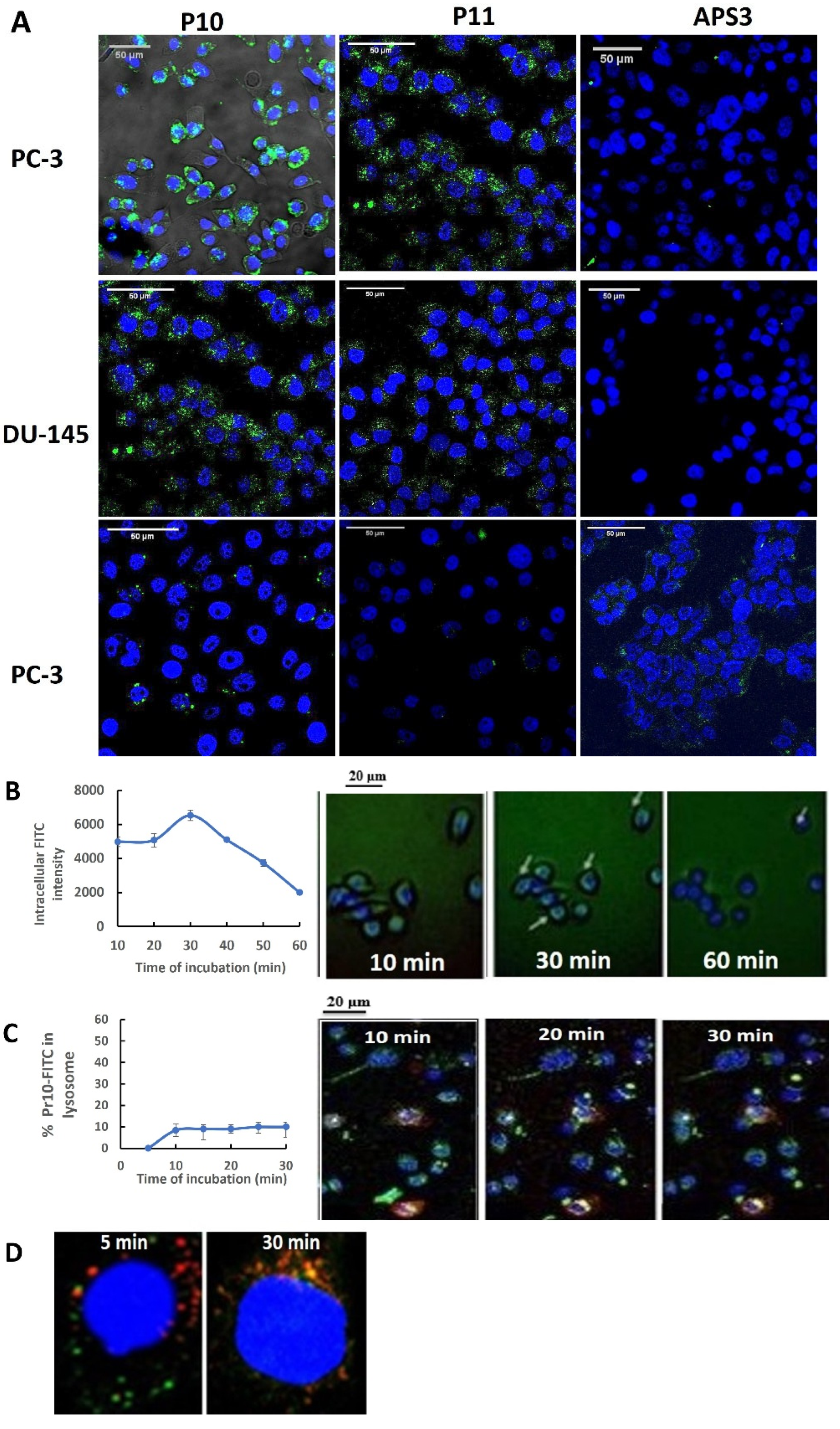
RWPE-1, PC3, LNCaP, 22Rv1, and DU145 cells were seeded onto 8-well MilliCell® EZ slides (Millipore, Mercury, Rosh Hain, Israel) at 2.5 × 104 cells/mL and cultured for 24 h. The cells were washed twice with PBS, and then cultured in fresh RPMI containing 2% FBS, and 10 µM of FITC-peptide was added for 5–60 min. The cells were washed twice with PBS, fixed with 4% PFA (pH = 7.4) for 20 min at RT in the dark, washed with PBS 3 times, stained with DAPI (VECTOR laboratories, Inc., Zotal, Tel Aviv, Israel), and imaged with an inverted Zeiss Axio-observer Z1 confocal microscope. The color channels were merged with ImageJ 1.53a software (RRID:SCR_003070).
To measure uptake kinetics, PC3 cells were seeded at a density of 5 × 105/200 µL onto the well of a glass slide (Millipore Millicell® EZ Slide, Mercury, Rosh Hain, Israel). The medium was replaced with RPMI 1640 medium containing 20 µM of P10–FITC and Hoechst 33342 to stain the DNA. After 20 min at 4 °C, the cells were washed with cold PBS and viewed by live microscopy (Olympus, Tokyo, Japan) at 37 °C in an environment containing 5% CO2. To accurately assess the kinetics of Pr10 uptake, intracellular fluorescence images were collected for the same group of cells every 10 min for 60 min. ImageJ 1.53a software (Fiji Win64) was used for immunofluorescence intensity analysis.
To visualize intracellular peptide localization, PC3 cells were seeded at a density of 5 × 105/200 µL into the wells of a glass slide (Millipore Millicell® EZ Slide). The following day, the cells were transduced at a confluence of 70% with 3 μL of Cell Light™ Lysosome-RFP reagent (BacMam 2.0, Rhenium, Modiin, Israel) by incubation for 16 h at 37 °C. Subsequently, the medium was replaced with RPMI 1640 medium without FBS but containing 20 µM of P10–FITC, and the cells were incubated at 37 °C in an environment containing 5% CO2. After each incubation, the medium was removed, and the cells were washed with PBS/0.05% Tween 20 and then with glycine HCl (0.1 M, pH = 2.2) for 10 min at room temperature. The cells were fixed in 4% paraformaldehyde for 10 min, washed with PBS, and stained with mounting medium containing DAPI (Vectashield). Fluorescence images were collected with a confocal microscope (ZEISS LSM700) using CellLight Lysosome-RFP (555/584 nm), DAPI, and PrC–FITC. ImageJ2 software (Fiji Win64) was used for immunofluorescence analysis.
2.4.3. Peptide Endocytic Pathways
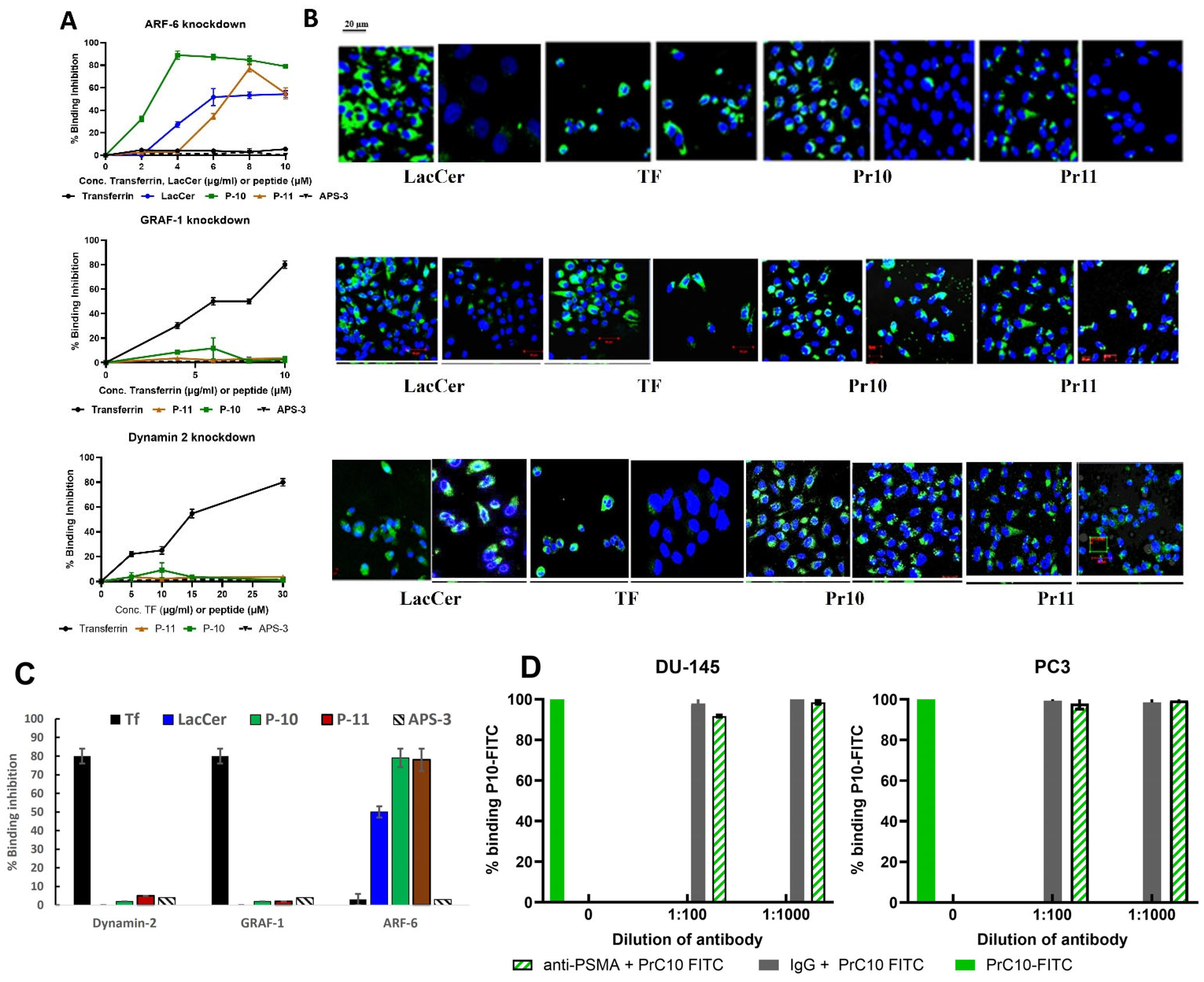
Preliminary experiments with chemical inhibitors of endocytic pathways (Dynasore, Pitstop, and MβCD) indicated that the uptake of P10 or P11 did not involve the caveolae, CIE, CDE, or DDE pathways (
Figure S6). To investigate peptide uptake more precisely, single siRNAs were used to knockdown specific proteins associated with different endocytic pathways: Dynamin 2 (si-Dyn2 5′-CCCUCAAGGAGGCGCUCA-3′), ARF-6 (si-ARF-6 5′-AGGACUUUACCAAGAGCUGUU-3′), GRAF-1 (si-GRAF-1 5′-UUUGAAACUGGUACAUCAUGAGUGGUU-3′), or the control protein GAPDH (si-GAPDH 5′-UGGUUUACAUGUUCCAACA-3′) (Dharmacon, Abu-Gosh, Israel). In preliminary experiments, the maximum knockdown of protein expression was achieved by transfecting cells at 37 °C for 72 h with 25 nM of siRNA using the TransIT–X2
® Dynamic Delivery System (Mirus Bio, Tel Aviv, Israel) according to the manufacturer’s protocol. β-Actin was used as a loading control. The immunoblot band density was quantified using ImageJ2 software, and the relative ratios of specific siRNA/control siRNA-treated cells were calculated.
Figure S7 shows that the maximum expression knockdown reached 60% for Dynamin 2 and 95% for ARF6 and GRAF1.
PC3 cells were plated overnight at a density of 5 × 105 cells/mL, and replicate cultures were transfected as described above and cultured for an additional 72 h. Then, 100 µL of 5, 10, 15, or 20 µg/mL of P10- or P11-FITC; 5, 10, 15, or 20 µg/mL of Transferrin-Alexa Fluor; or 4, 8, or 10 µg/mL of fluorescent Cholera toxin B was added to separate the wells of the PC3-knockdown cultures. The cells were divided into two aliquots. One sample was processed for confocal microscopy as previously described. The second aliquot was washed in ice-cold PBS and collected on ice by scraping it into lysis buffer (Cytobuster™) containing a protease inhibitor cocktail. Lysates were centrifuged at 13,000× g (4 °C) for 10 min, and the supernatant was stored at −20 °C or processed immediately. For processing, 1 Bolt™ LDS sample buffer (4×) was added to the samples. The mixture was heated for 10 min at 70 °C, separated by SDS-PAGE (Invitrogen™), and transferred to PVDF membranes which were blocked, probed with goat antibodies to Dynamin 2, ARF-6, GRAF-1, or β- actin followed by HRP-conjugated rabbit anti-goat antibodies, and then illuminated by the addition of Luminata™ Crescendo Western HRP substrate (Millipore, Burlington, MA, USA), using a Life Technologies blotting apparatus (Thermo Fisher). The membranes were photographed (ImageQuant™ LAS 4000 Mini) and analyzed using ImageJ2 software (Fiji Win64).
2.5. Analysis of PrC10 Binding to PSMA
PC3 cells were seeded for 24–48 h, washed with PBS, and recultured for 10 min in fresh medium containing a 1/100 or 1/1000 dilution of rabbit anti-human PSMA or non-relevant rabbit IgG antibody. The cells were washed with PBS and incubated in fresh medium supplemented with 10 µM of PrC10-FITC for 20 min. Following incubation, the cells were washed twice with PBS, gently scraped from the wells, and resuspended in 1% BSA. The cell suspension was immediately analyzed by a flow cytometer to measure the fluorescence intensity of PrC10-FITC.
2.6. PDC Synthesis and Stability
2.7. Cell Cytotoxicity of Free Drugs and PDCs
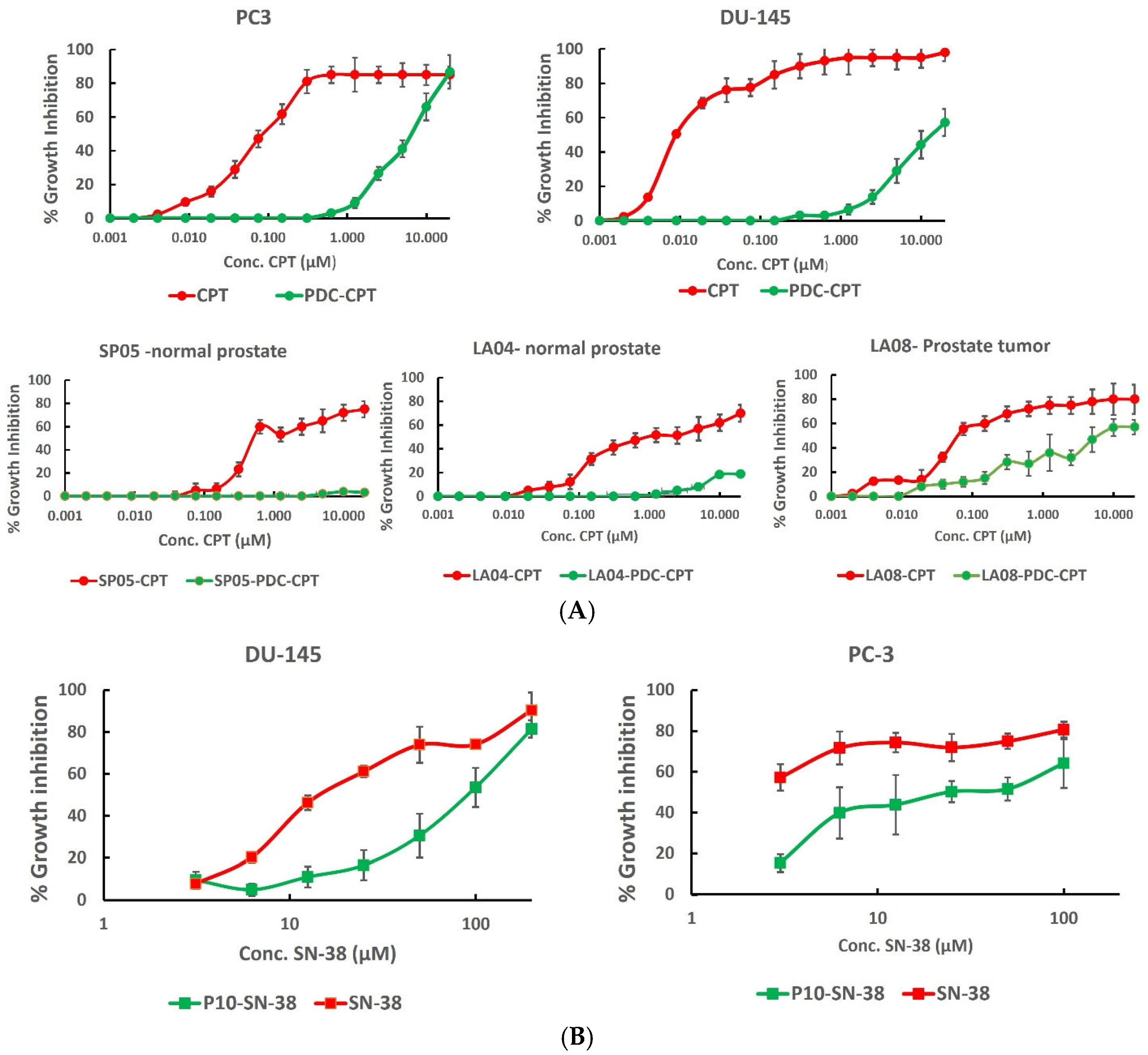
To test the cytotoxicity of free and P10-conjugated CPT, cells were seeded onto 96-well microplates (Nunc) at 5 × 104 cells/mL for 24 h. The medium was refreshed either alone (control) or supplemented with increasing drug concentrations (0.0625–10 µM of CPT, or 0–50 µM of PDC), and the cells were cultured for 24 h. To test the cytotoxicity of free or conjugated SN-38, cells were cultured for 6 h with increasing drug doses. The cells were then washed in PBS and recultured in drug-free medium for 72 h. The cellular metabolism of cells following treatment, as determined with an XTT Cell Proliferation Kit (Satorius, Beit Haemek, Israel), was used as a measure of cell viability. Absorbances were measured with a TECAN Infinite M200 ELISA reader (Tecan Life Sciences, Mannedorf, Switzerland). The specific absorbance readings at 680 nm (nonspecific measurements) minus the readings at 480 nm were used to calculate the % growth inhibition (GI) of the surviving cells exposed to the drug compared to that of the surviving cells not exposed to the drug.
2.8. In Vivo Tumor Targeting
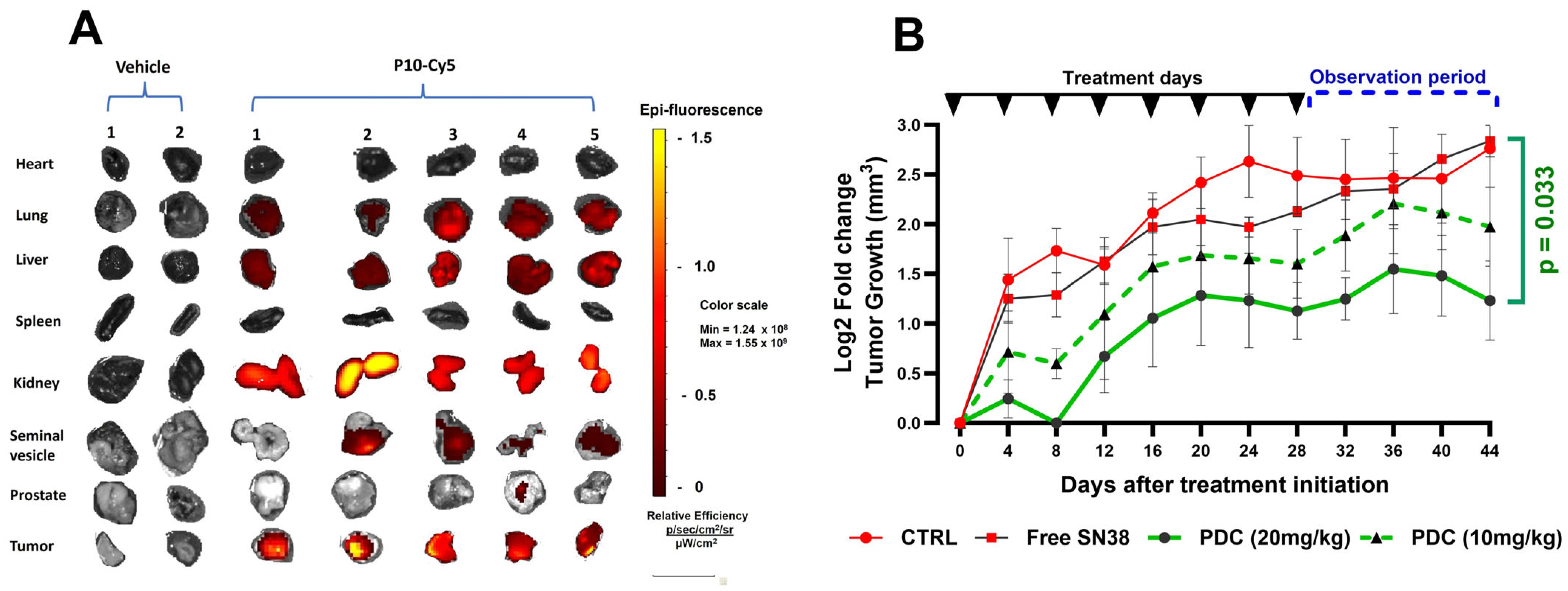
All animal experiments were performed in accordance with standard procedures for laboratory animals approved by the Institutional Animal Care and Use Committee of Ariel University (#AU-IL-2401-104). Seven 8-week-old male athymic, nu/nu mice were inoculated subcutaneously with 1.5 × 106 PC3 cells suspended in Matrigel. On day 12 post-injection, 3 mg/kg of P-10-Cy5 peptide conjugate in PBS was administered via tail vein injection to five mice, while two mice received only vehicle. After 48 h, the mice were euthanized via CO2 inhalation, and major organs were collected, placed on Petri dishes, and fluorescence imaging taken using the IVIS Spectrum CT system (PerkinElmer, Waltham, MA, USA) (excitation filter: 640 nm; emission filter: 680 nm; exposure time: 0.5 s; binning: medium (8)).
2.9. Xenograft Study to Evaluate Anti-Tumor Effect of PrC10-SN38
As described above, a xenograft tumor model was established by injecting mice with PC3 cells. Tumor growth was monitored regularly, and mice were randomized into one of four groups (n = 5 per group) when their tumor volume reached approximately 140 mm3 (approx. day 16 post-injection) so as to ensure the equal distribution of tumor burden among the groups. The mice were treated twice weekly for four weeks via tail vein injection as follows. Group 1 (Control) received vehicle only; Group 2 (free SN-38) received SN38 at a dose of 5 mg/kg; Group 3 (PDC 10 mg/kg) received PrC10-SN38 at a dose of 10 mg/kg; Group 4 (PDC 20 mg/kg) received PrC10-SN38 at a dose of 20 mg/kg. Tumor volume was measured regularly until day 44 from the initiation of treatment, after which all mice were euthanized via CO2 inhalation.
2.10. Statistics
The statistical significance of differences between groups was determined using Student’s t test. A p value ≤ 0.05 was considered to indicate statistical significance. A comparison of the IC50 values to the specified values was performed with the Wilcoxon signed-rank test. The percentage of positive antibody binding in flow cytometry was calculated with the Overton cumulative histogram subtraction algorithm in FlowJo software.
4. Discussion
There is an increasing interest in using peptides as drug carriers in targeted drug delivery systems, including delivery to prostate cancer cells [
19], but to date, only two PDCs have received FDA approval [
20]. One reason for this limited success is that most studies have selected candidate peptides based on their selective binding to tumor-associated receptors known to be overexpressed on the cancer cells [
21], even though those receptors may not be optimal candidates for use in targeted drug delivery. An example of one such receptor is CD33, also known as Siglec-3, which was originally used for targeted drug delivery to Acute Myelogenous Leukemia (AML) cells [
22]. A more holistic approach to selecting peptides that fulfill additional important functional requirements may result in the discovery of more effective drug carriers for targeted drug delivery to cancer cells. To accomplish this goal, we began by using a phage display peptide library preabsorbed in vivo on normal tissue to screen for peptides that internalized into PrC cell lines and PrC patient tumor cells, without a priori knowledge of the target receptor (unbiased) (
Figure 1). The peptides displayed by these phages were subjected to a set of biochemical, biological, and functional in vitro and in vivo tests to ensure that they would be appropriate candidates for pre-clinical targeted drug delivery studies to effectively treat mPrC.
Several key conclusions can be drawn from our results. First, from the initial six phage clones chosen from the library screen, only two clones displayed peptides, P10 and P11, which in their free form retained significant binding above both negative control cell lines (
Figure 2A). This result highlights the important point that the activity of a phage-displayed peptide does not necessarily predict the activity of the free peptide. This may be due to structural changes in the peptide when displayed as an attachment to a large protein (in the present case, to the P3 tail protein of the M13 phage) compared to the free protein, which would affect binding activity [
23].
Second, it is critical to ensure that the target-positive phage clones also internalize into drug-resistant cells (
Figure 2B,C). This is particularly important, as initial applications of PDC therapy will presumably not be for front-line therapy but rather for treating patients whose cancer cells are already drug-resistant.
Third, following the demonstration of target cell specificity in vitro, it is necessary to show that the candidate peptide targets tumor cells in vivo. This was demonstrated for P10 (
Figure 6A). Although epifluorescence imaging of internal organs also showed an accumulation of Cy5 labels in the liver and kidney, this is presumably due to filtration and excretion.
Fourth, it is important to acquire initial information on the nature of peptide uptake and its intracellular trafficking, as this can have important implications for PDC breakdown and drug release following conjugate uptake. Little attention has been given to defining the mechanism of drug carrier uptake in drug delivery systems [
12,
24]. Initially, we studied the uptake of P10 and P11 by first using the chemical inhibitors of endocytic pathways, MβCD, Pitstop 2, and Dynasore. We found that Dynasore did not inhibit peptide uptake, suggesting that neither P10 nor P11 utilizes dynamin-dependent pathways (
Figure 5A-left). In contrast, Pitstop 2, which inhibits both the CIE and CDE pathways, blocked P11 uptake, while MβCD, which inhibits the Caveolae + CIE pathways, blocked P10 uptake (
Figure S6), suggesting that these two peptides might utilize different uptake pathways and presumably bind different receptors.
Although the chemical inhibitors provided us with general indications of the pathways used by these peptides, they could not provide sufficient detail. For this reason, siRNAs were used to knockdown the expression of specific protein components of certain endocytic pathways. These experiments confirmed that P10 and P11 do not use dynamin-dependent pathways, nor is their uptake dependent on the expression of GRAF1 (a GTPase regulator associated with focal adhesion kinase-1) (
Figure 4A). These results are in line with what is known about GRAF-1, an essential component of clathrin-independent endocytic pathways, which colocalizes with dynamin during the organization of molecular networks and membrane dynamics [
25]. These results also indicate that once endocytosed, the peptides are not transported to the Golgi apparatus [
26,
27]. In contrast, the siRNA-mediated knockdown of ARF6 completely inhibited the uptake of P10 (94%) and P11 (90%), albeit at different concentrations (
Figure 4A). ARF6 (ADP-ribosylation factor 6) is involved in various functions associated with trafficking and plasma membrane protein recycling to the cell surface [
28,
29]. Internalization via ARF-6 is clathrin- and dynamin-independent but cholesterol-dependent [
26]. ARF6 has been associated with several cell-surface receptors expressed on cancer cells, such as CD147 [
30], CD44, CD98 [
31], CD55, and CD59 [
32]. In particular, the expression of CD55 and CD59 on PrC cells may be associated with promoting cell survival and contributing to the metastatic potential of prostate cancer cells [
33]. Recently, it was shown that ARF-6 is involved in the recycling of CIE proteins (CD98 and CD147), which are rapidly removed from endosomes after entry into recycling tubules and thus do not move on to lysosomes [
34]. These data may help explain the results of
Figure 3D, which show, by double-label fluorescent tracking, that P10-FITC is not taken up into lysosomes. Indeed, uptake into lysosomes has been recognized as a major limiting factor in the effective delivery of nanomedicines [
35].
Fifth, using the unbiased exposure of the phage library to the target cells rather than to a pre-determined receptor can lead to the discovery of peptides targeting receptors not previously recognized as potential targets. We found that our lead peptides do not bind PSMA using this approach. While this receptor is abundant on most PrC cells, radiolabeled ligands targeting it can lead to significant side effects [
7,
8].
The purpose of carrying out these initial functional assays was to select peptide candidates that would effectively deliver drug payloads into target cells and kill them. As peptide P10 fulfilled these functional criteria, it was selected for chemical conjugation to the Topo I inhibitor drugs CPT and SN-38 and subjected to the second testing stage. This stage initially involves the demonstration of selective in vitro target cell cytotoxicity. As predicted from the binding results (
Figure 2C,D), the P10-PDCs (P1-CPT and P10-SN-38, see
Supplementary) specifically killed PrC cells but not healthy prostate tissue (
Figure 5). The final functional test is in vivo efficacy in a murine cancer model.
Figure 6B shows that the P10-SN-38 PDC significantly halted tumor progression in a dose-dependent manner. From day 36, tumor sizes in the PDC-treated mice began to regress, while those in the control and free SN-38-treated mice continued to increase.


 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}