

Carboxylesterase Factors Influencing the Therapeutic Activity of Common Antiviral Medications Used for SARS-CoV-2 Infection



Abstract
1. Introduction
2. Background
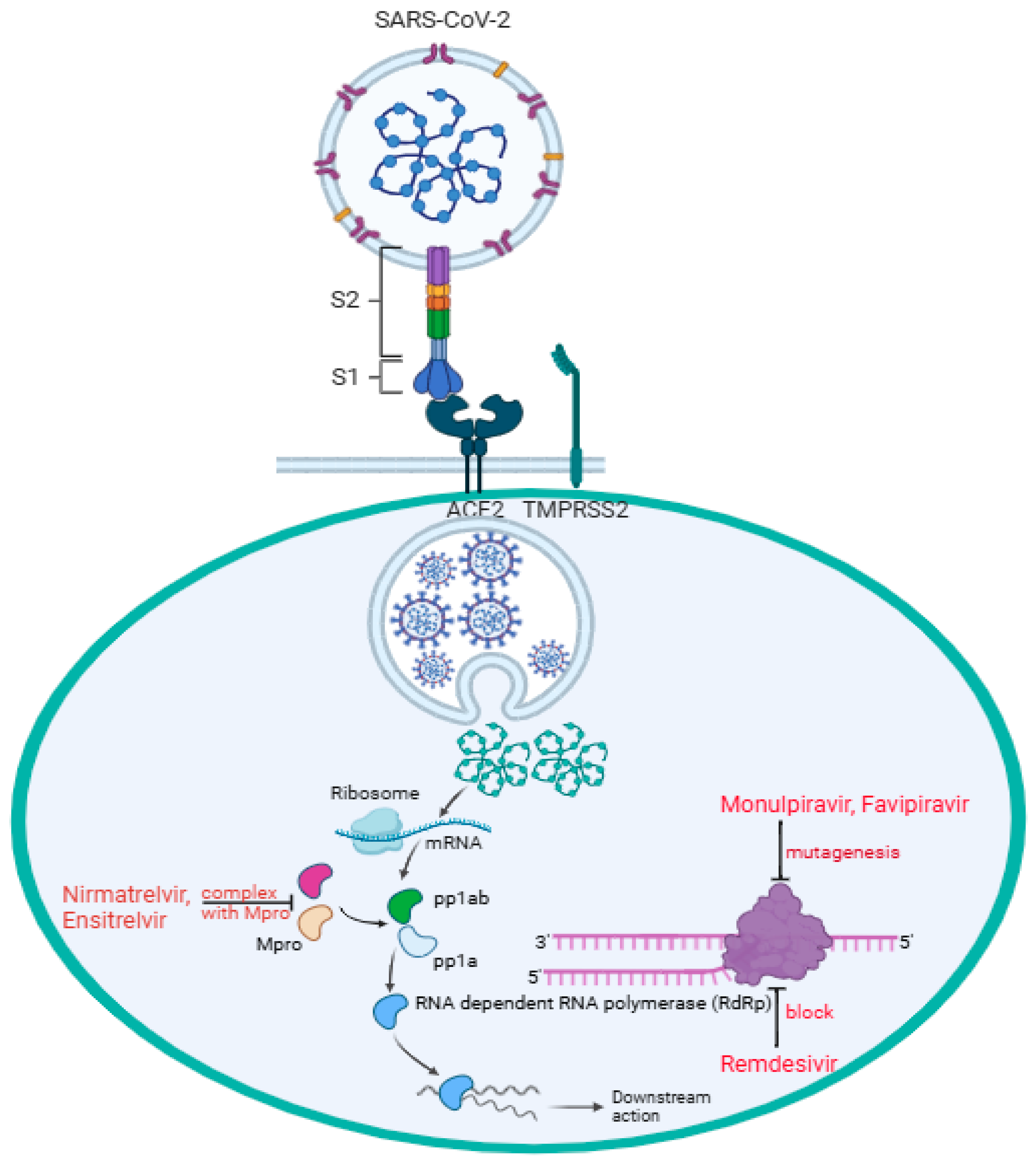
2.1. Antiviral Drugs for the Treatment of COVID-19

{kind=link}
{kind=link}
{kind=link}
| Drug Name | Administration Route | Prodrug Activation | Mechanism of Action | Note |
|---|---|---|---|---|
| Remdesivir (Veklury®) | Intravenously infusion | remdesivir triphosphate (RTP) | RdRp inhibitor | |
| Nirmatrelvir/Ritonavir (Paxlovid™) 1 | Oral | Nirmatrelvir is not a prodrug | Protease inhibitor | Emergency Use Authorization (EUA) in Dec. 2021, later with full approval |
| Molnupiravir (LAGEVRIO™) | Oral | NHC triphosphate | RdRp inhibitor | EUA 2 |
| Favifpiravir | Oral | favipiravir ribofuranosyl-5′-triphostphate | RdRp inhibitor | |
| Ensitrelvir | Oral | Ensitrelvir is not a prodrug | Protease inhibitor |
2.2. Enzymes Involved in Antiviral Drug Activation and Metabolism: Carboxylesterases
3. Carboxylesterase 1 (CES1)
3.1. CES1 Expression and Substrate Specificity
3.2. CES1 Pharmacogenetic Variability
4. Carboxylesterase 2 (CES2)
4.1. CES2 Expression and Substrate Specificity
4.2. CES2 Pharmacogenetic Variability
5. Drug Metabolism on the Therapeutic Activity of SARS-CoV-2 Antiviral Drugs
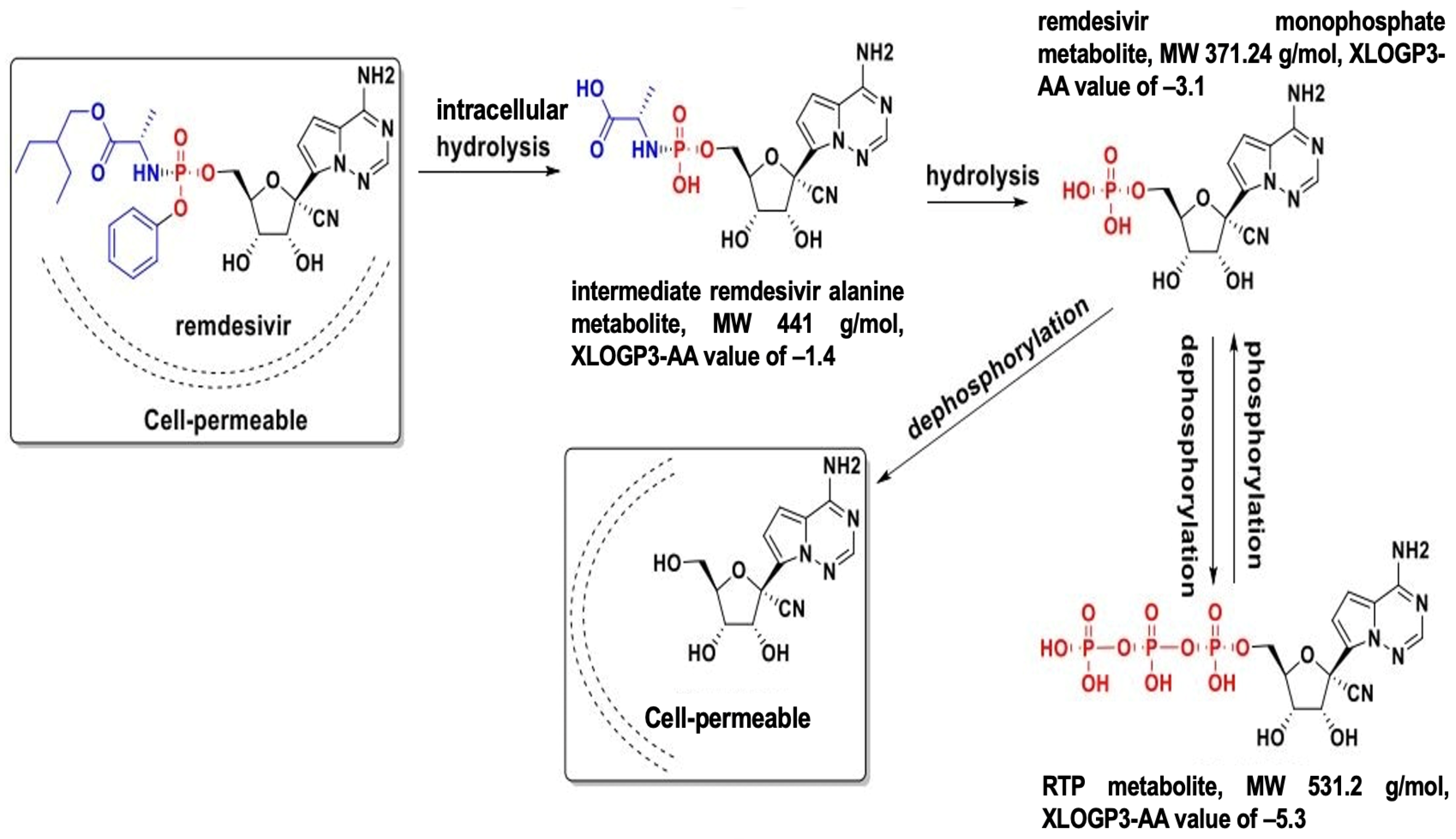
5.1. Remdesivir
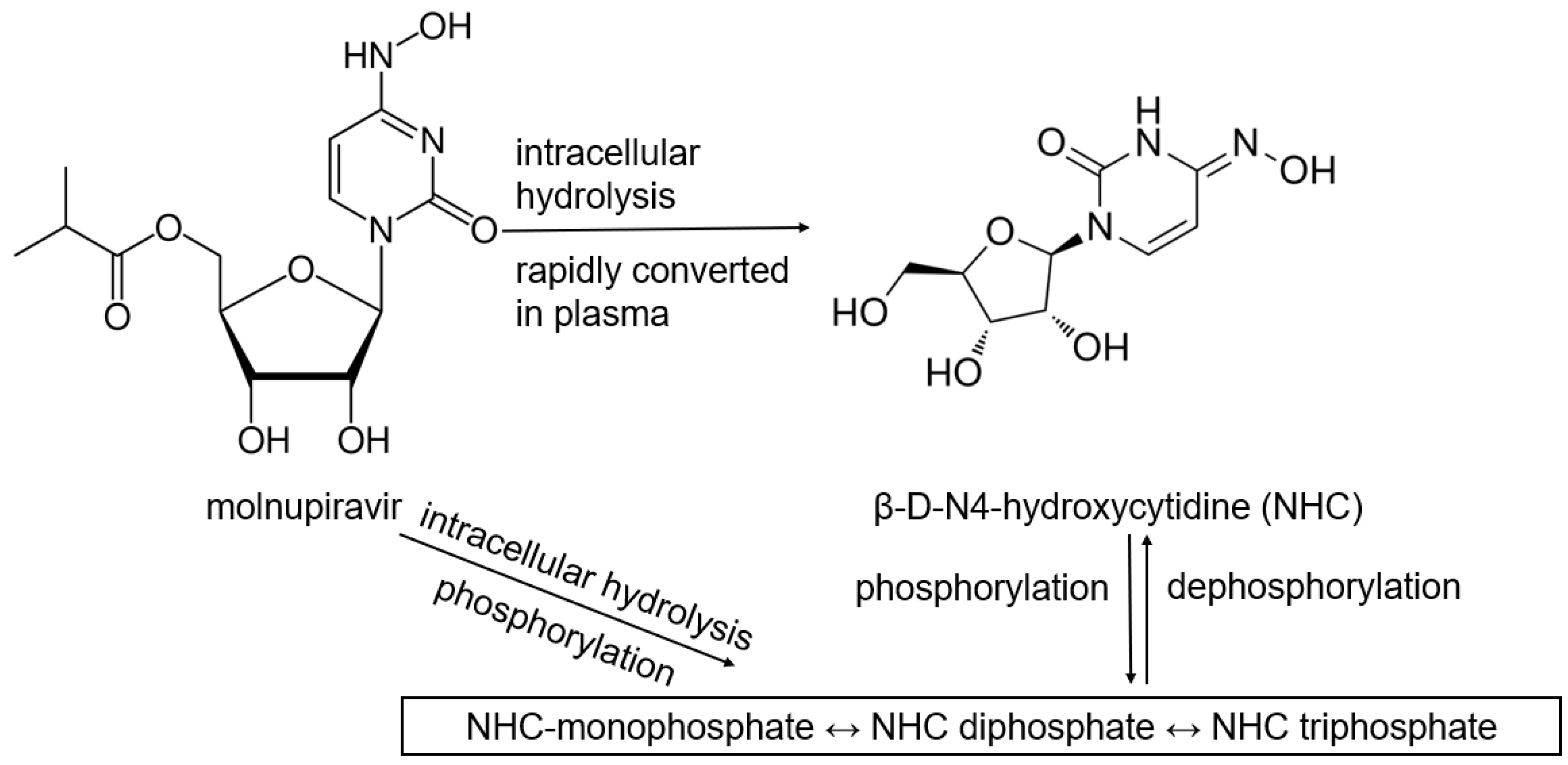
5.2. Molnupiravir
5.3. Nirmaltrevil
6. Implications and Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| SARS-CoV-2 | severe acute respiratory syndrome coronavirus 2 |
| ACE2 | angiotensin-converting enzyme 2 |
| Mpro | main protease |
| 3CLpro | 3-chymotrypsin-like proteases |
| RdRp | RNA-dependent RNA polymerase |
| CES | carboxylesterases |
| CYP | cytochrome P450 |
| S | spike |
| RBD | receptor-biding domain |
| TMPRSS2 | transmembrane protease, serine 2 |
| FCS | furin cleavage site |
| GI | gastrointestinal |
| NHC | β-D-N4-hydroxycytidine |
| RTP | remdesivir triphosphate |
| EUA | Emergency Use Authorization |
| ORF | open reading frames |
| nsps | non-structure proteins |
| P-gp | P-glycoprotein |
| SNP | single nucleotide polymorphism |
| TAF | tenofovir alafenamide |
| TFV | tenofovir |
| PBMC | peripheral blood mononuclear cells |
| SLC | solute carrier |
| OATP | organic anion transporting polypeptide |
| ENT | equilibrative nucleoside transporter |
| MDCK | Madin-Darby canine kidney |
| CNT | concentrative nucleoside transporter |
References
- Pollard, C.A.; Morran, M.P.; Nestor-Kalinoski, A.L. The COVID-19 pandemic: A global health crisis. Physiol. Genomics 2020, 52, 549–557. [Google Scholar] [CrossRef] [PubMed]
- Beyerstedt, S.; Casaro, E.B.; Rangel, É.B. COVID-19: Angiotensin-converting enzyme 2 (ACE2) expression and tissue susceptibility to SARS-CoV-2 infection. Eur. J. Clin. Microbiol. Infect. Dis. 2021, 40, 905–919. [Google Scholar] [CrossRef] [PubMed]
- Zang, R.; Gomez Castro, M.F.; McCune, B.T.; Zeng, Q.; Rothlauf, P.W.; Sonnek, N.M.; Liu, Z.; Brulois, K.F.; Wang, X.; Greenberg, H.B.; et al. TMPRSS2 and TMPRSS4 promote SARS-CoV-2 infection of human small intestinal enterocytes. Sci. Immunol. 2020, 5, eabc3582. [Google Scholar] [CrossRef] [PubMed]
- Xue, S.; Han, Y.; Wu, F.; Wang, Q. Mutations in the SARS-CoV-2 spike receptor binding domain and their delicate balance between ACE2 affinity and antibody evasion. Protein Cell 2024, 15, 403–418. [Google Scholar] [CrossRef]
- Lavie, M.; Dubuisson, J.; Belouzard, S. SARS-CoV-2 Spike Furin Cleavage Site and S2’ Basic Residues Modulate the Entry Process in a Host Cell-Dependent Manner. J. Virol. 2022, 96, e0047422. [Google Scholar] [CrossRef]
- Peacock, T.P.; Goldhill, D.H.; Zhou, J.; Baillon, L.; Frise, R.; Swann, O.C.; Kugathasan, R.; Penn, R.; Brown, J.C.; Sanchez-David, R.Y.; et al. The furin cleavage site in the SARS-CoV-2 spike protein is required for transmission in ferrets. Nat. Microbiol. 2021, 6, 899–909. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, L.; Wu, J.; Yu, Y.; Liu, S.; Li, T.; Li, Q.; Ding, R.; Wang, H.; Nie, J.; et al. A second functional furin site in the SARS-CoV-2 spike protein. Emerg. Microbes Infect. 2022, 11, 182–194. [Google Scholar] [CrossRef]
- Zhao, X.; Chen, H.; Wang, H. Glycans of SARS-CoV-2 Spike Protein in Virus Infection and Antibody Production. Front. Mol. Biosci. 2021, 8, 629873. [Google Scholar] [CrossRef]
- Aloor, A.; Aradhya, R.; Venugopal, P.; Gopalakrishnan Nair, B.; Suravajhala, R. Glycosylation in SARS-CoV-2 variants: A path to infection and recovery. Biochem. Pharmacol. 2022, 206, 115335. [Google Scholar] [CrossRef]
- Markovic, M.; Ben-Shabat, S.; Dahan, A. Prodrugs for Improved Drug Delivery: Lessons Learned from Recently Developed and Marketed Products. Pharmaceutics 2020, 12, 1031. [Google Scholar] [CrossRef]
- Gordon, C.J.; Tchesnokov, E.P.; Woolner, E.; Perry, J.K.; Feng, J.Y.; Porter, D.P.; Götte, M. Remdesivir is a direct-acting antiviral that inhibits RNA-dependent RNA polymerase from severe acute respiratory syndrome coronavirus 2 with high potency. J. Biol. Chem. 2020, 295, 6785–6797. [Google Scholar] [CrossRef]
- Eastman, R.T.; Roth, J.S.; Brimacombe, K.R.; Simeonov, A.; Shen, M.; Patnaik, S.; Hall, M.D. Remdesivir: A Review of Its Discovery and Development Leading to Emergency Use Authorization for Treatment of COVID-19. ACS Cent. Sci. 2020, 6, 672–683. [Google Scholar] [CrossRef] [PubMed]
- Tian, L.; Pang, Z.; Li, M.; Lou, F.; An, X.; Zhu, S.; Song, L.; Tong, Y.; Fan, H.; Fan, J. Molnupiravir and Its Antiviral Activity Against COVID-19. Front. Immunol. 2022, 13, 855496. [Google Scholar] [CrossRef]
- Hashemian, S.M.R.; Pourhanifeh, M.H.; Hamblin, M.R.; Shahrzad, M.K.; Mirzaei, H. RdRp inhibitors and COVID-19: Is molnupiravir a good option? Biomed. Pharmacother. 2022, 146, 112517. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; He, G.; Huang, W. A novel model of molnupiravir against SARS-CoV-2 replication: Accumulated RNA mutations to induce error catastrophe. Signal Transduct. Target. Ther. 2021, 6, 410. [Google Scholar] [CrossRef]
- Yasri, S.; Wiwanitki, V. Molnupiravir, favipiravir and other antiviral drugs with proposed potentials for management of COVID-19: A concern on antioxidant aspect. Int. J. Biochem. Mol. Biol. 2022, 13, 1–4. [Google Scholar] [PubMed]
- Alamer, A.; Alrashed, A.A.; Alfaifi, M.; Alosaimi, B.; AlHassar, F.; Almutairi, M.; Howaidi, J.; Almutairi, W.; Mohzari, Y.; Sulaiman, T.; et al. Effectiveness and safety of favipiravir compared to supportive care in moderately to critically ill COVID-19 patients: A retrospective study with propensity score matching sensitivity analysis. Curr. Med. Res. Opin. 2021, 37, 1085–1097. [Google Scholar] [CrossRef]
- Gandhi, S.; Klein, J.; Robertson, A.J.; Peña-Hernández, M.A.; Lin, M.J.; Roychoudhury, P.; Lu, P.; Fournier, J.; Ferguson, D.; Mohamed Bakhash, S.A.K.; et al. De novo emergence of a remdesivir resistance mutation during treatment of persistent SARS-CoV-2 infection in an immunocompromised patient: A case report. Nat. Commun. 2022, 13, 1547. [Google Scholar] [CrossRef]
- Strizki, J.M.; Gaspar, J.M.; Howe, J.A.; Hutchins, B.; Mohri, H.; Nair, M.S.; Kinek, K.C.; McKenna, P.; Goh, S.L.; Murgolo, N. Molnupiravir maintains antiviral activity against SARS-CoV-2 variants and exhibits a high barrier to the development of resistance. Antimicrob. Agents Chemother. 2024, 68, e0095323. [Google Scholar] [CrossRef]
- Kabinger, F.; Stiller, C.; Schmitzová, J.; Dienemann, C.; Kokic, G.; Hillen, H.S.; Höbartner, C.; Cramer, P. Mechanism of molnupiravir-induced SARS-CoV-2 mutagenesis. Nat. Struct. Mol. Biol. 2021, 28, 740–746. [Google Scholar] [CrossRef]
- Joyce, R.P.; Hu, V.W.; Wang, J. The history, mechanism, and perspectives of nirmatrelvir (PF-07321332): An orally bioavailable main protease inhibitor used in combination with ritonavir to reduce COVID-19-related hospitalizations. Med. Chem. Res. 2022, 31, 1637–1646. [Google Scholar] [CrossRef]
- Ohmagari, N.; Yotsuyanagi, H.; Doi, Y.; Yamato, M.; Imamura, T.; Sakaguchi, H.; Yamanaka, H.; Imaoka, R.; Fukushi, A.; Ichihashi, G.; et al. Efficacy and Safety of Ensitrelvir for Asymptomatic or Mild COVID-19: An Exploratory Analysis of a Multicenter, Randomized, Phase 2b/3 Clinical Trial. Influenza Other Respir. Viruses 2024, 18, e13338. [Google Scholar] [CrossRef]
- Sacco, M.D.; Hu, Y.; Gongora, M.V.; Meilleur, F.; Kemp, M.T.; Zhang, X.; Wang, J.; Chen, Y. The P132H mutation in the main protease of Omicron SARS-CoV-2 decreases thermal stability without compromising catalysis or small-molecule drug inhibition. Cell Res. 2022, 32, 498–500. [Google Scholar] [CrossRef] [PubMed]
- Bouzidi, H.S.; Driouich, J.S.; Klitting, R.; Bernadin, O.; Piorkowski, G.; Amaral, R.; Fraisse, L.; Mowbray, C.E.; Scandale, I.; Escudié, F.; et al. Generation and evaluation of protease inhibitor-resistant SARS-CoV-2 strains. Antivir. Res. 2024, 222, 105814. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Cao, R.; Zhang, L.; Yang, X.; Liu, J.; Xu, M.; Shi, Z.; Hu, Z.; Zhong, W.; Xiao, G. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res. 2020, 30, 269–271. [Google Scholar] [CrossRef] [PubMed]
- Pruijssers, A.J.; George, A.S.; Schäfer, A.; Leist, S.R.; Gralinksi, L.E.; Dinnon, K.H., III; Yount, B.L.; Agostini, M.L.; Stevens, L.J.; Chappell, J.D.; et al. Remdesivir Inhibits SARS-CoV-2 in Human Lung Cells and Chimeric SARS-CoV Expressing the SARS-CoV-2 RNA Polymerase in Mice. Cell Rep. 2020, 32, 107940. [Google Scholar] [CrossRef]
- Ko, W.C.; Rolain, J.M.; Lee, N.Y.; Chen, P.L.; Huang, C.T.; Lee, P.I.; Hsueh, P.R. Arguments in favour of remdesivir for treating SARS-CoV-2 infections. Int. J. Antimicrob. Agents 2020, 55, 105933. [Google Scholar] [CrossRef]
- Macip, G.; Garcia-Segura, P.; Mestres-Truyol, J.; Saldivar-Espinoza, B.; Pujadas, G.; Garcia-Vallvé, S. A Review of the Current Landscape of SARS-CoV-2 Main Protease Inhibitors: Have We Hit the Bullseye Yet? Int. J. Mol. Sci. 2021, 23, 259. [Google Scholar] [CrossRef]
- Marzolini, C.; Kuritzkes, D.R.; Marra, F.; Boyle, A.; Gibbons, S.; Flexner, C.; Pozniak, A.; Boffito, M.; Waters, L.; Burger, D.; et al. Recommendations for the Management of Drug-Drug Interactions Between the COVID-19 Antiviral Nirmatrelvir/Ritonavir (Paxlovid) and Comedications. Clin. Pharmacol. Ther. 2022, 112, 1191–1200. [Google Scholar] [CrossRef]
- Yan, B. Carboxylesterases. Part II: Enzyme Systems Involved in Drug Metabolism and Interactions in Animals and Humans. In Encyclopedia of Drug Metabolism and Interactions, 1st ed.; Lyubimov, A.V., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2012. [Google Scholar] [CrossRef]
- Wang, D.; Zou, L.; Jin, Q.; Hou, J.; Ge, G.; Yang, L. Human carboxylesterases: A comprehensive review. Acta Pharm. Sin. B 2018, 8, 699–712. [Google Scholar] [CrossRef]
- Shen, Y.; Eades, W.; Yan, B. The COVID-19 Medicine Remdesivir Is Therapeutically Activated by Carboxylesterase-1, and Excessive Hydrolysis Increases Cytotoxicity. Hepatol. Commun. 2021, 5, 1622–1623. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Liclican, A.; Xu, Y.; Pitts, J.; Niu, C.; Zhang, J.; Kim, C.; Zhao, X.; Soohoo, D.; Babusis, D.; et al. Key Metabolic Enzymes Involved in Remdesivir Activation in Human Lung Cells. Antimicrob. Agents Chemother. 2021, 65, e0060221. [Google Scholar] [CrossRef]
- Shen, Y.; Eades, W.; Liu, W.; Yan, B. The COVID-19 Oral Drug Molnupiravir Is a CES2 Substrate: Potential Drug-Drug Interactions and Impact of CES2 Genetic Polymorphism In Vitro. Drug Metab. Dispos. 2022, 50, 1151–1160. [Google Scholar] [CrossRef]
- Shen, Y.; Eades, W.; Yan, B. Remdesivir potently inhibits carboxylesterase-2 through covalent modifications: Signifying strong drug-drug interactions. Fundam. Clin. Pharmacol. 2021, 35, 432–434. [Google Scholar] [CrossRef] [PubMed]
- Sokolova, A.S.; Okhina, A.A.; Shtro, A.A.; Klabukov, A.M.; Galochkina, A.V.; Nikolaeva, Y.V.; Petukhova, G.D.; Yarovaya, O.I.; Rogachev, A.D.; Baev, D.S.; et al. Biostability, in vivo antiviral activity against respiratory syncytial virus, and pharmacokinetic profiles of (-)-borneol esters. Eur. J. Pharmacol. 2025, 996, 177567. [Google Scholar] [CrossRef] [PubMed]
- Eisner, H.; Riegler-Berket, L.; Gamez, C.F.R.; Sagmeister, T.; Chalhoub, G.; Darnhofer, B.; Jazleena, P.J.; Birner-Gruenberger, R.; Pavkov-Keller, T.; Haemmerle, G.; et al. The Crystal Structure of Mouse Ces2c, a Potential Ortholog of Human CES2, Shows Structural Similarities in Substrate Regulation and Product Release to Human CES1. Int. J. Mol. Sci. 2022, 23, 13101. [Google Scholar] [CrossRef]
- Elens, L.; Langman, L.J.; Hesselink, D.A.; van Gelder, T. The Impact of COVID-19 on Drug Metabolism and Pharmacokinetics. Clin. Pharmacokinet. 2020, 59, 1357–1365. [Google Scholar]
- Gene Expression Omnibus (GEO) Database. GSE150316. 2020. Available online: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE150316 (accessed on 31 May 2025).
- Su, Y.; Chen, D.; Yuan, D.; Lausted, C.; Choi, J.; Dai, C.L.; Voillet, V.; Duvvuri, V.R.; Scherler, K.; Troisch, P.; et al. Multi-Omics Resolves a Sharp Disease-State Shift between Mild and Moderate COVID-19. Nature 2020, 588, 147–152. [Google Scholar] [CrossRef] [PubMed]
- Nie, X.; Qian, L.; Sun, R.; Huang, B.; Dong, X.; Xiao, Q.; Zhang, Q.; Lu, T.; Yue, L.; Chen, S.; et al. Multi-Omics Analyses Reveal Systemic Insights into COVID-19 Pathophysiology. Cell 2021, 184, 3165–3179. [Google Scholar]
- Liu, Y.; Li, J.; Zhu, H.J. Regulation of carboxylesterases and its impact on pharmacokinetics and pharmacodynamics: An up-to-date review. Expert Opin. Drug Metab. Toxicol. 2024, 20, 377–397. [Google Scholar] [CrossRef]
- Lewis, J.P.; Horenstein, R.B.; Ryan, K.; O’Connell, J.R.; Gibson, Q.; Mitchell, B.D.; Tanner, K.; Chai, S.; Bliden, K.P.; Tantry, U.S.; et al. The functional G143E variant of carboxylesterase 1 is associated with increased clopidogrel active metabolite levels and greater clopidogrel response. Pharmacogenet Genom. 2013, 23, 1–8. [Google Scholar] [CrossRef]
- Wang, X.; Her, L.; Xiao, J.; Shi, J.; Wu, A.H.; Bleske, B.E.; Zhu, H.J. Impact of carboxylesterase 1 genetic polymorphism on trandolapril activation in human liver and the pharmacokinetics and pharmacodynamics in healthy volunteers. Clin. Transl. Sci. 2021, 14, 1380–1389. [Google Scholar] [CrossRef] [PubMed]
- Xiao, J.; Shi, J.; Thompson, B.R.; Smith, D.E.; Zhang, T.; Zhu, H.J. Physiologically-Based Pharmacokinetic Modeling to Predict Methylphenidate Exposure Affected by Interplay Among Carboxylesterase 1 Pharmacogenetics, Drug-Drug Interactions, and Sex. J. Pharm. Sci. 2022, 111, 2606–2613. [Google Scholar] [CrossRef] [PubMed]
- Her, L.H.; Wang, X.; Shi, J.; Choi, H.J.; Jung, S.M.; Smith, L.S.; Wu, A.H.; Bleske, B.E.; Zhu, H.J. Effect of CES1 genetic variation on enalapril steady-state pharmacokinetics and pharmacodynamics in healthy subjects. Br. J. Clin. Pharmacol. 2021, 87, 4691–4700. [Google Scholar] [CrossRef] [PubMed]
- Ikonnikova, A.; Rodina, T.; Dmitriev, A.; Melnikov, E.; Kazakov, R.; Nasedkina, T. The Influence of the CES1 Genotype on the Pharmacokinetics of Enalapril in Patients with Arterial Hypertension. J. Pers. Med. 2022, 12, 580. [Google Scholar] [CrossRef]
- Nemoda, Z.; Angyal, N.; Tarnok, Z.; Gadoros, J.; Sasvari-Szekely, M. Carboxylesterase 1 gene polymorphism and methylphenidate response in ADHD. Neuropharmacology 2009, 57, 731–733. [Google Scholar] [CrossRef]
- de With, M.; van Doorn, L.; Maasland, D.C.; Mulder, T.A.M.; Oomen-de Hoop, E.; Mostert, B.; Homs, M.Y.V.; El Bouazzaoui, S.; Mathijssen, R.H.J.; van Schaik, R.H.N.; et al. Capecitabine-induced hand-foot syndrome: A pharmacogenetic study beyond DPYD. Biomed. Pharmacother. 2023, 159, 114232. [Google Scholar] [CrossRef]
- Ji, Q.; Zhang, C.; Xu, Q.; Wang, Z.; Li, X.; Lv, Q. The impact of ABCB1 and CES1 polymorphisms on dabigatran pharmacokinetics and pharmacodynamics in patients with atrial fibrillation. Br. J. Clin. Pharmacol. 2021, 87, 2247–2255. [Google Scholar] [CrossRef]
- Shnayder, N.A.; Petrova, M.M.; Shesternya, P.A.; Savinova, A.V.; Bochanova, E.N.; Zimnitskaya, O.V.; Pozhilenkova, E.A.; Nasyrova, R.F. Using Pharmacogenetics of Direct Oral Anticoagulants to Predict Changes in Their Pharmacokinetics and the Risk of Adverse Drug Reactions. Biomedicines 2021, 9, 451. [Google Scholar] [CrossRef]
- Rodríguez-Lopez, A.; Ochoa, D.; Soria-Chacartegui, P.; Martín-Vilchez, S.; Navares-Gómez, M.; González-Iglesias, E.; Luquero-Bueno, S.; Román, M.; Mejía-Abril, G.; Abad-Santos, F. An Investigational Study on the Role of CYP2D6, CYP3A4 and UGTs Genetic Variation on Fesoterodine Pharmacokinetics in Young Healthy Volunteers. Pharmaceuticals 2024, 17, 1236. [Google Scholar] [CrossRef]
- Yan, D.; Yan, B. Viral target and metabolism-based rationale for combined use of recently authorized small molecule COVID-19 medicines: Molnupiravir, nirmatrelvir, and remdesivir. Fundam. Clin. Pharmacol. 2023, 37, 726–738. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.; Lee, S.; Lee, H.; Cho, J.Y.; Yoon, S.H.; Jang, I.J.; Yu, K.S.; Lim, K.S. The novel carboxylesterase 1 variant c.662A>G may decrease the bioactivation of oseltamivir in humans. PLoS ONE 2017, 12, e0176320. [Google Scholar] [CrossRef]
- Gu, Z.C.; Ma, X.W.; Zheng, X.Y.; Shen, L.; Shi, F.H.; Li, H. Left Atrial Appendage Thrombus Formation in a Patient on Dabigatran Therapy Associated With ABCB1 and CES-1 Genetic Defect. Front. Pharmacol. 2018, 9, 491. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.T.; Beery, N.; Taran, A.; Stevens, T.; Henzler, C.; Badalamenti, J.; Regal, R.; McCarty, C.A. Associations between CES1 variants and dosing and adverse effects in children taking methylphenidate. Front. Pediatr. 2023, 10, 958622. [Google Scholar] [CrossRef] [PubMed]
- Hodges, L.M.; Markova, S.M.; Chinn, L.W.; Gow, J.M.; Kroetz, D.L.; Klein, T.E.; Altman, R.B. Very important pharmacogene summary: ABCB1 (MDR1, P-glycoprotein). Pharmacogenet Genom. 2011, 21, 152–161. [Google Scholar] [CrossRef]
- Laizure, S.C.; Parker, R.B.; Herring, V.L.; Hu, Z.Y. Identification of carboxylesterase-dependent dabigatran etexilate hydrolysis. Drug Metab. Dispos. 2014, 42, 201–206. [Google Scholar] [CrossRef]
- Ning, R.; Wang, X.P.; Zhan, Y.R.; Qi, Q.; Huang, X.F.; Hu, G.; Guo, Q.L.; Liu, W.; Yang, J. Gambogic acid potentiates clopidogrel-induced apoptosis and attenuates irinotecan-induced apoptosis through down-regulating human carboxylesterase 1 and -2. Xenobiotica 2016, 46, 816–824. [Google Scholar] [CrossRef]
- Eades, W.; Liu, W.; Shen, Y.; Shi, Z.; Yan, B. Covalent CES2 Inhibitors Protect against Reduced Formation of Intestinal Organoids by the Anticancer Drug Irinotecan. Curr. Drug Metab. 2022, 23, 1000–1010. [Google Scholar] [CrossRef]
- Liu, S.; Wang, Z.; Tian, X.; Cai, W. Predicting the Effects of CYP2C19 and Carboxylesterases on Vicagrel, a Novel P2Y12 Antagonist, by Physiologically Based Pharmacokinetic/Pharmacodynamic Modeling Approach. Front. Pharmacol. 2020, 11, 591854. [Google Scholar] [CrossRef]
- Xiao, D.; Shi, D.; Yang, D.; Barthel, B.; Koch, T.H.; Yan, B. Carboxylesterase-2 is a highly sensitive target of the antiobesity agent orlistat with profound implications in the activation of anticancer prodrugs. Biochem. Pharmacol. 2013, 85, 439–447. [Google Scholar] [CrossRef]
- Fujiyama, N.; Miura, M.; Satoh, S.; Inoue, K.; Kagaya, H.; Saito, M.; Habuchi, T.; Suzuki, T. Influence of carboxylesterase 2 genetic polymorphisms on mycophenolic acid pharmacokinetics in Japanese renal transplant recipients. Xenobiotica 2009, 39, 407–414. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.R.; Sai, K.; Tanaka-Kagawa, T.; Jinno, H.; Ozawa, S.; Kaniwa, N.; Saito, Y.; Akasawa, A.; Matsumoto, K.; Saito, H.; et al. Haplotypes and a novel defective allele of CES2 found in a Japanese population. Drug Metab. Dispos. 2007, 35, 1865–1872. [Google Scholar] [CrossRef] [PubMed]
- Cura, Y.; Sánchez-Martín, A.; Márquez-Pete, N.; González-Flores, E.; Martínez-Martínez, F.; Pérez-Ramírez, C.; Jiménez-Morales, A. Association of Single-Nucleotide Polymorphisms in Capecitabine Bioactivation Pathway with Adjuvant Therapy Safety in Colorectal Cancer Patients. Pharmaceutics 2023, 15, 2548. [Google Scholar] [CrossRef]
- Maslarinou, A.; Manolopoulos, V.G.; Ragia, G. Pharmacogenomic-guided dosing of fluoropyrimidines beyond DPYD: Time for a polygenic algorithm? Front. Pharmacol. 2023, 14, 1184523. [Google Scholar] [CrossRef]
- Schiel, M.A. Human Carboxylesterase 2 Splice Variants: Expression, Activity, and Role in the Metabolism of Irinotecan and Capecitabine. Ph.D. Thesis, Department of Biochemistry & Molecular Biology, Indiana University, Bloomington, IN, USA, February 2009. [Google Scholar]
- Tang, M.; Mukundan, M.; Yang, J.; Charpentier, N.; LeCluyse, E.L.; Black, C.; Yang, D.; Shi, D.; Yan, B. Antiplatelet agents aspirin and clopidogrel are hydrolyzed by distinct carboxylesterases, and clopidogrel is transesterificated in the presence of ethyl alcohol. Pharmacol. Exp. Ther. 2006, 319, 1467–1476. [Google Scholar] [CrossRef] [PubMed]
- Kubo, T.; Kim, S.R.; Sai, K.; Saito, Y.; Nakajima, T.; Matsumoto, K.; Saito, H.; Shirao, K.; Yamamoto, N.; Minami, H.; et al. Functional characterization of three naturally occurring single nucleotide polymorphisms in the CES2 gene encoding carboxylesterase 2 (HCE-2). Drug Metab. Dispos. 2005, 33, 1482–1487. [Google Scholar] [CrossRef]
- Birkus, G.; Bam, R.A.; Willkom, M.; Frey, C.R.; Tsai, L.; Stray, K.M.; Yant, S.R.; Cihlar, T. Intracellular Activation of Tenofovir Alafenamide and the Effect of Viral and Host Protease Inhibitors. Antimicrob. Agents Chemother. 2015, 60, 316–322. [Google Scholar] [CrossRef]
- Li, Y.; Cao, L.; Li, G.; Cong, F.; Li, Y.; Sun, J.; Luo, Y.; Chen, G.; Li, G.; Wang, P.; et al. Remdesivir Metabolite GS-441524 Effectively Inhibits SARS-CoV-2 Infection in Mouse Models. J. Med. Chem. 2022, 65, 2785–2793. [Google Scholar] [CrossRef]
- Cox, R.M.; Wolf, J.D.; Lieber, C.M.; Sourimant, J.; Lin, M.J.; Babusis, D.; DuPont, V.; Chan, J.; Barrett, K.T.; Lye, D.; et al. Oral prodrug of remdesivir parent GS-441524 is efficacious against SARS-CoV-2 in ferrets. Nat. Commun. 2021, 12, 6415. [Google Scholar] [CrossRef]
- Gandhi, Z.; Mansuri, Z.; Bansod, S. Potential Interactions of Remdesivir with Pulmonary Drugs: A COVID-19 Perspective. SN Compr. Clin. Med. 2020, 2, 1707–1708. [Google Scholar] [CrossRef]
- Yang, K. What Do We Know About Remdesivir Drug Interactions? Clin. Transl. Sci. 2020, 13, 842–844. [Google Scholar] [CrossRef] [PubMed]
- Deb, S.; Reeves, A.A.; Hopefl, R.; Bejusca, R. ADME and Pharmacokinetic Properties of Remdesivir: Its Drug Interaction Potential. Pharmaceuticals 2021, 14, 655. [Google Scholar] [CrossRef] [PubMed]
- Leegwater, E.; Moes, D.J.A.R.; Bosma, L.B.E.; Ottens, T.H.; van der Meer, I.M.; van Nieuwkoop, C.; Wilms, E.B. Population Pharmacokinetics of Remdesivir and GS-441524 in Hospitalized COVID-19 Patients. Antimicrob. Agents Chemother. 2022, 66, e0025422. [Google Scholar] [CrossRef] [PubMed]
- Choe, P.G.; Jeong, S.I.; Kang, C.K.; Yang, L.; Lee, S.; Cho, J.Y.; Han, S.S.; Kim, D.K.; Lee, S.M.; Park, W.B.; et al. Exploration for the effect of renal function and renal replacement therapy on pharmacokinetics of remdesivir and GS-441524 in patients with COVID-19: A limited case series. Clin. Transl. Sci. 2022, 15, 732–740. [Google Scholar] [CrossRef]
- Xu, Y.; Barauskas, O.; Kim, C.; Babusis, D.; Murakami, E.; Kornyeyev, D.; Lee, G.; Stepan, G.; Perron, M.; Bannister, R.; et al. Off-Target In Vitro Profiling Demonstrates that Remdesivir Is a Highly Selective Antiviral Agent. Antimicrob. Agents Chemother. 2021, 65, e02237-20. [Google Scholar] [CrossRef]
- Hu, B.; Huang, S.; Yin, L. The cytokine storm and COVID-19. J. Med. Virol. 2021, 93, 250–256. [Google Scholar] [CrossRef]
- Miller, S.R.; McGrath, M.E.; Zorn, K.M.; Ekins, S.; Wright, S.H.; Cherrington, N.J. Remdesivir and EIDD-1931 Interact with Human Equilibrative Nucleoside Transporters 1 and 2: Implications for Reaching SARS-CoV-2 Viral Sanctuary Sites. Mol. Pharmacol. 2021, 100, 548–557. [Google Scholar] [CrossRef]
- Wang, A.Q.; Hagen, N.R.; Padilha, E.C.; Yang, M.; Shah, P.; Chen, C.Z.; Huang, W.; Terse, P.; Sanderson, P.; Zheng, W.; et al. Preclinical Pharmacokinetics and In Vitro Properties of GS-441524, a Potential Oral Drug Candidate for COVID-19 Treatment. Front. Pharmacol. 2022, 13, 918083. [Google Scholar] [CrossRef]
- Akinci, E.; Cha, M.; Lin, L.; Yeo, G.; Hamilton, M.C.; Donahue, C.J.; Bermudez-Cabrera, H.C.; Zanetti, L.C.; Chen, M.; Barkal, S.A.; et al. Elucidation of remdesivir cytotoxicity pathways through genome-wide CRISPR-Cas9 screening and transcriptomics. bioRxiv 2020. [Google Scholar] [CrossRef]
- Painter, W.P.; Holman, W.; Bush, J.A.; Almazedi, F.; Malik, H.; Eraut, N.C.J.E.; Morin, M.J.; Szewczyk, L.J.; Painter, G.R. Human Safety, Tolerability, and Pharmacokinetics of Molnupiravir, a Novel Broad-Spectrum Oral Antiviral Agent with Activity Against SARS-CoV-2. Antimicrob. Agents Chemother. 2021, 65, e02428-20. [Google Scholar] [CrossRef]
- Khiali, S.; Khani, E.; B Rouy, S.; Entezari-Maleki, T. Comprehensive review on molnupiravir in COVID-19: A novel promising antiviral to combat the pandemic. Future Microbiol. 2022, 17, 377–391. [Google Scholar] [CrossRef]
- Rossi, Á.D.; de Araújo, J.L.F.; de Almeida, T.B.; Ribeiro-Alves, M.; de Almeida Velozo, C.; Almeida, J.M.; de Carvalho Leitão, I.; Ferreira, S.N.; da Silva Oliveira, J.; Alves, H.J.; et al. Association between ACE2 and TMPRSS2 nasopharyngeal expression and COVID-19 respiratory distress. Sci. Rep. 2021, 11, 9658. [Google Scholar] [CrossRef] [PubMed]
- Li, L.Q.; Huang, T.; Wang, Y.Q.; Wang, Z.P.; Liang, Y.; Huang, T.B.; Zhang, H.Y.; Sun, W.; Wang, Y. COVID-19 patients’ clinical characteristics, discharge rate, and fatality rate of meta-analysis. J. Med. Virol. 2020, 92, 577–583. [Google Scholar] [CrossRef] [PubMed]
- Harmer, D.; Gilbert, M.; Borman, R.; Clark, K.L. Quantitative mRNA expression profiling of ACE 2, a novel homologue of angiotensin converting enzyme. FEBS Lett. 2002, 532, 107–110. [Google Scholar] [CrossRef]
- Lin, L.; Zeng, F.; Mai, L.; Gao, M.; Fang, Z.; Wu, B.; Huang, S.; Shi, H.; He, J.; Liu, Y.; et al. Expression of ACE2, TMPRSS2, and SARS-CoV-2 nucleocapsid protein in gastrointestinal tissues from COVID-19 patients and association with gastrointestinal symptoms. Am. J. Med. Sci. 2023, 366, 430–437. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Mady Traore, M.D.; Li, R.; Yuan, H.; He, M.; Wen, B.; Gao, W.; Jonsson, C.B.; Fitzpatrick, E.A.; Sun, D. Optimization of the Prodrug Moiety of Remdesivir to Improve Lung Exposure/Selectivity and Enhance Anti-SARS-CoV-2 Activity. J. Med. Chem. 2022, 65, 12044–12054. [Google Scholar] [CrossRef]
- Bakos, É.; Temesszentandrási-Ambrus, C.; Özvegy-Laczka, C.; Gáborik, Z.; Sarkadi, B.; Telbisz, Á. Interactions of the Anti-SARS-CoV-2 Agents Molnupiravir and Nirmatrelvir/Paxlovid with Human Drug Transporters. Int. J. Mol. Sci. 2023, 24, 11237. [Google Scholar] [CrossRef]
- Ravi, N.; Cortade, D.L.; Ng, E.; Wang, S.X. Diagnostics for SARS-CoV-2 detection: A comprehensive review of the FDA-EUA COVID-19 testing landscape. Biosens. Bioelectron. 2020, 165, 112454. [Google Scholar] [CrossRef]
- Loos, N.H.C.; Beijnen, J.H.; Schinkel, A.H. The inhibitory and inducing effects of ritonavir on hepatic and intestinal CYP3A and other drug-handling proteins. Biomed. Pharmacother. 2023, 162, 114636. [Google Scholar] [CrossRef]
- Loos, N.H.C.; Beijnen, J.H.; Schinkel, A.H. The Mechanism-Based Inactivation of CYP3A4 by Ritonavir: What Mechanism? Int. J. Mol. Sci. 2022, 23, 9866. [Google Scholar] [CrossRef]
- Ratain, M.J.; Greenblatt, D.J. Drug Interactions With a Short Course of Nirmatrelvir and Ritonavir: Prescribers and Patients Beware. J. Clin. Pharmacol. 2022, 62, 925–927. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.K.H.; Lau, J.J.; Au, I.C.H.; Lau, K.T.K.; Hung, I.F.N.; Peiris, M.; Leung, G.M.; Wu, J.T. Optimal timing of nirmatrelvir/ritonavir treatment after COVID-19 symptom onset or diagnosis: Target trial emulation. Nat. Commun. 2023, 14, 8377. [Google Scholar] [CrossRef] [PubMed]
| Variant | Prevalence | Drug Metabolism | Note | |
|---|---|---|---|---|
| G143E | Amino acid glycine (Gly) at position 143 is replaced by glutamic acid (Glu) | Relatively low frequency of G143E heterozygotes [43,44,45,46,47,48]. G143E is rare in most populations but shows measurable frequencies in Europeans (3–4%) and Hispanics (1–2%) | Potentially influence remdesivir activation and efficacy, thus, requiring dose adjustment based on individual genetic profile for better efficacy or to avoid adverse reactions. | |
| rs2244613 (c.1165-33C>A) | Single-nucleotide polymorphism (SNP) in the CES1 gene that involves a change of C to A at position 1165-33. | Low frequency of rs2244613 heterozygotes and homozygotes [47,49,50,51] Approximately 20–25% of Europeans, 15–20% of East Asians, and 10–15% of Africans have rs2244613. | ||
| c.662A>G | SNP in the CES1 gene, which involves a change of A to G at position 662. | Relatively common, with the homozygous for c.662AA being the most prevalent [54]. |
| |
| rs8192935 | SNP variants in the CES1 gene | Relatively common |
| |
| rs4784563 | Fairly common | |||
| rs4580160 | Fairly common |
| ||
| rs4122238 | Fairly common, but rs4122238 can exhibit different minor allele frequency values depending on the specific population studied [55,56] | |||
| Variant | Prevalence | Drug Metabolism | Note | |
|---|---|---|---|---|
| rs2241409 (c.1613-108G>A) | SNP in the CES2 gene, which involves a change of G to A at position c.1613-108. | Relatively low frequency of rs2241409 homozygotes. rs2241409 is more frequent in Asian populations. |
| Potentially influence remdesivir and molnupiravir activation and efficacy |
| rs11075646 (c.-806C>G, previously referred to as c.-823C>G) | SNP in the CES2 gene, which involves a change of C to G in the 5′-untranslated region. | Very low frequency |
| |
| ∆458-473 | Deletion of residues 458–473 in the CES2 enzyme. |
| ||
| A139T and F458V | Two specific nonsynonymous SNPs in CES2 |
| ||
| rs72547531 (c.100C>T, R34W) | SNP in the CES2 gene, which involves a change of C to T. |
| ||
| rs72547532 (c.424G>A, V142M) | SNP in the CES2 gene, which involves a change of G to A. | |||
| IVS8-2A>G | Deletion of 32 base pairs in exon 9 at the splice acceptor site in intron 8, resulting in aberrant splicing, producing truncated CES2 proteins |
| ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shen, Y.; Eades, W.; Dinh, L.; Yan, B. Carboxylesterase Factors Influencing the Therapeutic Activity of Common Antiviral Medications Used for SARS-CoV-2 Infection. Pharmaceutics 2025, 17, 832. https://doi.org/10.3390/pharmaceutics17070832
Shen Y, Eades W, Dinh L, Yan B. Carboxylesterase Factors Influencing the Therapeutic Activity of Common Antiviral Medications Used for SARS-CoV-2 Infection. Pharmaceutics. 2025; 17(7):832. https://doi.org/10.3390/pharmaceutics17070832
Chicago/Turabian StyleShen, Yue, William Eades, Linh Dinh, and Bingfang Yan. 2025. "Carboxylesterase Factors Influencing the Therapeutic Activity of Common Antiviral Medications Used for SARS-CoV-2 Infection" Pharmaceutics 17, no. 7: 832. https://doi.org/10.3390/pharmaceutics17070832
APA StyleShen, Y., Eades, W., Dinh, L., & Yan, B. (2025). Carboxylesterase Factors Influencing the Therapeutic Activity of Common Antiviral Medications Used for SARS-CoV-2 Infection. Pharmaceutics, 17(7), 832. https://doi.org/10.3390/pharmaceutics17070832