
Preparation and Evaluation of Long-Acting Injectable Levocetirizine Prodrug Formulation


Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
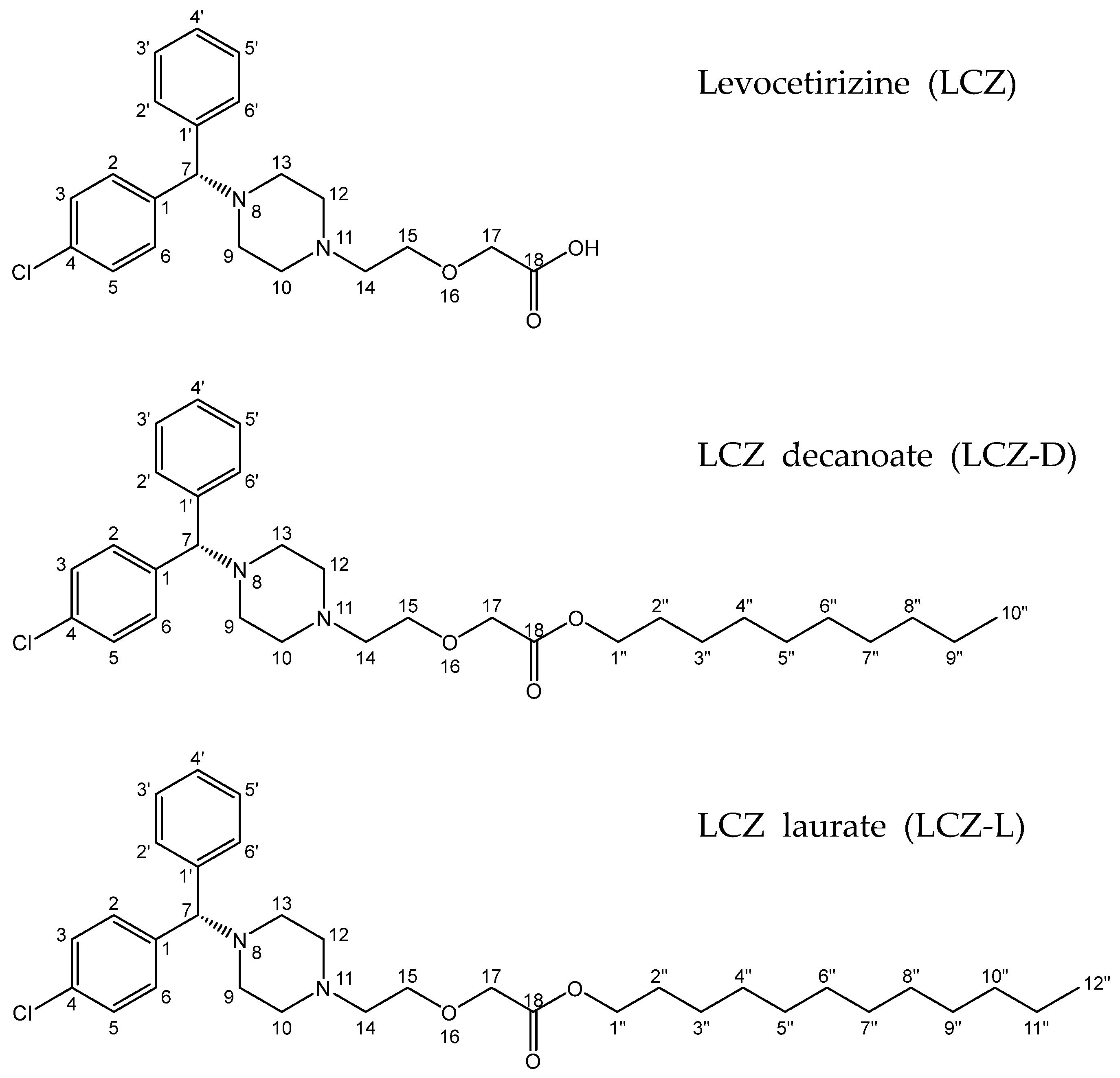
2.2. Synthesis of Levocetirizine Prodrugs
2.3. Nuclear Magnetic Resonance Spectroscopy
2.4. Fourier Transformed Infrared Spectroscopy
2.5. Differential Scanning Calorimetry
2.6. Hydrolysis of Prodrug in Porcine Liver Esterase (PLE)
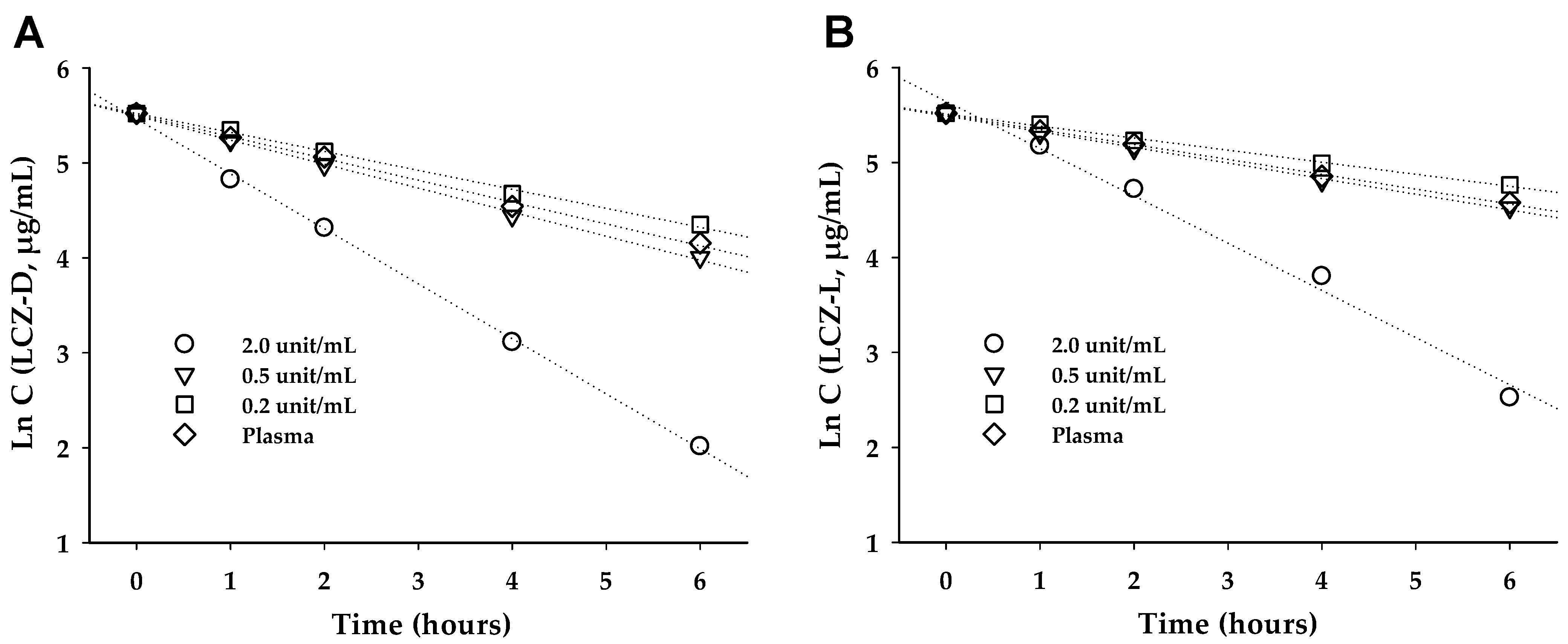
2.7. Hydrolysis of Prodrugs in Rat Plasma
2.8. Preparation of Formulation
2.9. Pharmacokinetic Study
2.10. Stability Test
2.11. HPLC, LC-MS/MS Condition and Data Analysis
3. Results
3.1. Nuclear Magnetic Resonance Spectroscopy
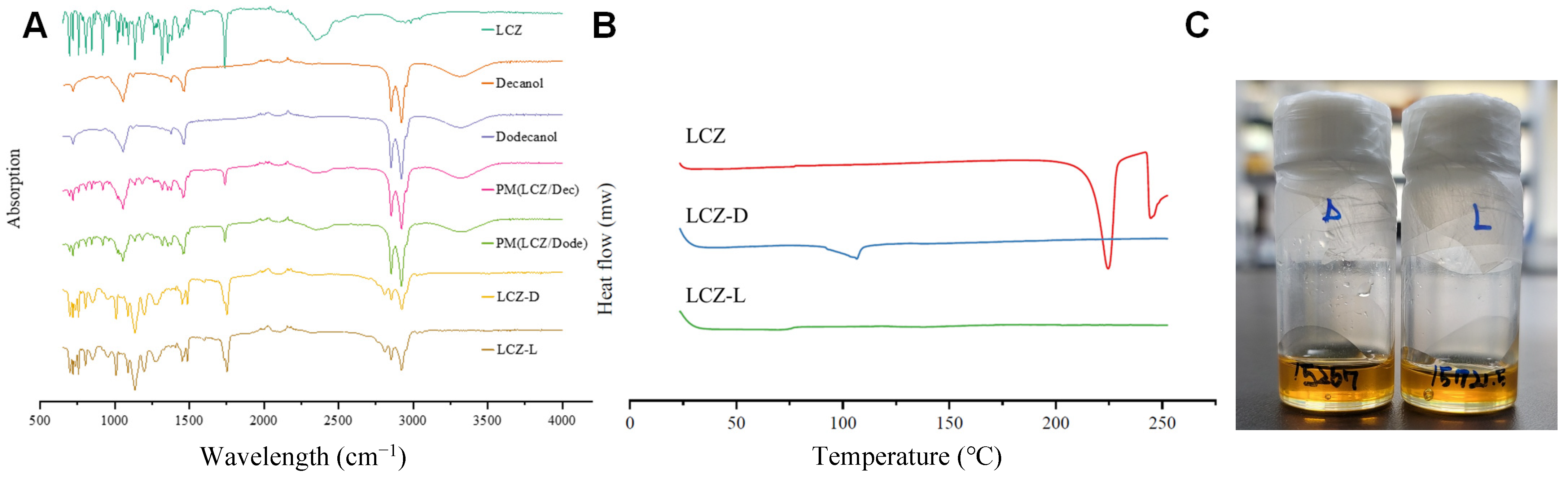
3.2. Fourier Transform Infrared Spectroscopy
3.3. Differential Scanning Calorimetry (DSC) and Appearance
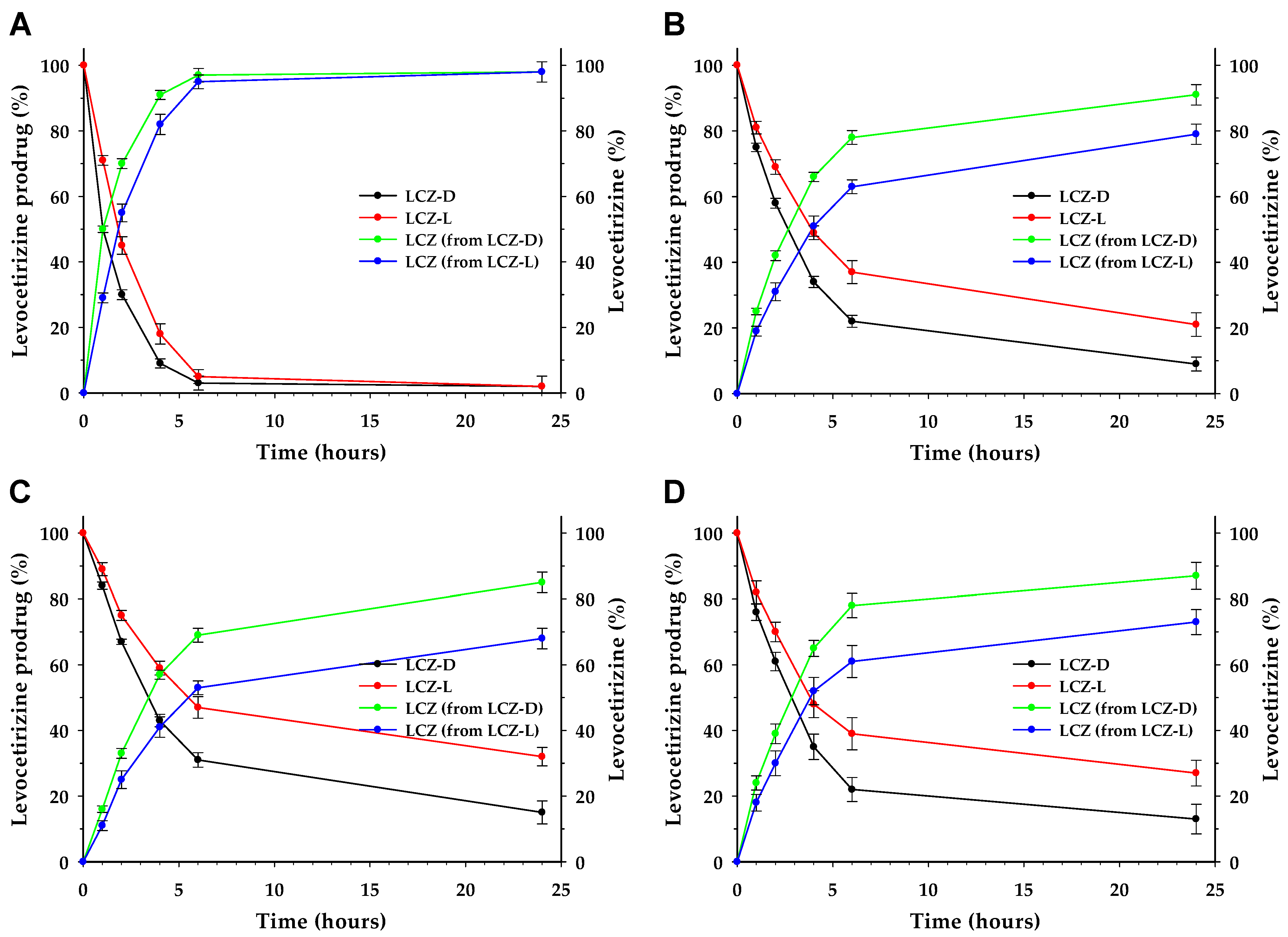
3.4. LCZ Prodrug Hydrolysis
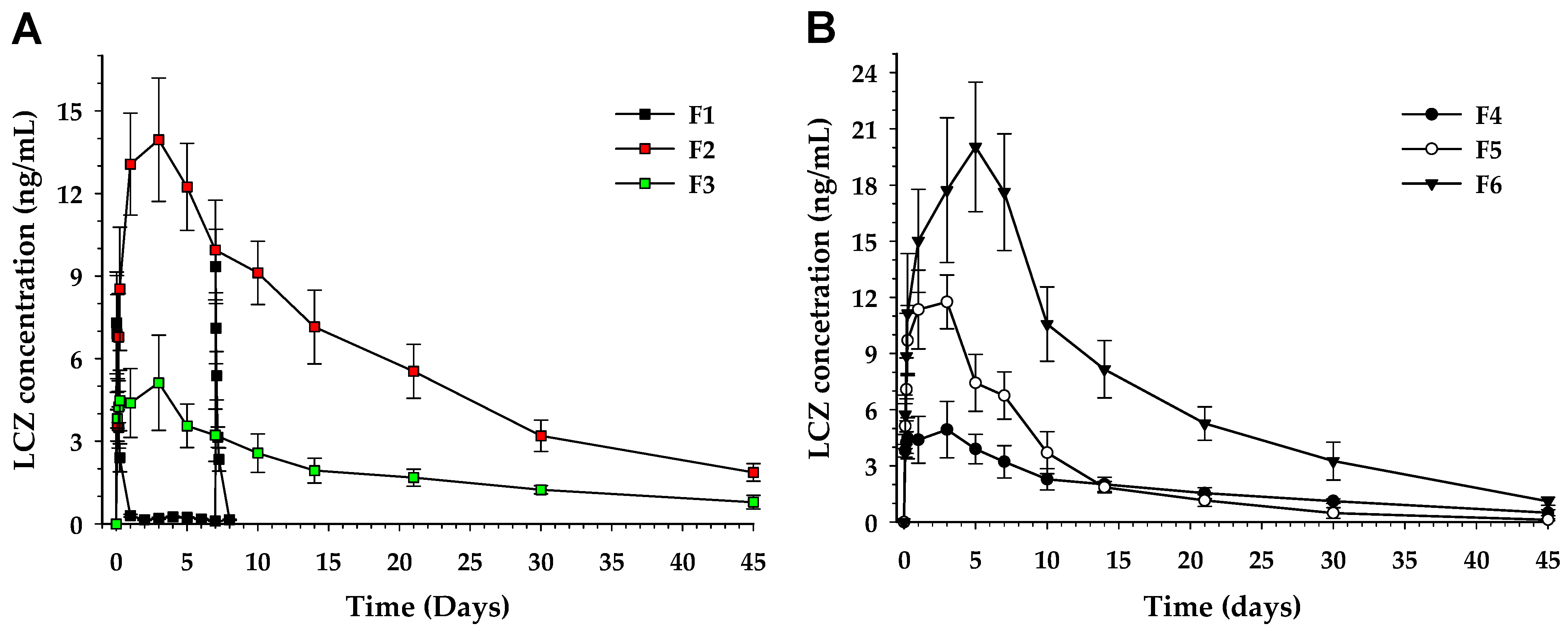
3.5. Pharmacokinetics
3.6. Stability
4. Discussion
5. Conclusions
Supplementary Materials
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ARIA | Allergic Rhinitis and its Impact on Asthma |
| LCZ | Levocetirizine |
| LCZ-D | Levocetirizine decanoate |
| LCZ-L | Levocetirizine laurate |
| BB | Benzyl benzoate |
| BA | Benzyl alcohol |
| TW80 | Polysorbate 80 |
| DMSO | dimethylsulfoxide |
| PLE | Porcine liver esterase |
| BHT | Butylated hydroxytoluene |
| CO | Castor oil |
| MC | Chloromethane |
| SD | Sprague Dawley |
| TMS | Tetramethylsilane |
| MeOH | Methanol |
| DSC | Differential scanning calorimetry |
| Eq | Equivalent |
| HPLC | High-Performance Liquid Chromatography |
| EC50 | Half Maximal Effective Concentration |
| HED | Human equivalent dose |
References
- Bousquet, J.; Anto, J.M.; Bachert, C.; Baiardini, I.; Bosnic-Anticevich, S.; Canonica, G.W.; Melén, E.; Palomares, O.; Scadding, G.K.; Togias, A.; et al. Allergic rhinitis. Nat. Rev. Dis. Primers 2020, 6, 95. [Google Scholar] [CrossRef]
- Kim, S.Y.; Yoon, S.-J.; Jo, M.-W.; Kim, E.-J.; Kim, H.-J.; Oh, I.-H. Economic burden of allergic rhinitis in Korea. Am. J. Rhinol. Allergy 2010, 24, e110-3. [Google Scholar] [CrossRef] [PubMed]
- Blaiss, M.S.; Bernstein, J.A.; Kessler, A.; Pines, J.M.; Camargo, C.A.; Fulgham, P.; Haumschild, R.; Rupp, K.; Tyler, T.; Moellman, J. The Role of Cetirizine in the Changing Landscape of IV Antihistamines: A Narrative Review. Adv. Ther. 2022, 39, 178–192. [Google Scholar] [CrossRef]
- Kim, B.K.; Kim, J.-Y.; Kang, M.-K.; Yang, M.-S.; Park, H.-W.; Min, K.-U.; Cho, S.-H.; Kang, H.-R. Allergies are still on the rise? A 6-year nationwide population-based study in Korea. Allergol. Int. 2016, 65, 186–191. [Google Scholar]
- Dierick, B.J.; van der Molen, T.; Blok, B.M.J.F.-D.; Muraro, A.; Postma, M.J.; Kocks, J.W.H.; van Boven, J.F.M. Burden and socioeconomics of asthma, allergic rhinitis, atopic dermatitis and food allergy. Expert Rev. Pharmacoeconomics Outcomes Res. 2020, 20, 437–453. [Google Scholar] [CrossRef] [PubMed]
- Schuler Iv, C.F.; Montejo, J.M. Allergic Rhinitis in Children and Adolescents. Pediatr. Clin. North Am. 2019, 66, 981–993. [Google Scholar] [CrossRef] [PubMed]
- Bousquet, J.J.; Schünemann, H.J.; Togias, A.; Erhola, M.; Hellings, P.W.; Zuberbier, T.; Agache, I.; Ansotegui, I.J.; Anto, J.M.; Bachert, C.; et al. Next-generation ARIA care pathways for rhinitis and asthma: A model for multimorbid chronic diseases. Clin. Transl. Allergy 2019, 9, 44. [Google Scholar] [CrossRef]
- Siddiqui, Z.; Walker, A.; Pirwani, M.; Tahiri, M.; Syed, I. Allergic rhinitis: Diagnosis and management. Br. J. Hosp. Med. 2022, 83, 1–9. [Google Scholar] [CrossRef]
- Bernstein, J.A.; Bernstein, J.S.; Makol, R.; Ward, S. Allergic Rhinitis: A Review. JAMA 2024, 331, 866–877. [Google Scholar] [CrossRef]
- Branco, A.; Yoshikawa, F.S.Y.; Pietrobon, A.J.; Sato, M.N. Role of Histamine in Modulating the Immune Response and Inflammation. Mediators Inflamm. 2018, 2018, 9524075. [Google Scholar] [CrossRef]
- Brozek, J.L.; Bousquet, J.; Agache, I.; Agarwal, A.; Bachert, C.; Bosnic-Anticevich, S.; Brignardello-Petersen, R.; Canonica, G.W.; Casale, T.; Chavannes, N.H.; et al. Allergic Rhinitis and its Impact on Asthma (ARIA) guidelines-2016 revision. J. Allergy Clin. Immunol. 2017, 140, 950–958. [Google Scholar] [CrossRef] [PubMed]
- Goniotakis, I.; Perikleous, E.; Fouzas, S.; Steiropoulos, P.; Paraskakis, E. A Clinical Approach of Allergic Rhinitis in Children. Children 2023, 10, 1571. [Google Scholar] [CrossRef] [PubMed]
- Liva, G.A.; Karatzanis, A.D.; Prokopakis, E.P. Review of Rhinitis: Classification, Types, Pathophysiology. J. Clin. Med. 2021, 10, 3183. [Google Scholar] [CrossRef] [PubMed]
- Hiraoka, K.; Tashiro, M.; Grobosch, T.; Maurer, M.; Oda, K.; Toyohara, J.; Ishii, K.; Ishiwata, K.; Yanai, K. Brain histamine H1 receptor occupancy measured by PET after oral administration of levocetirizine, a non-sedating antihistamine. Expert Opin. Drug Saf. 2015, 14, 199–206. [Google Scholar] [CrossRef]
- Wang, S.; Liu, R.; Fu, Y.; Kao, W.J. Release mechanisms and applications of drug delivery systems for extended-release. Expert Opin. Drug Deliv. 2020, 17, 1289–1304. [Google Scholar] [CrossRef]
- Malaya, E.; Piątkowska, A.; Panek, M.; Kuna, P.; Kupczyk, M.; Kardas, G. Medication adherence in allergic diseases and asthma: A literature review. Front. Pharmacol. 2024, 15, 1488665. [Google Scholar] [CrossRef]
- Fox, M.G.; Cass, L.M.; Sykes, K.J.; Cummings, E.L.; Fassas, S.N.; Nallani, R.; Smith, J.B.; Chiu, A.G.; Villwock, J.A. Factors affecting adherence to intranasal treatment for allergic rhinitis: A qualitative study. Laryngoscope Investig. Otolaryngol. 2023, 8, 40–45. [Google Scholar] [CrossRef]
- Zuberbier, T.; Lötvall, J.; Simoens, S.; Subramanian, S.V.; Church, M.K. Economic burden of inadequate management of allergic diseases in the European Union: A GA2LEN review. Allergy 2014, 69, 1275–1279. [Google Scholar] [CrossRef]
- Rishovd, A. Pediatric intramuscular injections: Guidelines for best practice. MCN Am. J. Matern. Child Nurs. 2014, 39, 107–112, quiz 113-4. [Google Scholar] [CrossRef]
- Yang, J.; Zeng, H.; Luo, Y.; Chen, Y.; Wang, M.; Wu, C.; Hu, P. Recent Applications of PLGA in Drug Delivery Systems. Polymers 2024, 16, 2606. [Google Scholar] [CrossRef]
- Hu, F.; Qi, J.; Lu, Y.; He, H.; Wu, W. PLGA-based implants for sustained delivery of peptides/proteins: Current status, challenge and perspectives. Chin. Chem. Lett. 2023, 34, 108250. [Google Scholar] [CrossRef]
- Rahmani, F.; Naderpour, S.; Nejad, B.G.; Rahimzadegan, M.; Ebrahimi, Z.N.; Kamali, H.; Nosrati, R. The recent insight in the release of anticancer drug loaded into PLGA microspheres. Med. Oncol. 2023, 40, 229. [Google Scholar] [CrossRef] [PubMed]
- Nele, V.; Campani, V.; Moosavian, S.A.; De Rosa, G. Lipid nanoparticles for RNA delivery: Self-assembling vs driven-assembling strategies. Adv. Drug Deliv. Rev. 2024, 208, 115291. [Google Scholar] [CrossRef]
- Masson, P.; Shaihutdinova, Z.; Lockridge, O. Drug and pro-drug substrates and pseudo-substrates of human butyrylcholinesterase. Biochem. Pharmacol. 2023, 218, 115910. [Google Scholar] [CrossRef]
- Perez, C.; Daniel, K.B.; Cohen, S.M. Evaluating prodrug strategies for esterase-triggered release of alcohols. ChemMedChem 2013, 8, 1662–1667. [Google Scholar] [CrossRef] [PubMed]
- Markovic, M.; Ben-Shabat, S.; Dahan, A. Prodrugs for Improved Drug Delivery: Lessons Learned from Recently Developed and Marketed Products. Pharmaceutics 2020, 12, 1031. [Google Scholar] [CrossRef]
- Al-Arafi, N.; Salimon, J. Production of Oleic Acid Based Wax Ester Using Acidic Homogeneous Catalysts. J. Chem. 2012, 9, 99–106. [Google Scholar] [CrossRef]
- Morris, A.P.; Brain, K.R.; Heard, C.M. Synthesis of haloperidol prodrugs and their hydrolysis by porcine liver esterase. Drug Metab. Lett. 2008, 2, 275–279. [Google Scholar] [CrossRef]
- U.S. Food and Drug Administration. AVEED (Testosterone Undecanoate) Injection Label. 2014. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2014/022219s000lbl.pdf (accessed on 15 June 2025).
- U.S. Food and Drug Administration. FASLODEX (Fulvestrant) Injection Label. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2002/21344lbl.pdf (accessed on 25 April 2002).
- Kumar, M.; Visth, S.; Ali, S.; Agarwal, S.; Bhola, A. Preparation and evaluation of fast dissolving drug delivery system containing levocetrizine HCl. Int. J. Pharm. Pharm. Sci. 2010, 2, 109–111. [Google Scholar]
- Sharma, V.; Chopra, H. Formulation and Evaluation of Taste Masked Mouth Dissolving Tablets of Levocetirizine Hydrochloride. Iran. J. Pharm. Res. IJPR 2012, 11, 457–463. [Google Scholar]
- Reissmann, S.; Greiner, G. Hydrolysis of peptide esters by different enzymes. Int. J. Pept. Protein Res. 1992, 40, 110–113. [Google Scholar] [CrossRef] [PubMed]
- Srividhya, J.; Schnell, S. Why substrate depletion has apparent first-order kinetics in enzymatic digestion. Comput. Biol. Chem. 2006, 30, 209–214. [Google Scholar] [CrossRef]
- Segel, I.H. Enzyme Kinetics: Behavior and Analysis of Rapid Equilibrium and Steady State Enzyme Systems; Wiley Classics Library, Ed.; Wiley: New York, NY, USA, 1993; p. 957. [Google Scholar]
- Eilertsen, J.; Schnell, S.; Walcher, S. The Michaelis-Menten Reaction at Low Substrate Concentrations: Pseudo-First-Order Kinetics and Conditions for Timescale Separation. Bull. Math. Biol. 2024, 86, 68. [Google Scholar] [CrossRef]
- Petri, Y.D.; Verresen, R.; Gutierrez, C.S.; Kojasoy, V.; Zhang, E.; Abularrage, N.S.; Wralstad, E.C.; Weiser, K.R.; Raines, R.T. Mammalian Esterase Activity: Implications for Peptide Prodrugs. Biochemistry 2024, 63, 2580–2593. [Google Scholar] [CrossRef] [PubMed]
- Weidolf, L.; Andersson, T.; Bercu, J.P.; Brink, A.; Glowienke, S.; Harvey, J.; Hayes, M.A.; Jacques, P.; Lu, C.; Manevski, N.; et al. Qualification of impurities based on metabolite data. Regul. Toxicol. Pharmacol. 2020, 110, 104524. [Google Scholar] [CrossRef]
- Kalicharan, R.W.; Baron, P.; Oussoren, C.; Bartels, L.; Vromans, H. Spatial distribution of oil depots monitored in human muscle using MRI. Int. J. Pharm. 2016, 505, 52–60. [Google Scholar] [CrossRef] [PubMed]
- Yáñez, J.A.; Remsberg, C.M.; Sayre, C.L.; Forrest, M.L.; Davies, N.M. Flip-flop pharmacokinetics--delivering a reversal of disposition: Challenges and opportunities during drug development. Ther. Deliv. 2011, 2, 643–672. [Google Scholar] [CrossRef]
- Simons, F.E.R.; Simons, K.J. Levocetirizine: Pharmacokinetics and pharmacodynamics in children age 6 to 11 years. J. Allergy Clin. Immunol. 2005, 116, 355–361. [Google Scholar] [CrossRef]
- Iwatsubo, T.; Suzuki, H.; Sugiyama, Y. Prediction of Species Differences (Rats, Dogs, Humans) in the In Vivo Metabolic Clearance of YM796 by the Liver from In Vitro Data. J. Pharmacol. Exp. Ther. 1997, 283, 462–469. [Google Scholar] [CrossRef]
- Sharma, V.; McNeill, J.H. To scale or not to scale: The principles of dose extrapolation. Br. J. Pharmacol. 2009, 157, 907–921. [Google Scholar] [CrossRef]
- Reagan-Shaw, S.; Nihal, M.; Ahmad, N. Dose translation from animal to human studies revisited. Faseb J. 2008, 22, 659–661. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Formulation | Composition | |||
|---|---|---|---|---|
| Drug | Excipient | |||
| F1 | LCZ 2HCl | 150.0 mg | Distilled water | 850.0 mg |
| F2 | LCZ-D | 171.9 mg | BB:CO (6:4) | 828.1 mg |
| F3 | LCZ-L | 181.0 mg | BB:CO (6:4) | 819.0 mg |
| F4 | LCZ-L | 181.0 mg | BB:CO (6:4) | 319.0 mg |
| F5 | LCZ-L | 181.0 mg | BB:CO (6:4), TW80 | 804.0 mg, 15.0 mg |
| F6 | LCZ-L | 181.0 mg | BB:CO (6:4), BA | 789.0 mg, 30.0 mg |
| F7 | LCZ-L | 181.0 mg | BB:CO (6:4), BHT | 818.7 mg, 0.3 mg |
| Formulation | Parameter | ||||
|---|---|---|---|---|---|
| Cmax (ng/mL) | Tmax (day) | AUC1d (h·ng/mL) | AUC0–30d (h·ng/mL) | AUC0–45d (h·ng/mL) | |
| F1 | 7.31 ± 1.85 (1d) 9.35 ± 1.32 (7d) | 0.083 | 52.72 ± 11.86 (0–1d) 47.78 ± 8.10 (7–8d) | (1581.75 *) (1433.33 *) | - |
| F2 | 13.95 ± 2.24 | 3 | - | 5510.53 ± 1063.11 | 6423.12 ± 1063.11 |
| F3 | 5.12 ± 1.73 | 3 | - | 1743.82 ± 497.95 | 2109.22 ± 497.96 |
| F4 | 4.94 ± 1.51 | 3 | - | 1697.05 ± 434.96 | 1987.75 ± 434.96 |
| F5 | 11.76 ± 2.44 | 3 | - | 2652.42 ± 596.15 | 2761.46 ± 596.15 |
| F6 | 20.03 ± 3.46 | 5 | - | 6832.15 ± 1539.69 | 7618.75 ± 1539.69 |
| Formulation | Condition (°C) | Impurity (=LCZ) | ||||
|---|---|---|---|---|---|---|
| 0 Week | 1 Week | 2 Week | 4 Week | 6 Week | ||
| F3 | 25 | 0% | 0.19% | 0.24% | 0.35% | 0.58% |
| 40 | 0% | 0.42% | 0.60% | 0.88% | 1.23% | |
| 60 | 0% | 0.65% | 1.15% | 2.67% | 4.72% | |
| F7 | 25 | 0% | 0.06% | 0.07% | 0.09% | 0.12% |
| 40 | 0% | 0.09% | 0.14% | 0.22% | 0.31% | |
| 60 | 0% | 0.23% | 0.41% | 0.73% | 1.17% | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahn, J.-h. Preparation and Evaluation of Long-Acting Injectable Levocetirizine Prodrug Formulation. Pharmaceutics 2025, 17, 806. https://doi.org/10.3390/pharmaceutics17070806
Ahn J-h. Preparation and Evaluation of Long-Acting Injectable Levocetirizine Prodrug Formulation. Pharmaceutics. 2025; 17(7):806. https://doi.org/10.3390/pharmaceutics17070806
Chicago/Turabian StyleAhn, Jun-hyun. 2025. "Preparation and Evaluation of Long-Acting Injectable Levocetirizine Prodrug Formulation" Pharmaceutics 17, no. 7: 806. https://doi.org/10.3390/pharmaceutics17070806
APA StyleAhn, J.-h. (2025). Preparation and Evaluation of Long-Acting Injectable Levocetirizine Prodrug Formulation. Pharmaceutics, 17(7), 806. https://doi.org/10.3390/pharmaceutics17070806