
Targeting S. aureus Extracellular Vesicles: A New Putative Strategy to Counteract Their Pathogenic Potential

 ,
,  , , , and
, , , and _Di_Marco.png)
Abstract
1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Bacterial Growth, Bacterial Suspensions, and Free Cells Supernatant (FCS) Preparation
2.3. BEVs Isolation
2.4. Proteomic Analysis of BEVs by SDS Page and LC-MS/MS
2.5. BEVs Treatment with HAc40, HA, and c40
2.6. Dynamic Light Scattering Method
2.7. Treated BEVs on HaCaT Cells
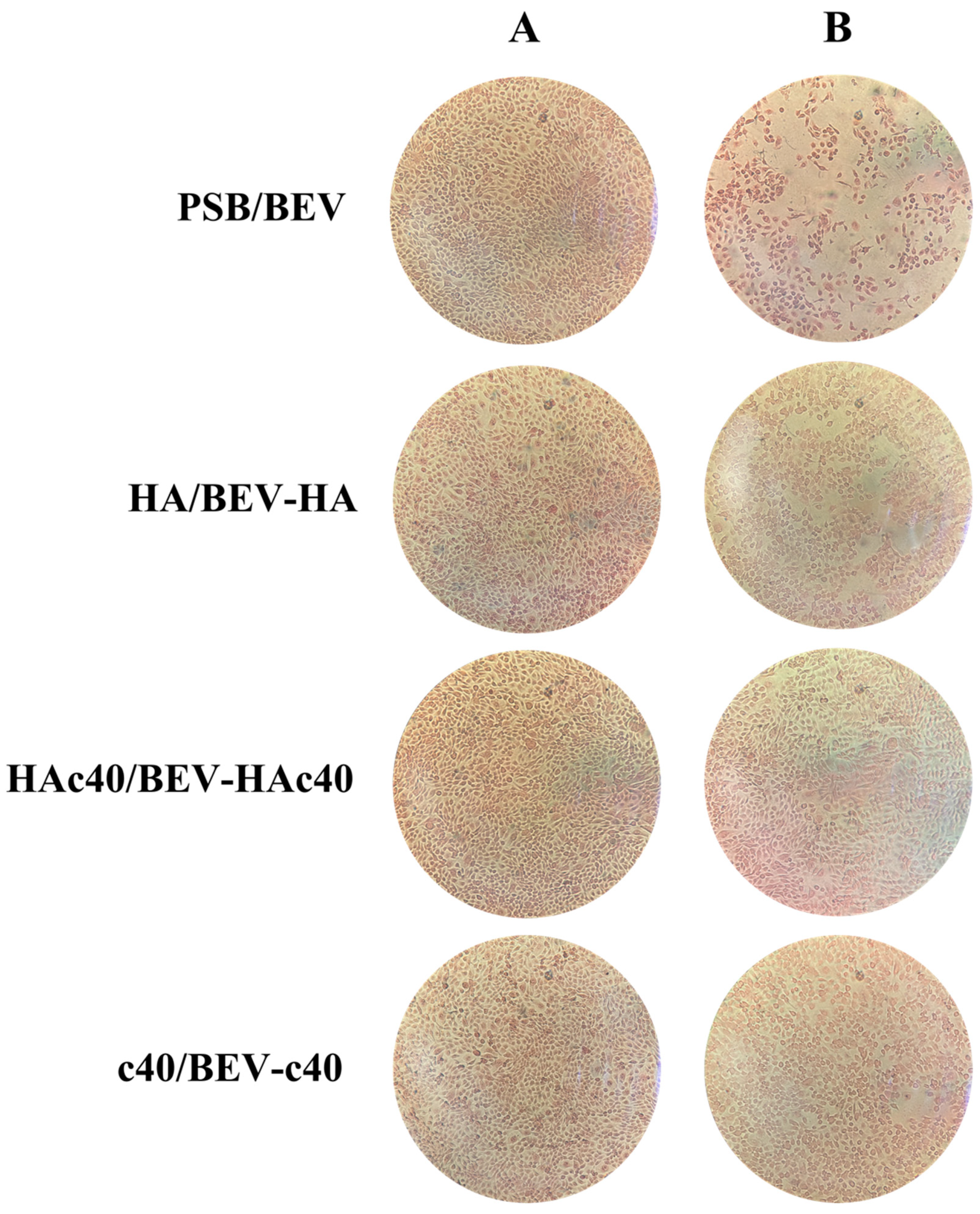
2.8. Microscopic and Image Analysis for Adherent Cell Evaluation
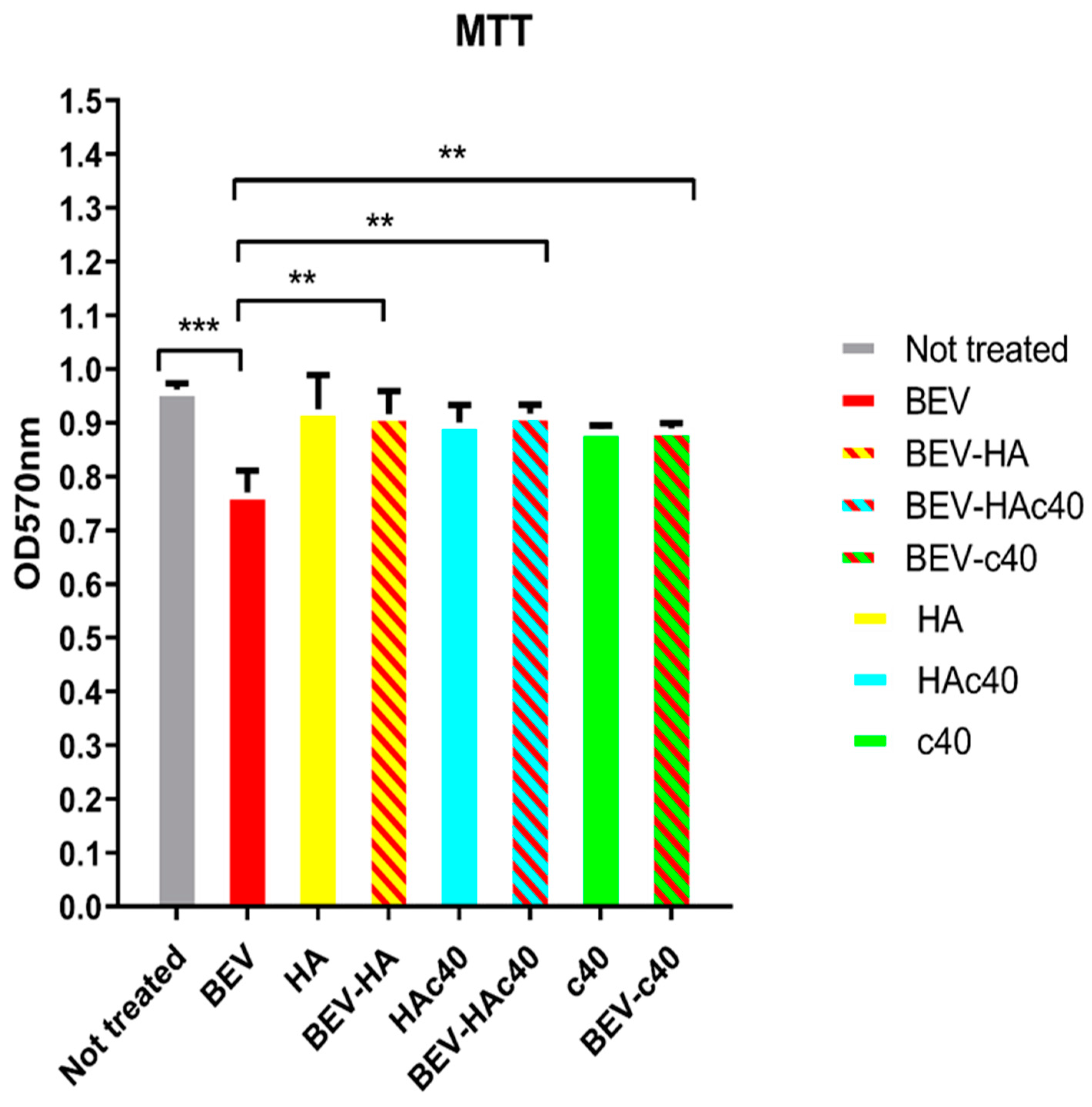
2.9. MTT Assay
2.10. RT-qPCR TJP1 ZO1 and Claudin 1
2.11. Statistical Analysis
3. Results
3.1. Protein Analysis
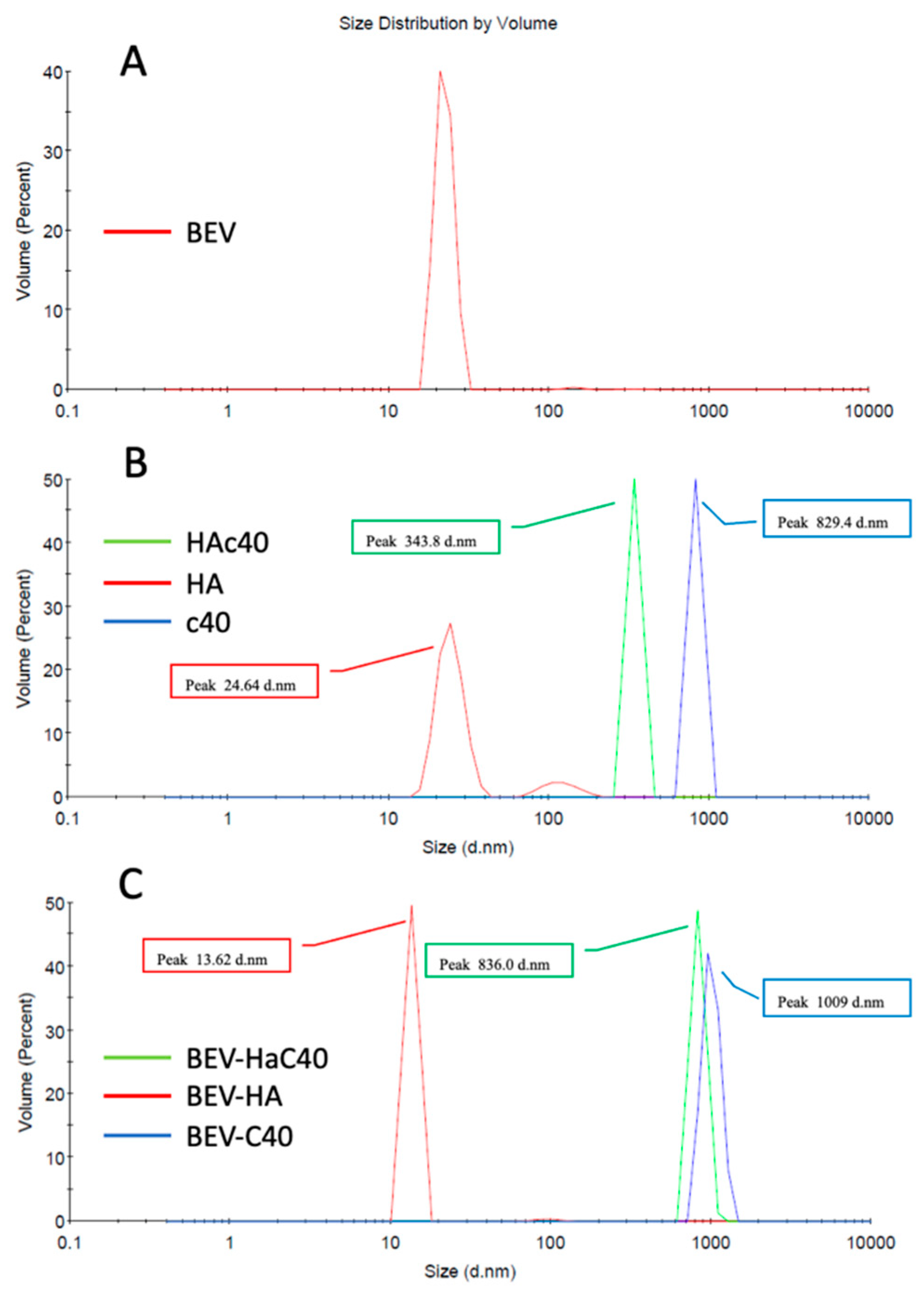
3.2. Dynamic Light Scattering Method (DLS)
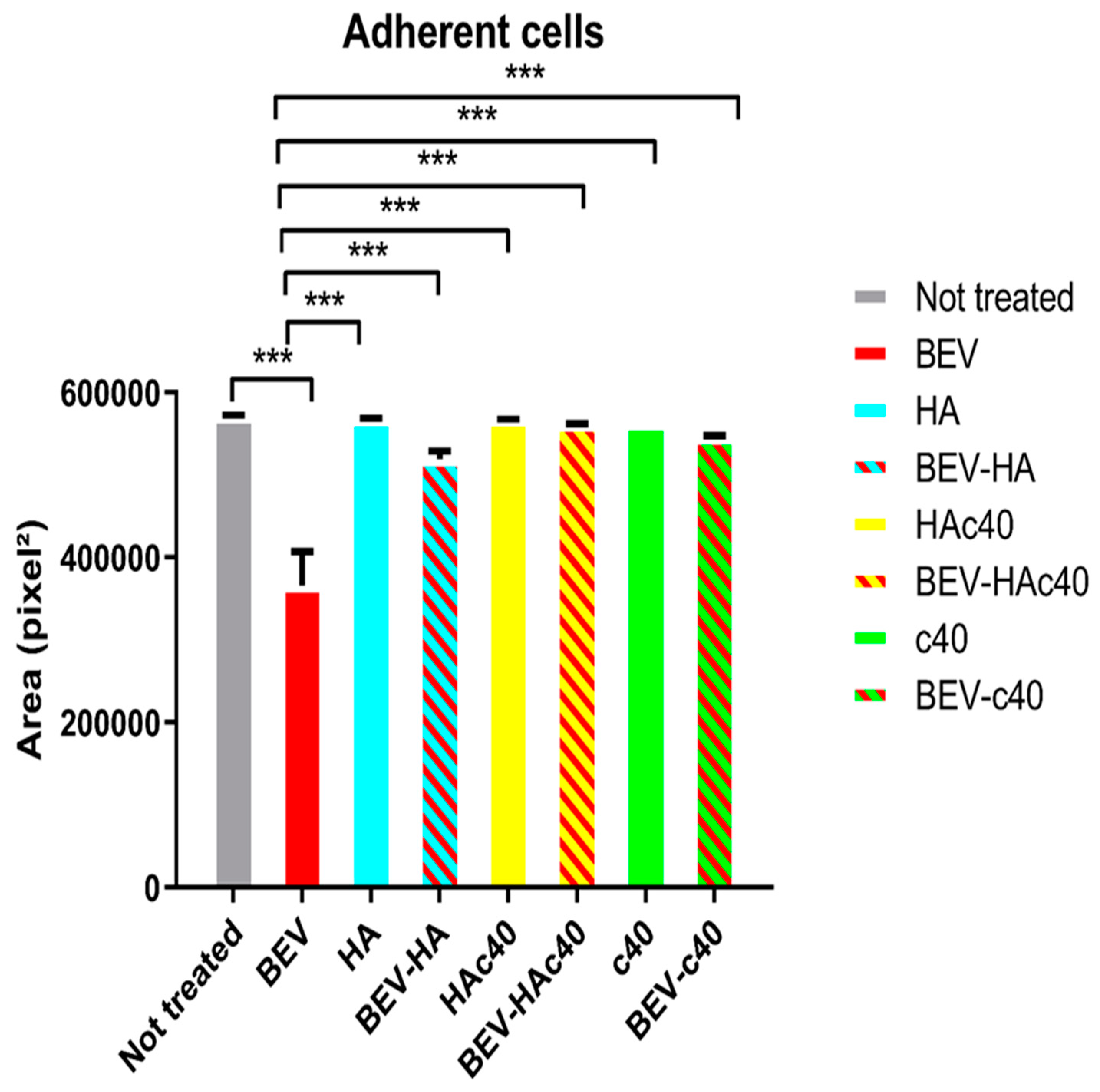
3.3. Assessment of Adherent Cells by Microscopic Evaluation and Image Analysis
3.4. MTT Assay
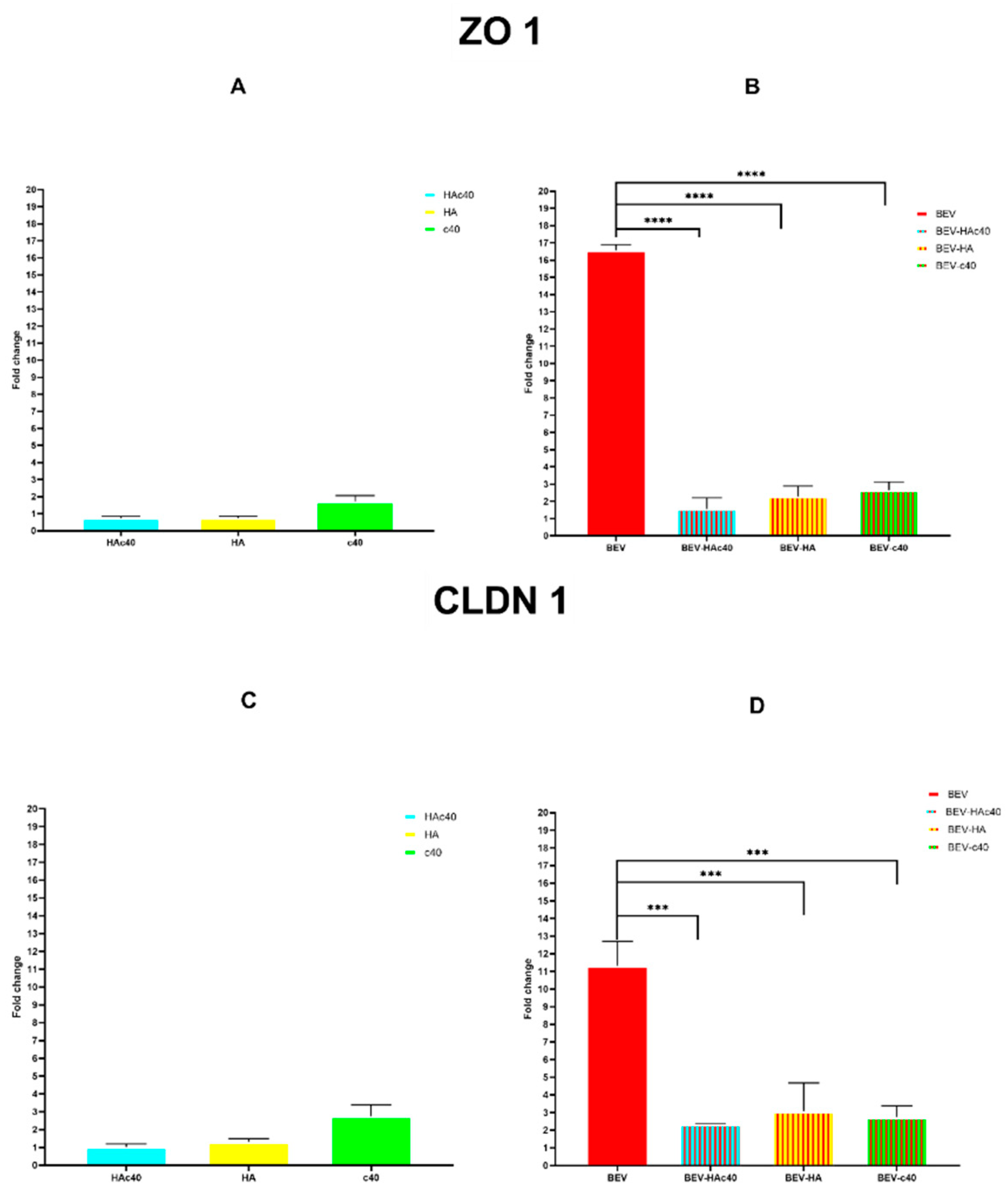
3.5. mRNA Expression Levels of ZO-1 and CLDN1
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Weidinger, S.; Beck, L.A.; Bieber, T.; Kabashima, K.; Irvine, A.D. Atopic Dermatitis. Nat. Rev. Dis. Primers 2018, 4, 1. [Google Scholar] [CrossRef]
- Geoghegan, J.A.; Irvine, A.D.; Foster, T.J. Staphylococcus aureus and Atopic Dermatitis: A Complex and Evolving Relationship. Trends Microbiol. 2018, 26, 484–497. [Google Scholar] [CrossRef] [PubMed]
- Magnifico, I.; Petronio Petronio, G.; Venditti, N.; Cutuli, M.A.; Pietrangelo, L.; Vergalito, F.; Mangano, K.; Zella, D.; Di Marco, R. Atopic Dermatitis as a Multifactorial Skin Disorder. Can the Analysis of Pathophysiological Targets Represent the Winning Therapeutic Strategy? Pharmaceuticals 2020, 13, 411. [Google Scholar] [CrossRef]
- Foster, T.J.; Geoghegan, J.A.; Ganesh, V.K.; Höök, M. Adhesion, Invasion and Evasion: The Many Functions of the Surface Proteins of Staphylococcus aureus. Nat. Rev. Microbiol. 2014, 12, 49–62. [Google Scholar] [CrossRef] [PubMed]
- Iwatsuki, K.; Yamasaki, O.; Morizane, S.; Oono, T. Staphylococcal Cutaneous Infections: Invasion, Evasion and Aggression. J. Dermatol. Sci. 2006, 42, 203–214. [Google Scholar] [CrossRef] [PubMed]
- Bukowski, M.; Wladyka, B.; Dubin, G. Exfoliative Toxins of Staphylococcus aureus. Toxins 2010, 2, 1148–1165. [Google Scholar] [CrossRef] [PubMed]
- Nakatsuji, T.; Chen, T.H.; Narala, S.; Chun, K.A.; Two, A.M.; Yun, T.; Shafiq, F.; Kotol, P.F.; Bouslimani, A.; Melnik, A.V.; et al. Antimicrobials from Human Skin Commensal Bacteria Protect against Staphylococcus aureus and Are Deficient in Atopic Dermatitis. Sci. Transl. Med. 2017, 9, eaah4680. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.-W.; Choi, E.-B.; Min, T.-K.; Kim, J.-H.; Kim, M.-H.; Jeon, S.G.; Lee, B.-J.; Gho, Y.S.; Jee, Y.-K.; Pyun, B.-Y.; et al. An Important Role of α-Hemolysin in Extracellular Vesicles on the Development of Atopic Dermatitis Induced by Staphylococcus aureus. PLoS ONE 2014, 9, e100499. [Google Scholar] [CrossRef]
- Tam, K.; Torres, V.J. Staphylococcus aureus Secreted Toxins and Extracellular Enzymes. Microbiol. Spectr. 2019, 7, 10–1128. [Google Scholar] [CrossRef]
- Lee, E.-Y.; Choi, D.-Y.; Kim, D.-K.; Kim, J.-W.; Park, J.O.; Kim, S.; Kim, S.-H.; Desiderio, D.M.; Kim, Y.-K.; Kim, K.-P.; et al. Gram-Positive Bacteria Produce Membrane Vesicles: Proteomics-Based Characterization of Staphylococcus aureus-Derived Membrane Vesicles. Proteomics 2009, 9, 5425–5436. [Google Scholar] [CrossRef]
- Jun, S.H.; Lee, J.H.; Kim, S.I.; Choi, C.W.; Park, T.I.; Jung, H.R.; Cho, J.W.; Kim, S.H.; Lee, J.C. Staphylococcus aureus-Derived Membrane Vesicles Exacerbate Skin Inflammation in Atopic Dermatitis. Clin. Exp. Allergy 2017, 47, 85–96. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.-P.; Han, S.B.; Kim, Y.K.; Park, E.E.; Doh, E.J.; Kim, K.H.; Lee, D.H.; Chung, J.H. Changes in Tight Junction Protein Expression in Intrinsic Aging and Photoaging in Human Skin in Vivo. J. Dermatol. Sci. 2016, 84, 99–101. [Google Scholar] [CrossRef] [PubMed]
- Svoboda, M.; Bílková, Z.; Muthný, T. Could Tight Junctions Regulate the Barrier Function of the Aged Skin? J. Dermatol. Sci. 2016, 81, 147–152. [Google Scholar] [CrossRef] [PubMed]
- Brandner, J.M.; Schulzke, J.D. Hereditary Barrier-Related Diseases Involving the Tight Junction: Lessons from Skin and Intestine. Cell Tissue Res. 2015, 360, 723–748. [Google Scholar] [CrossRef]
- Bäsler, K.; Bergmann, S.; Heisig, M.; Naegel, A.; Zorn-Kruppa, M.; Brandner, J.M. The Role of Tight Junctions in Skin Barrier Function and Dermal Absorption. J. Control. Release 2016, 242, 105–118. [Google Scholar] [CrossRef]
- Yokouchi, M.; Kubo, A. Maintenance of Tight Junction Barrier Integrity in Cell Turnover and Skin Diseases. Exp. Dermatol. 2018, 27, 876–883. [Google Scholar] [CrossRef]
- Herbig, M.E.; Houdek, P.; Gorissen, S.; Zorn-Kruppa, M.; Wladykowski, E.; Volksdorf, T.; Grzybowski, S.; Kolios, G.; Willers, C.; Mallwitz, H.; et al. A Custom Tailored Model to Investigate Skin Penetration in Porcine Skin and Its Comparison with Human Skin. Eur. J. Pharm. Biopharm. 2015, 95, 99–109. [Google Scholar] [CrossRef]
- Harris-Tryon, T.A.; Grice, E.A. Microbiota and Maintenance of Skin Barrier Function. Science 2022, 376, 940–945. [Google Scholar] [CrossRef]
- Luna, P.C. Skin Microbiome as Years Go By. Am. J. Clin. Dermatol. 2020, 21, 12–17. [Google Scholar] [CrossRef]
- Chen, P.; He, G.; Qian, J.; Zhan, Y.; Xiao, R. Potential Role of the Skin Microbiota in Inflammatory Skin Diseases. J. Cosmet. Dermatol. 2021, 20, 400–409. [Google Scholar] [CrossRef]
- Flowers, L.; Grice, E.A. The Skin Microbiota: Balancing Risk and Reward. Cell Host Microbe 2020, 28, 190–200. [Google Scholar] [CrossRef] [PubMed]
- Paller, A.S.; Kong, H.H.; Seed, P.; Naik, S.; Scharschmidt, T.C.; Gallo, R.L.; Luger, T.; Irvine, A.D. The Microbiome in Patients with Atopic Dermatitis. J. Allergy Clin. Immunol. 2019, 143, 26–35. [Google Scholar] [CrossRef] [PubMed]
- Ito, Y.; Amagai, M. Controlling Skin Microbiome as a New Bacteriotherapy for Inflammatory Skin Diseases. Inflamm. Regen. 2022, 42, 26. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Shi, L.; Ren, Y.; Tan, X.; Liu, W.; Liu, Z. Applications of Human Skin Microbiota in the Cutaneous Disorders for Ecology-Based Therapy. Front. Cell Infect. Microbiol. 2020, 10, 570261. [Google Scholar] [CrossRef] [PubMed]
- Magnifico, I.; Perna, A.; Cutuli, M.A.; Medoro, A.; Pietrangelo, L.; Guarnieri, A.; Foderà, E.; Passarella, D.; Venditti, N.; Vergalito, F.; et al. A Wall Fragment of Cutibacterium Acnes Preserves Junctional Integrity Altered by Staphylococcus aureus in an Ex Vivo Porcine Skin Model. Pharmaceutics 2023, 15, 1224. [Google Scholar] [CrossRef] [PubMed]
- Sacchetti, R.; Gregori, G.; Moggio, E.; Gobbo, L.; Bonzano, L.; Pellacani, G. HAc40 Is a Novel Microbiome Modulator, Effective on Atopic Dermatitis in Children: Data from Two Pilot Vehicle-Controlled Trials. J. Eur. Acad. Dermatol. Venereol. 2021, 35, e767–e768. [Google Scholar] [CrossRef] [PubMed]
- Pietrangelo, L.; Dattola, A.; Magnifico, I.; Petronio Petronio, G.; Cutuli, M.A.; Venditti, N.; Guarnieri, A.; Wollenberg, A.; Pellacani, G.; Di Marco, R. Efficacy and Microbiota Modulation Induced by LimpiAL 2.5%, a New Medical Device for the Inverse Psoriasis Treatment. Int. J. Mol. Sci. 2023, 24, 6339. [Google Scholar] [CrossRef]
- Pietrangelo, L.; Magnifico, I.; Guerrera, A.; Cutuli, M.A.; Petronio, G.P.; Venditti, N.; Covelli, M.; Buccieri, N.; Garofalo, S.; Di Marco, R. LimpiAD Foam and the Potential Control of the Pressure Ulcers Onset. Biomed. Pharmacother. 2021, 144, 112327. [Google Scholar] [CrossRef] [PubMed]
- Welsh, J.A.; Goberdhan, D.C.I.; O’Driscoll, L.; Buzas, E.I.; Blenkiron, C.; Bussolati, B.; Cai, H.; Di Vizio, D.; Driedonks, T.A.P.; Erdbrügger, U.; et al. Minimal Information for Studies of Extracellular Vesicles (MISEV2023): From Basic to Advanced Approaches. J. Extracell. Vesicles 2024, 13, e12404. [Google Scholar] [CrossRef]
- Wei, S.; Jiao, D.; Xing, W. A Rapid Method for Isolation of Bacterial Extracellular Vesicles from Culture Media Using Epsilon-Poly-L-Lysine That Enables Immunological Function Research. Front. Immunol. 2022, 13, 930510. [Google Scholar] [CrossRef]
- Sielaff, M.; Kuharev, J.; Bohn, T.; Hahlbrock, J.; Bopp, T.; Tenzer, S.; Distler, U. Evaluation of FASP, SP3, and iST Protocols for Proteomic Sample Preparation in the Low Microgram Range. J. Proteome Res. 2017, 16, 4060–4072. [Google Scholar] [CrossRef] [PubMed]
- Ngo, Q.V.; Faass, L.; Sähr, A.; Hildebrand, D.; Eigenbrod, T.; Heeg, K.; Nurjadi, D. Inflammatory Response Against Staphylococcus aureus via Intracellular Sensing of Nucleic Acids in Keratinocytes. Front. Immunol. 2022, 13, 828626. [Google Scholar] [CrossRef]
- Nicolosi, D.; Genovese, C.; Cutuli, M.A.; D’angeli, F.; Pietrangelo, L.; Davinelli, S.; Petronio, G.P.; Di Marco, R. Preliminary in Vitro Studies on Corynebacterium Urealyticum Pathogenetic Mechanisms, a Possible Candidate for Chronic Idiopathic Prostatitis? Microorganisms 2020, 8, 463. [Google Scholar] [CrossRef] [PubMed]
- Spandidos, A.; Wang, X.; Wang, H.; Seed, B. PrimerBank: A Resource of Human and Mouse PCR Primer Pairs for Gene Expression Detection and Quantification. Nucleic Acids Res. 2010, 38, D792–D799. [Google Scholar] [CrossRef]
- Kim, D.-K.; Kang, B.; Kim, O.Y.; Choi, D.-S.; Lee, J.; Kim, S.R.; Go, G.; Yoon, Y.J.; Kim, J.H.; Jang, S.C.; et al. EVpedia: An Integrated Database of High-Throughput Data for Systemic Analyses of Extracellular Vesicles. J. Extracell. Vesicles 2013, 2, 20384. [Google Scholar] [CrossRef]
- Danaei, M.; Dehghankhold, M.; Ataei, S.; Hasanzadeh Davarani, F.; Javanmard, R.; Dokhani, A.; Khorasani, S.; Mozafari, M. Impact of Particle Size and Polydispersity Index on the Clinical Applications of Lipidic Nanocarrier Systems. Pharmaceutics 2018, 10, 57. [Google Scholar] [CrossRef]
- Wu, L.; Zhang, J.; Watanabe, W. Physical and Chemical Stability of Drug Nanoparticles. Adv. Drug Deliv. Rev. 2011, 63, 456–469. [Google Scholar] [CrossRef]
- Lee, E.-Y.; Choi, D.-S.; Kim, K.-P.; Gho, Y.S. Proteomics in Gram-Negative Bacterial Outer Membrane Vesicles. Mass Spectrom. Rev. 2008, 27, 535–555. [Google Scholar] [CrossRef] [PubMed]
- Verma, D.D.; Verma, S.; Blume, G.; Fahr, A. Particle Size of Liposomes Influences Dermal Delivery of Substances into Skin. Int. J. Pharm. 2003, 258, 141–151. [Google Scholar] [CrossRef] [PubMed]
- Augustine, R.; Hasan, A.; Primavera, R.; Wilson, R.J.; Thakor, A.S.; Kevadiya, B.D. Cellular Uptake and Retention of Nanoparticles: Insights on Particle Properties and Interaction with Cellular Components. Mater. Today Commun. 2020, 25, 101692. [Google Scholar] [CrossRef]
- Miyake, R.; Iwamoto, K.; Sakai, N.; Matsunae, K.; Aziz, F.; Sugai, M.; Takahagi, S.; Tanaka, A.; Hide, M. Uptake of Staphylococcus aureus by Keratinocytes Is Reduced by Interferon-Fibronectin Pathway and Filaggrin Expression. J. Dermatol. 2022, 49, 1148–1157. [Google Scholar] [CrossRef] [PubMed]
- Al-Rayyan, N.; Steidl, O.; Starr, N.L.; Smith, J.; Singh, A.M. Effect of Staphylococcus aureus on the Keratinocytes in Atopic Dermatitis. J. Allergy Clin. Immunol. 2023, 151, AB145. [Google Scholar] [CrossRef]
- Kwak, Y.-K.; Vikström, E.; Magnusson, K.-E.; Vécsey-Semjén, B.; Colque-Navarro, P.; Möllby, R. The Staphylococcus aureus Alpha-Toxin Perturbs the Barrier Function in Caco-2 Epithelial Cell Monolayers by Altering Junctional Integrity. Infect. Immun. 2012, 80, 1670–1680. [Google Scholar] [CrossRef] [PubMed]
- Ohnemus, U.; Kohrmeyer, K.; Houdek, P.; Rohde, H.; Wladykowski, E.; Vidal, S.; Horstkotte, M.A.; Aepfelbacher, M.; Kirschner, N.; Behne, M.J.; et al. Regulation of Epidermal Tight-Junctions (TJ) during Infection with Exfoliative Toxin-Negative Staphylococcus Strains. J. Investig. Dermatol. 2008, 128, 906–916. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cellular Component/s | Gene Ontology (GO) | Numbers of Proteins | % (n. 422) |
|---|---|---|---|
| TOTAL | 178 | 42.18 | |
| Cytoplasm | 95 | 22.51 | |
| ribonucleoprotein | [GO:1990904] | 19 | |
| small ribosomal subunit | [GO:0015935] | 4 | |
| large ribosomal subunit | GO:0015934] | 7 | |
| cell division site | [GO:0032153] | 1 | |
| DNA polymerase III complex | [GO:0009360] | 1 | |
| DNA-directed RNA polymerase complex | [GO:0000428] | 2 | |
| nucleoid | [GO:0009295] | 1 | |
| Cytoplasmatic only | [GO:0005737] | 60 | |
| Membrane | 46 | 10.90 | |
| plasma membrane | [GO:0005886] | 13 | |
| membrane raft | [GO:0045121] | 1 | |
| proton-transporting ATP synthase complex | [GO:0045261] | 3 | |
| respirasome | [GO:0070469] | 2 | |
| membrane only | [GO:0016020] | 27 | |
| Extracellular region | 25 | 5.92 | |
| membrane | [GO:0016020] | 5 | |
| extracellular space | [GO:0005615] | 1 | |
| host cell plasma membrane; membrane | [GO:0020002] [GO:0005886] | 1 | |
| phosphopyruvate hydratase; cell surface | [GO:0000015] [GO:0009986] | 2 | |
| Extracellular region only | [GO:0005576] | 16 | |
| DNA-directed RNA polymerase complex | [GO:0000428] | 2 | 0.47 |
| Glycerol-3-phosphate dehydrogenase | [GO:0009331] | 2 | 0.47 |
| Cellular anatomical entity | [GO:0110165] | 2 | 0.47 |
| Riboflavin synthase complex | [GO:0009349] | 1 | 0.24 |
| HslUV protease complex | [GO:0009376] | 1 | 0.24 |
| ABC transporter complex | [GO:0043190] | 1 | 0.24 |
| Chromosome | [GO:0005694] | 1 | 0.24 |
| BEV | HA | HAc40 | c40 | BEV-HA | BEV-HAc40 | BEV-c40 | |
|---|---|---|---|---|---|---|---|
| Volume/Peak (nm) | 22.45 | 24.64 | 343.8 | 829.4 | 13.62 | 836.0 | 1009 |
| Z-Average (nm) | 312.3 | 81 | 825 | 1799 | 58.14 | 1090 | 2379 |
| Treated (T) | Mean Area (Pixel2 ± D.S.) | Untreated (UT) | Mean Area (Pixel2 ± D.S.) | Mean Area Percentage Difference (T-UT)/T × 100 | p-Value |
|---|---|---|---|---|---|
| BEV | 366,445 ± 15.26 | PBS | 571,017.33 ± 24.35 | −55.83 | <0.001 |
| BEV-HAc40 | 561,058.66 ± 17 | BEV | 366,445 ± 15.26 | 34.69 | |
| BEV-HA | 519,178.66 ± 14.31 | 29.42 | |||
| BEV-c40 | 545,091.33 ± 13.76 | 32.77 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Petronio Petronio, G.; Di Naro, M.; Venditti, N.; Guarnieri, A.; Cutuli, M.A.; Magnifico, I.; Medoro, A.; Foderà, E.; Passarella, D.; Nicolosi, D.; et al. Targeting S. aureus Extracellular Vesicles: A New Putative Strategy to Counteract Their Pathogenic Potential. Pharmaceutics 2024, 16, 789. https://doi.org/10.3390/pharmaceutics16060789
Petronio Petronio G, Di Naro M, Venditti N, Guarnieri A, Cutuli MA, Magnifico I, Medoro A, Foderà E, Passarella D, Nicolosi D, et al. Targeting S. aureus Extracellular Vesicles: A New Putative Strategy to Counteract Their Pathogenic Potential. Pharmaceutics. 2024; 16(6):789. https://doi.org/10.3390/pharmaceutics16060789
Chicago/Turabian StylePetronio Petronio, Giulio, Maria Di Naro, Noemi Venditti, Antonio Guarnieri, Marco Alfio Cutuli, Irene Magnifico, Alessandro Medoro, Emanuele Foderà, Daniela Passarella, Daria Nicolosi, and et al. 2024. "Targeting S. aureus Extracellular Vesicles: A New Putative Strategy to Counteract Their Pathogenic Potential" Pharmaceutics 16, no. 6: 789. https://doi.org/10.3390/pharmaceutics16060789
APA StylePetronio Petronio, G., Di Naro, M., Venditti, N., Guarnieri, A., Cutuli, M. A., Magnifico, I., Medoro, A., Foderà, E., Passarella, D., Nicolosi, D., & Di Marco, R. (2024). Targeting S. aureus Extracellular Vesicles: A New Putative Strategy to Counteract Their Pathogenic Potential. Pharmaceutics, 16(6), 789. https://doi.org/10.3390/pharmaceutics16060789