
Pharmacokinetic Modeling to Guide Preclinical Development of an Islatravir-Eluting Reservoir-Style Biodegradable Implant for Long-Acting HIV PrEP

 , ,
, ,  , , , ,
, , , , 
Abstract
1. Introduction
2. Materials and Methods
2.1. Drug Preparation
2.2. Preclinical Islatravir PK Study
2.3. Dose Administration and Sparse Sampling Schema
2.4. Islatravir Quantification
2.5. Noncompartmental Analysis
2.6. Pharmacokinetic Analysis
2.7. Deconvolution of In Vivo Absorption
2.8. In Vivo Drug Absorption Models
3. Results
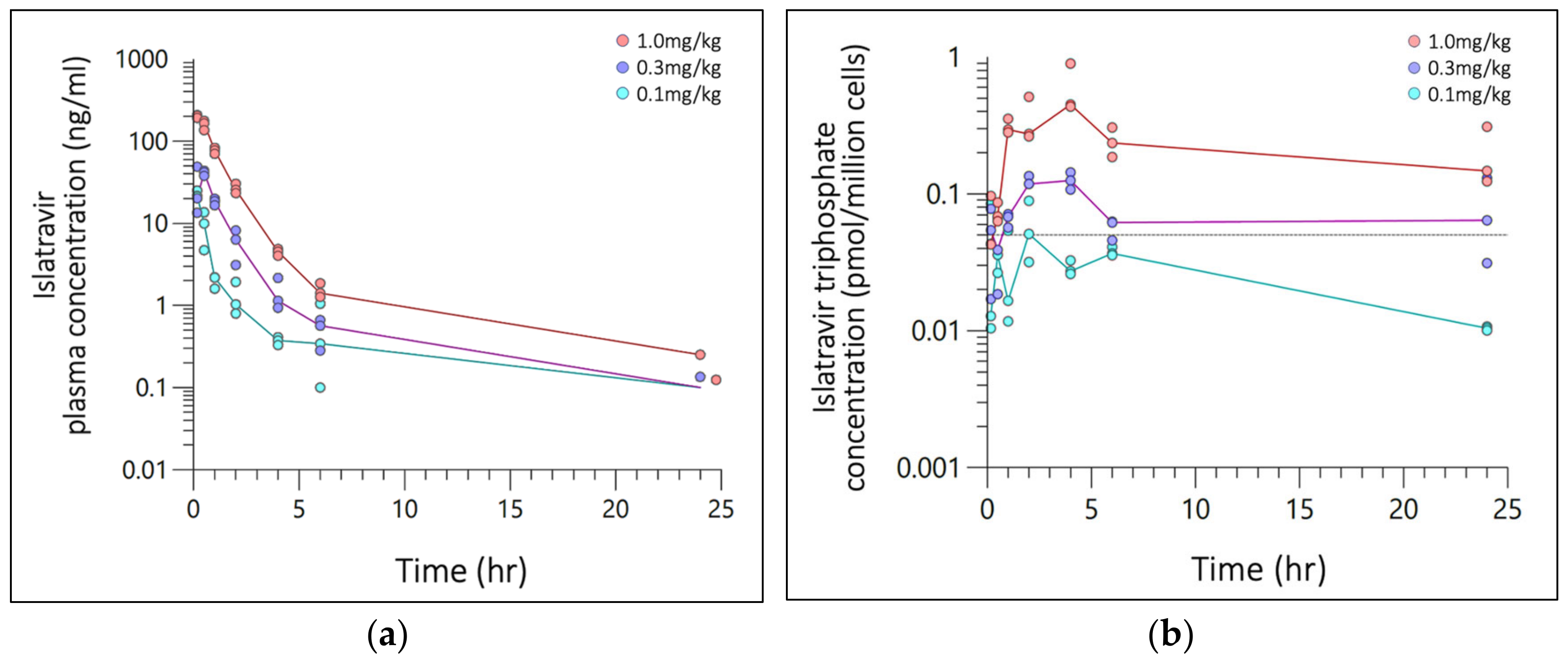
3.1. Noncompartmental PK Analysis
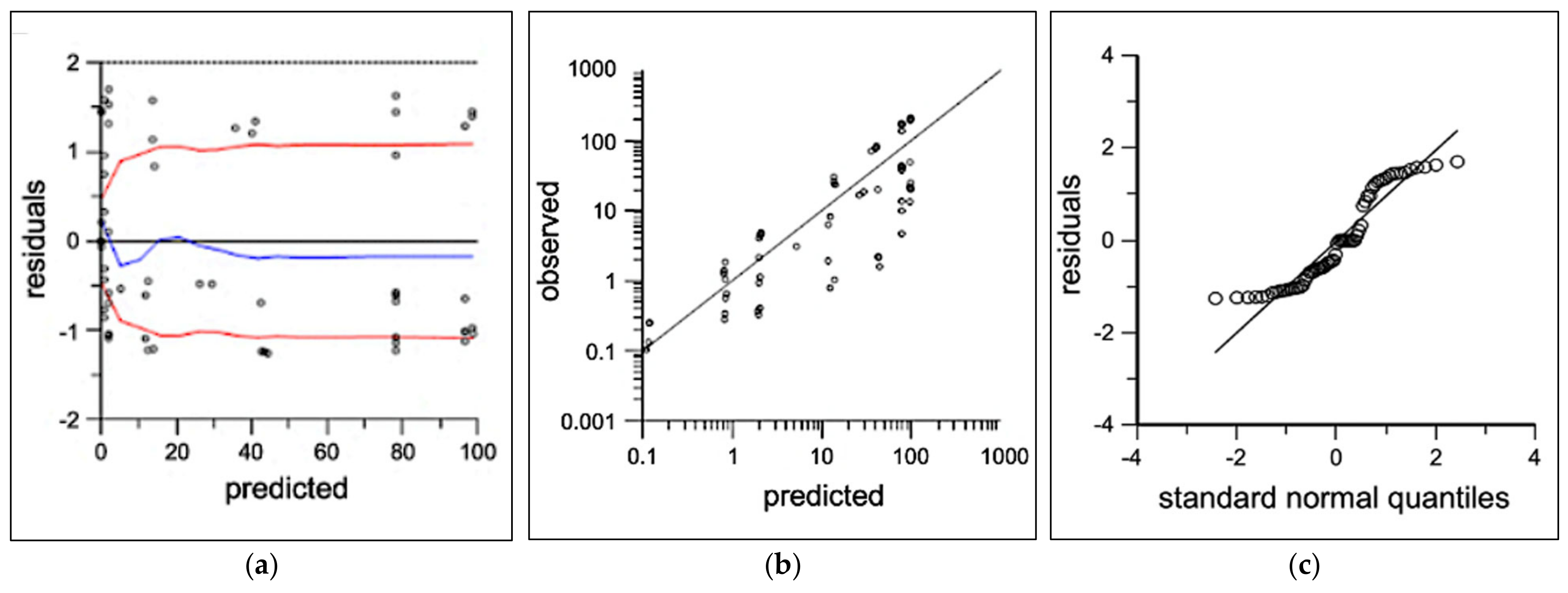
3.2. Pharmacokinetic Modeling
3.3. In Vivo Absorption
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Grant, R.M.; Lama, J.R.; Anderson, P.L.; McMahan, V.; Liu, A.Y.; Vargas, L.; Goicochea, P.; Casapía, M.; Guanira-Carranza, J.V.; Ramirez-Cardich, M.E.; et al. iPrEx Study Team Preexposure chemoprophylaxis for HIV prevention in men who have sex with men. N. Engl. J. Med. 2010, 363, 2587–2599. [Google Scholar] [CrossRef] [PubMed]
- Mayer, K.H.; Molina, J.-M.; Thompson, M.A.; Anderson, P.L.; Mounzer, K.C.; De Wet, J.J.; DeJesus, E.; Jessen, H.; Grant, R.M.; Ruane, P.J.; et al. Emtricitabine and tenofovir alafenamide vs. emtricitabine and tenofovir disoproxil fumarate for HIV pre-exposure prophylaxis (DISCOVER): Primary results from a randomised, double-blind, multicentre, active-controlled, phase 3, non-inferiority trial. Lancet 2020, 396, 239–254. [Google Scholar] [CrossRef] [PubMed]
- Van Damme, L.; Corneli, A.; Ahmed, K.; Agot, K.; Lombaard, J.; Kapiga, S.; Malahleha, M.; Owino, F.; Manongi, R.; Onyango, J.; et al. FEM-PrEP Study Group Preexposure prophylaxis for HIV infection among African women. N. Engl. J. Med. 2012, 367, 411–422. [Google Scholar] [CrossRef] [PubMed]
- Marrazzo, J.M.; Ramjee, G.; Richardson, B.A.; Gomez, K.; Mgodi, N.; Nair, G.; Palanee, T.; Nakabiito, C.; van der Straten, A.; Noguchi, L.; et al. VOICE Study Team Tenofovir-based preexposure prophylaxis for HIV infection among African women. N. Engl. J. Med. 2015, 372, 509–518. [Google Scholar] [CrossRef]
- Landovitz, R.J.; Donnell, D.; Clement, M.E.; Hanscom, B.; Cottle, L.; Coelho, L.; Cabello, R.; Chariyalertsak, S.; Dunne, E.F.; Frank, I.; et al. HPTN 083 Study Team Cabotegravir for HIV prevention in cisgender men and transgender women. N. Engl. J. Med. 2021, 385, 595–608. [Google Scholar] [CrossRef]
- Delany-Moretlwe, S.; Hughes, J.P.; Bock, P.; Ouma, S.G.; Hunidzarira, P.; Kalonji, D.; Kayange, N.; Makhema, J.; Mandima, P.; Mathew, C.; et al. HPTN 084 study group Cabotegravir for the prevention of HIV-1 in women: Results from HPTN 084, a phase 3, randomised clinical trial. Lancet 2022, 399, 1779–1789. [Google Scholar] [CrossRef] [PubMed]
- APRETUDE (Cabotegravir Extended-Release Injectable Suspension) Prescribing Information, ViiV Healthcare. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/215499s000lbl.pdf (accessed on 18 November 2023).
- Landovitz, R.J.; Li, S.; Eron, J.J.; Grinsztejn, B.; Dawood, H.; Liu, A.Y.; Magnus, M.; Hosseinipour, M.C.; Panchia, R.; Cottle, L.; et al. Tail-phase safety, tolerability, and pharmacokinetics of long-acting injectable cabotegravir in HIV-uninfected adults: A secondary analysis of the HPTN 077 trial. Lancet HIV 2020, 7, e472–e481. [Google Scholar] [CrossRef]
- Adams, K.; Beal, M.W. Implanon: A review of the literature with recommendations for clinical management. J. Midwifery Women’s Health 2009, 54, 142–149. [Google Scholar] [CrossRef]
- Brown, M.S.; Hanif, H.; Little, K.M.; Clark, M.R.; Thurman, A.R.; Flomen, L.; Doncel, G.F. End-user research in support of long-acting systemic antiretroviral delivery systems: Insights from qualitative research with providers and target users in South Africa. BMC Infect. Dis. 2022, 22, 919. [Google Scholar] [CrossRef]
- Humphries, H.; Upfold, M.; Mahlase, G.; Mdladla, M.; Gengiah, T.N.; Abdool Karim, Q. Implants for HIV prevention in young women: Provider perceptions and lessons learned from contraceptive implant provision. PLoS ONE 2022, 17, e0262043. [Google Scholar] [CrossRef]
- Markowitz, M.; Sarafianos, S.G. 4’-Ethynyl-2-fluoro-2’-deoxyadenosine, MK-8591: A novel HIV-1 reverse transcriptase translocation inhibitor. Curr. Opin. HIV AIDS 2018, 13, 294–299. [Google Scholar] [CrossRef] [PubMed]
- Markowitz, M.; Gettie, A.; St Bernard, L.; Andrews, C.D.; Mohri, H.; Horowitz, A.; Grasperge, B.F.; Blanchard, J.L.; Niu, T.; Sun, L.; et al. Once-Weekly Oral Dosing of MK-8591 Protects Male Rhesus Macaques from Intrarectal Challenge with SHIV109CP3. J. Infect. Dis. 2020, 221, 1398–1406. [Google Scholar] [CrossRef] [PubMed]
- Schürmann, D.; Rudd, D.J.; Zhang, S.; De Lepeleire, I.; Robberechts, M.; Friedman, E.; Keicher, C.; Hüser, A.; Hofmann, J.; Grobler, J.A.; et al. Safety, pharmacokinetics, and antiretroviral activity of islatravir (ISL, MK-8591), a novel nucleoside reverse transcriptase translocation inhibitor, following single-dose administration to treatment-naive adults infected with HIV-1: An open-label, phase 1b, consecutive-panel trial. Lancet HIV 2020, 7, e164–e172. [Google Scholar] [CrossRef] [PubMed]
- Matthews, R.P.; Jackson Rudd, D.; Zhang, S.; Fillgrove, K.L.; Sterling, L.M.; Grobler, J.A.; Vargo, R.C.; Stoch, S.A.; Iwamoto, M. Safety and Pharmacokinetics of Once-Daily Multiple-Dose Administration of Islatravir in Adults without HIV. J. Acquir. Immune Defic. Syndr. 2021, 88, 314–321. [Google Scholar] [CrossRef] [PubMed]
- Patel, M.; Zang, X.; Matthews, R.; Plank, R.; Sklar, P.; Grobler, J.; Robertson, M.; Vargo, R. Islatravir PK threshold & dose selection for monthly oral HIV-1 PrEP 2021. In Proceedings of the Conference on Retroviruses and Opportunistic Infection, Virtual, 7–10 March 2021. [Google Scholar]
- Matthews, R.P.; Patel, M.; Barrett, S.E.; Haspeslagh, L.; Reynders, T.; Zhang, S.; Rottey, S.; Goodey, A.; Vargo, R.C.; Grobler, J.A.; et al. Safety and pharmacokinetics of islatravir subdermal implant for HIV-1 pre-exposure prophylaxis: A randomized, placebo-controlled phase 1 trial. Nat. Med. 2021, 27, 1712–1717. [Google Scholar] [CrossRef] [PubMed]
- Matthews, R.P.; Zang, X.; Barrett, S.E.; Koynov, A.; Goodey, A.; Heimbach, T.; Weissler, V.L.; Leyssens, C.; Reynders, T.; Xu, Z.; et al. A Randomized, Double-Blind, Placebo-Controlled, Phase 1 Trial of Radiopaque Islatravir-Eluting Subdermal Implants for Pre-exposure Prophylaxis Against HIV-1 Infection. J. Acquir. Immune Defic. Syndr. 2023, 92, 310–316. [Google Scholar] [CrossRef] [PubMed]
- Matthews, R.P.; Ankrom, W.; Friedman, E.; Jackson Rudd, D.; Liu, Y.; Mogg, R.; Panebianco, D.; De Lepeleire, I.; Petkova, M.; Grobler, J.A.; et al. Safety, tolerability, and pharmacokinetics of single- and multiple-dose administration of islatravir (MK-8591) in adults without HIV. Clin. Transl. Sci. 2021, 14, 1935–1944. [Google Scholar] [CrossRef]
- Merck Announces Clinical Holds on Studies Evaluating Islatravir for the Treatment and Prevention of HIV-1 Infection—Merck.com. Available online: https://www.merck.com/news/merck-announces-clinical-holds-on-studies-evaluating-islatravir-for-the-treatment-and-prevention-of-hiv-1-infection/ (accessed on 9 September 2023).
- Squires, K.; Correll, T.; Robertson, M.; Klopfer, S.; May Tan Hwang, P.; Zhou, Y.-P.; Rhee, E. Effect of Islatravir on Total Lymphocyte and Lymhocyte Subset Counts. Available online: https://www.natap.org/2023/CROI/croi_35.htm (accessed on 10 September 2023).
- Li, L.; Gatto, G.J.; Brand, R.M.; Krovi, S.A.; Cottrell, M.L.; Norton, C.; van der Straten, A.; Johnson, L.M. Long-acting biodegradable implant for sustained delivery of antiretrovirals (ARVs) and hormones. J. Control. Release 2021, 340, 188–199. [Google Scholar] [CrossRef]
- National Research Council. The National Academies Guide for the Care and Use of Laboratory Animals: Eighth Edition, Copyright 2011, NAS|OLAW. Available online: https://olaw.nih.gov/resources/publications/guide-care-2011.htm (accessed on 20 February 2023).
- Sykes, C.; Van Horne, B.; Jones, J.; Kashuba, A.D.M.; Gatto, G.; Van Der Straten, A.; Johnson, L.; Cottrell, M.L. Intracellular islatravir pharmacology differs between species in an in vitro model: Implications for preclinical study design. J. Antimicrob. Chemother. 2022, 77, 1000–1004. [Google Scholar] [CrossRef]
- Beal, S.L. Ways to fit a PK model with some data below the quantification limit. J. Pharmacokinet. Pharmacodyn. 2001, 28, 481–504. [Google Scholar] [CrossRef]
- Bruschi Mathematical models of drug release. In Strategies to Modify the Drug Release from Pharmaceutical Systems; Elsevier: Amsterdam, The Netherlands, 2015; pp. 63–86. ISBN 9780081000922.
- Costa, P.; Sousa Lobo, J.M. Evaluation of mathematical models describing drug release from estradiol transdermal systems. Drug Dev. Ind. Pharm. 2003, 29, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Spinelli, M.A.; Grinsztejn, B.; Landovitz, R.J. Promises and challenges: Cabotegravir for preexposure prophylaxis. Curr. Opin. HIV AIDS 2022, 17, 186–191. [Google Scholar] [CrossRef] [PubMed]
- Organon & Co.; Jersey City, NJ, U.S.; Food and Drug Administration. Nexplanon (Etonogestrel Implant) Prescribing Information. Available online: https://www.organon.com/product/usa/pi_circulars/n/nexplanon/nexplanon_pi.pdf (accessed on 18 February 2023).
- Barrett, S.E.; Teller, R.S.; Forster, S.P.; Li, L.; Mackey, M.A.; Skomski, D.; Yang, Z.; Fillgrove, K.L.; Doto, G.J.; Wood, S.L.; et al. Extended-Duration MK-8591-Eluting Implant as a Candidate for HIV Treatment and Prevention. Antimicrob. Agents Chemother. 2018, 62, e01058-18. [Google Scholar] [CrossRef] [PubMed]
- Ritger, P.L.; Peppas, N.A. A simple equation for description of solute release II. Fickian and anomalous release from swellable devices. J. Control. Release 1987, 5, 37–42. [Google Scholar] [CrossRef]
- Macha, S.; Yong, C.-L.; Darrington, T.; Davis, M.S.; MacGregor, T.R.; Castles, M.; Krill, S.L. In vitro-in vivo correlation for nevirapine extended release tablets. Biopharm. Drug Dispos. 2009, 30, 542–550. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Manda, P.; Pavurala, N.; Khan, M.A.; Krishnaiah, Y.S.R. Development and validation of in vitro-in vivo correlation (IVIVC) for estradiol transdermal drug delivery systems. J. Control. Release 2015, 210, 58–66. [Google Scholar] [CrossRef]
- FDA Duidance for Industry: Extended Release Oral Dosage Forms: Development, Evaluation, and Application of In Vitro/In Vivo Correlations. Available online: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/extended-release-oral-dosage-forms-development-evaluation-and-application-vitroin-in-vivo-correlations (accessed on 27 September 2023).
- European Medicines Agency: Guideline on the Pharmacokinetic and Clinical Evaluation of Modified Release Dosage Forms. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-pharmacokinetic-clinical-evaluation-modified-release-dosage-forms_en.pdf (accessed on 26 September 2023).



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Zero-Order r2 | First-Order r2 | Higuchi r2 | Ritger–Peppas b r2 |
|---|---|---|---|---|
| ISL_only | 0.9680 | −6.6068 | 0.7054 | N.R. a |
| ISL_only | 0.8933 | −4.0118 | 0.5972 | 0.9909 |
| ISL_only | 0.9409 | −4.5061 | 0.6568 | 0.9944 |
| ISL_only | 0.9776 | −3.0757 | 0.7388 | 0.9908 |
| ISL_only | 0.9888 | −4.3850 | 0.7672 | 0.9942 |
| ISL_plus | 0.9805 | −6.5246 | 0.8384 | 0.9855 |
| ISL_plus | 0.8667 | −4.2326 | 0.5859 | 0.9844 |
| ISL_plus | 0.8971 | −4.7319 | 0.6481 | 0.9399 |
| ISL_plus | 0.9825 | −4.8870 | 0.7619 | 0.9878 |
| ISL_plus | 0.9615 | −4.6075 | 0.6997 | 0.9978 |
| Mean | 0.9457 | −4.7569 | 0.6999 | 0.9851 |
| Standard deviation | 0.04 | 1.08 | 0.08 | 0.02 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kinsale, T.S.; Cottrell, M.L.; Li, L.; Brand, R.; Gatto, G.; Luecke, E.; Norton, C.; Krovi, A.; Dumond, J.B.; Rao, G.; et al. Pharmacokinetic Modeling to Guide Preclinical Development of an Islatravir-Eluting Reservoir-Style Biodegradable Implant for Long-Acting HIV PrEP. Pharmaceutics 2024, 16, 201. https://doi.org/10.3390/pharmaceutics16020201
Kinsale TS, Cottrell ML, Li L, Brand R, Gatto G, Luecke E, Norton C, Krovi A, Dumond JB, Rao G, et al. Pharmacokinetic Modeling to Guide Preclinical Development of an Islatravir-Eluting Reservoir-Style Biodegradable Implant for Long-Acting HIV PrEP. Pharmaceutics. 2024; 16(2):201. https://doi.org/10.3390/pharmaceutics16020201
Chicago/Turabian StyleKinsale, Talisa S., Mackenzie L. Cottrell, Linying Li, Rhonda Brand, Greg Gatto, Ellen Luecke, Chasity Norton, Archana Krovi, Julie B. Dumond, Gauri Rao, and et al. 2024. "Pharmacokinetic Modeling to Guide Preclinical Development of an Islatravir-Eluting Reservoir-Style Biodegradable Implant for Long-Acting HIV PrEP" Pharmaceutics 16, no. 2: 201. https://doi.org/10.3390/pharmaceutics16020201
APA StyleKinsale, T. S., Cottrell, M. L., Li, L., Brand, R., Gatto, G., Luecke, E., Norton, C., Krovi, A., Dumond, J. B., Rao, G., Yeshwante, S., Van Horne, B., Van Der Straten, A., Kashuba, A. D. M., & Johnson, L. M. (2024). Pharmacokinetic Modeling to Guide Preclinical Development of an Islatravir-Eluting Reservoir-Style Biodegradable Implant for Long-Acting HIV PrEP. Pharmaceutics, 16(2), 201. https://doi.org/10.3390/pharmaceutics16020201