

Effects of Hyperlipidemia on the Pharmacokinetics of Tofacitinib, a JAK 1/3 Inhibitor, in Rats

Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Animals
2.3. Induction of Hyperlipidemia
2.4. Preliminary Study
2.5. Plasma Protein Binding of Tofacitinib
2.6. Intravenous and Oral Administration of Tofacitinib
2.7. Distribution of Tofacitinib in Tissues
2.8. Measurement of Vmax, Km, and CLint
2.9. Immunoblot Analysis
2.10. HPLC Analysis
2.11. Pharmacokinetic Analysis
2.12. Statistical Analysis
3. Results
3.1. Preliminary Study
3.2. Plasma Protein Binding of Tofacitinib
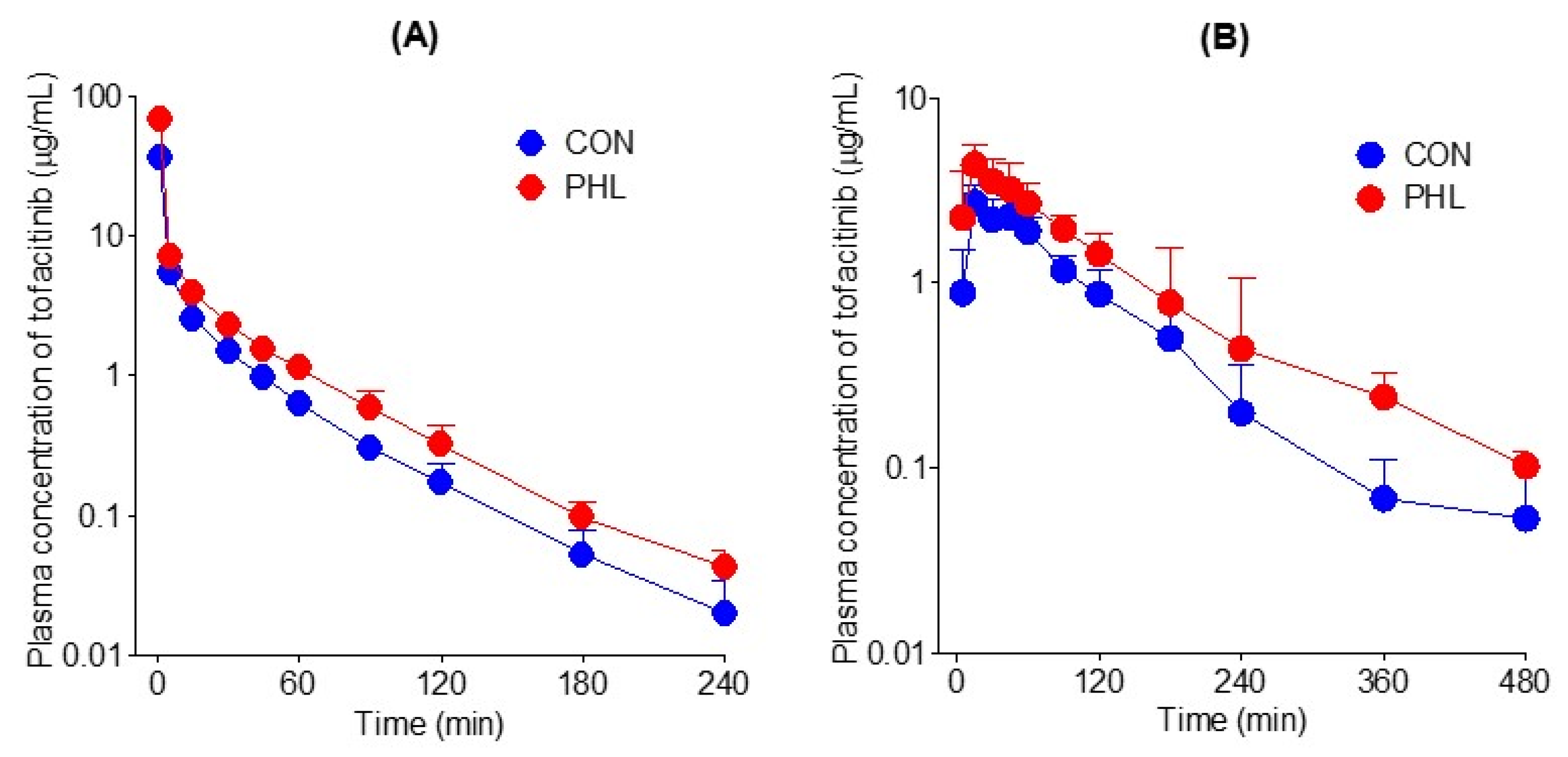
3.3. Pharmacokinetics of Tofacitinib following Intravenous and Oral Administration
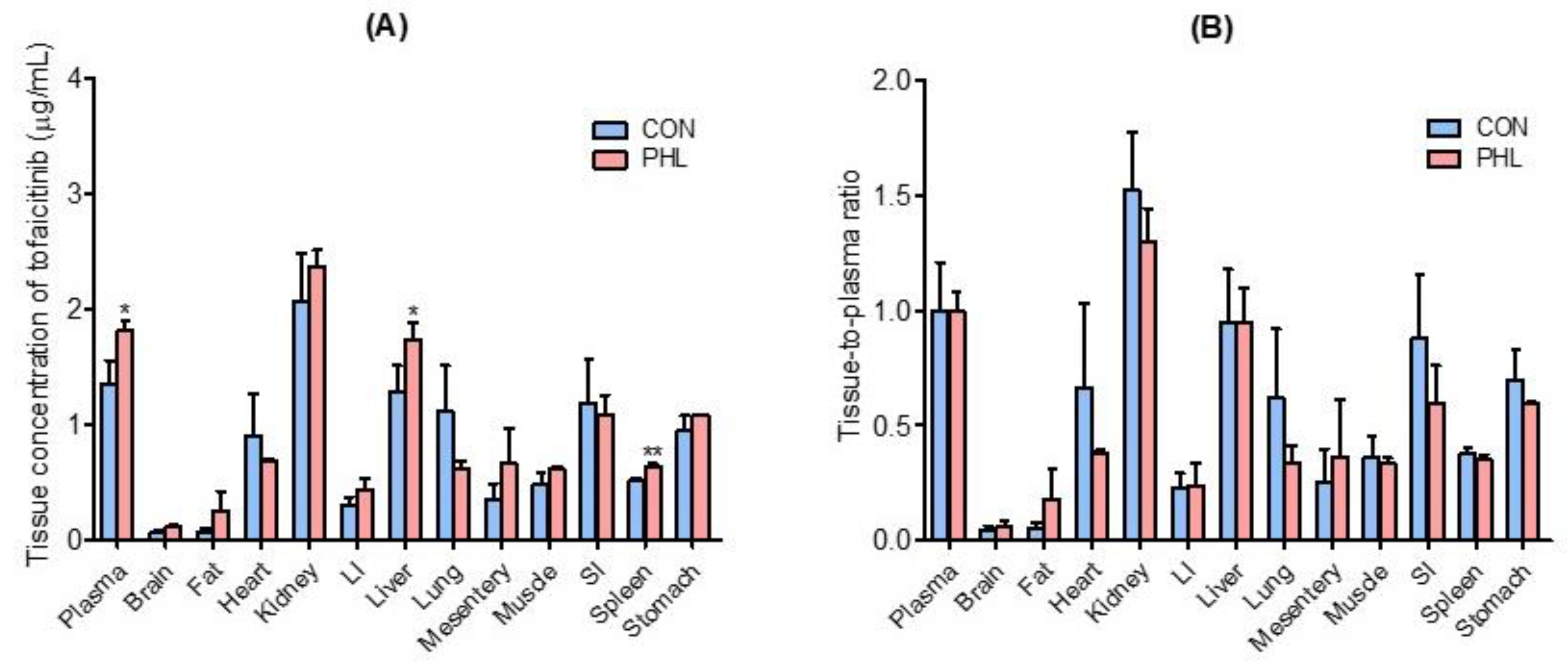
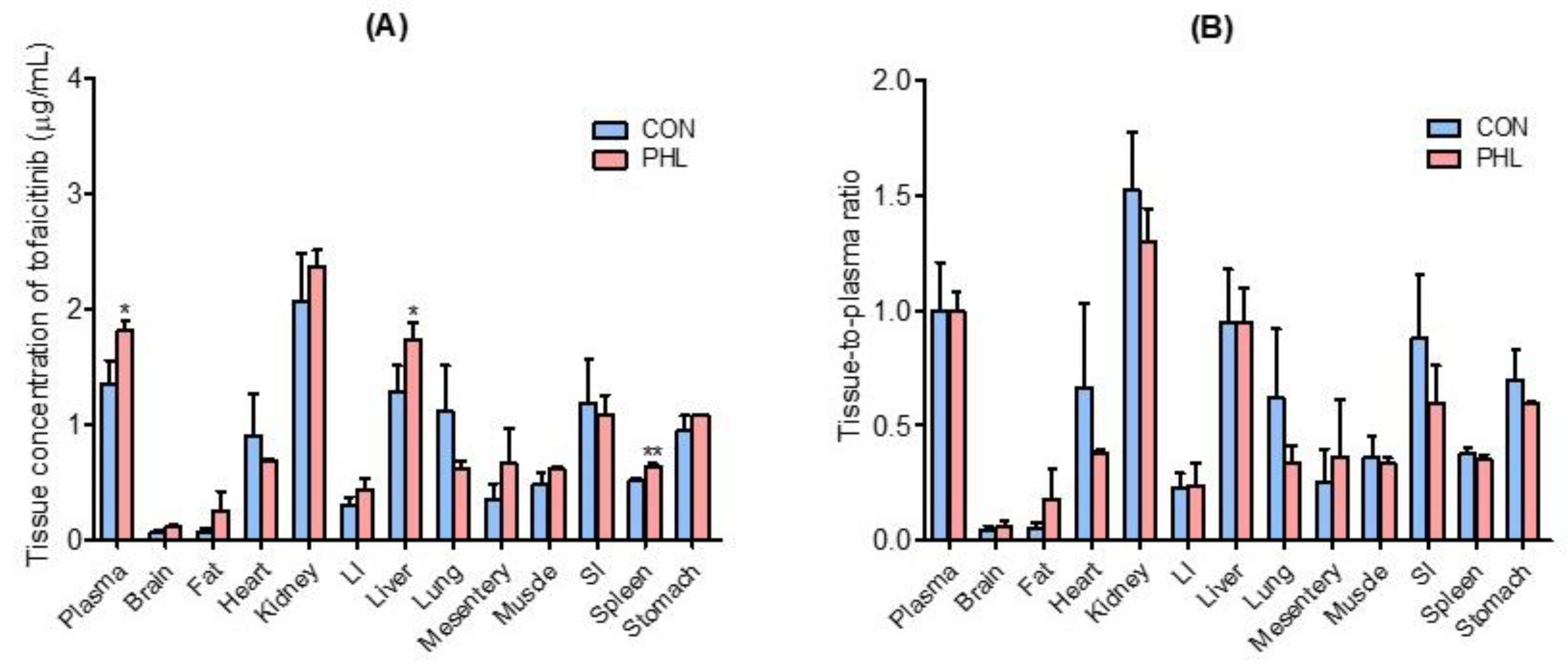
3.4. Distribution of Tofacitinib in Tissues
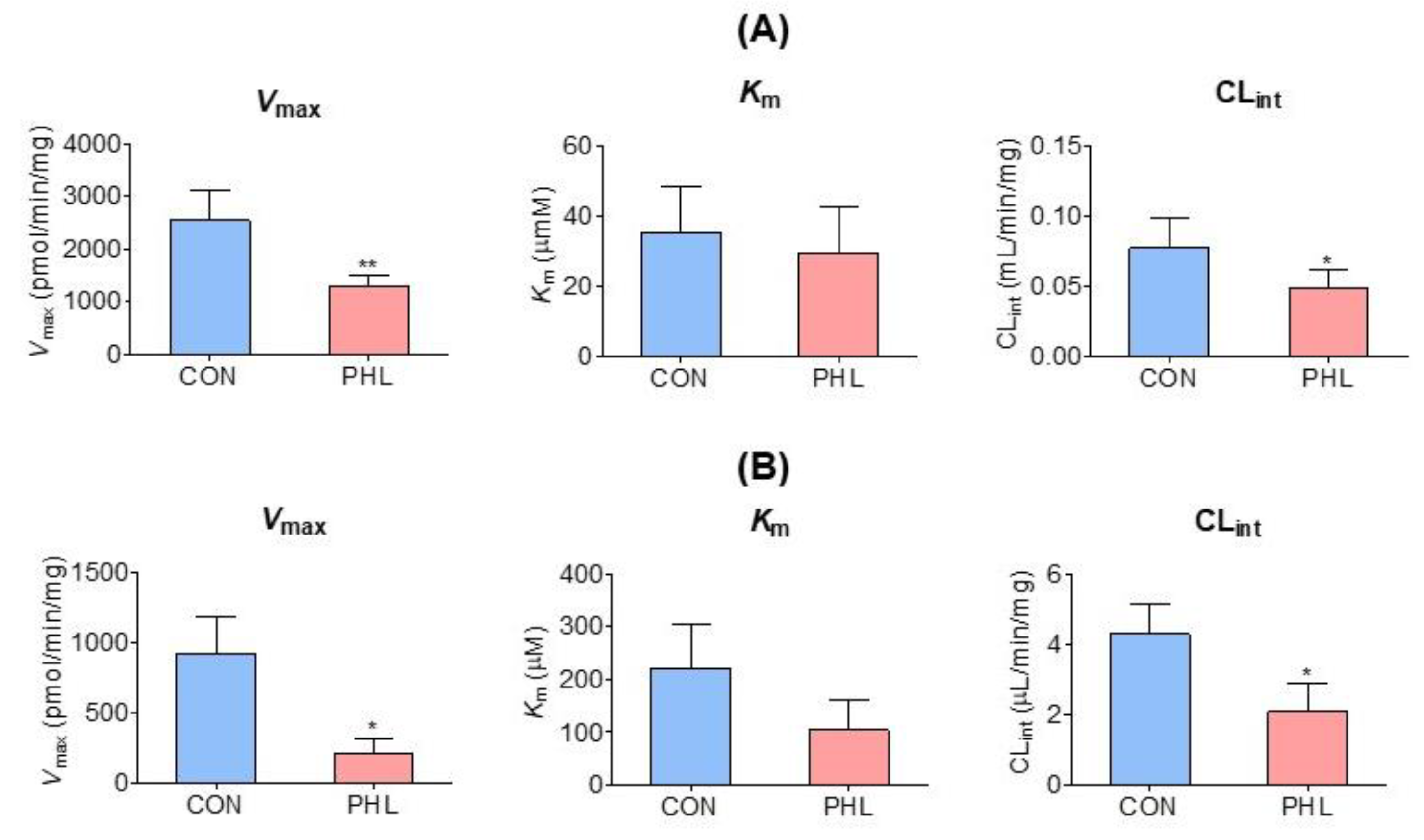
3.5. Measurement of Km, Vmax, and CLint of Tofacitinib
3.6. Protein Expression of CYP Isozymes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Claxton, L.; Taylor, M.; Soonasra, A.; Bourret, J.A.; Gerber, R.A. An economic evaluation of tofacitinib treatment in rheumatoid arthritis after methotrexate or after 1 or 2 TNF inhibitors from a U.S. payer perspective. J. Manag. Care Spec. Pharm. 2018, 24, 1010–1017. [Google Scholar] [CrossRef] [PubMed]
- Sandborn, W.J.; Peyrin-Biroulet, L.; Sharara, A.I.; Su, C.; Modesto, I.; Mundayat, R.; Gunay, L.M.; Salese, L.; Sands, B.E. Efficacy and safety of tofacitinib in ulcerative colitis based on prior tumor necrosis factor inhibitor failure status. Clin. Gastroenterol. Hepatol. 2022, 20, 591–601. [Google Scholar] [CrossRef]
- Changelian, P.S.; Moshinsky, D.; Kuhn, C.F.; Flanagan, M.E.; Munchhof, M.J.; Harris, T.M.; Whipple, D.A.; Doty, J.L.; Sun, J.; Kent, C.R.; et al. The specificity of JAK3 kinase inhibitors. Blood 2008, 111, 2155–2157. [Google Scholar] [CrossRef] [PubMed]
- Flanagan, M.E.; Blumenkopf, T.A.; Brissette, W.H.; Brown, M.F.; Casavant, J.M.; Shang-Poa, C.; Doty, J.L.; Elliott, E.A.; Fisher, M.B.; Hines, M.; et al. Discovery of CP-690,550: A potent and selective Janus kinase (JAK) inhibitor for the treatment of autoimmune diseases and organ transplant rejection. J. Med. Chem. 2010, 53, 8468–8484. [Google Scholar] [CrossRef]
- Fukuda, T.; Naganuma, M.; Kanai, T. Current new challenges in the management of ulcerative colitis. Intest. Res. 2019, 17, 36–44. [Google Scholar] [CrossRef]
- Dowty, M.E.; Lin, J.; Ryder, T.F.; Wang, W.; Walker, G.S.; Vaz, A.; Prakash, C. The pharmacokinetics, metabolism, and clearance mechanisms of tofacitinib, a janus kinase inhibitor, in humans. Drug Metab. Dispos. 2014, 42, 759–773. [Google Scholar] [CrossRef] [PubMed]
- Cada, D.J.; Demaris, K.; Levien, T.L.; Baker, D.E. Tofacitinib. Hosp. Pharm. 2013, 48, 413–424. [Google Scholar] [CrossRef]
- Scott, L.J. Tofacitinib: A review of its use in adult patients with rheumatoid arthritis. Drugs 2013, 73, 857–874. [Google Scholar] [CrossRef]
- Mohanakrishnan, R.; Beier, S.; Deodhar, A. Tofacitinib for the treatment of active ankylosing spondylitis in adults. Expert Rev. Clin. Immunol. 2022, 18, 273–280. [Google Scholar] [CrossRef]
- Ma, G.; Xie, R.; Strober, B.; Langley, R.; Ito, K.; Krishnaswami, S.; Wolk, R.; Valdez, H.; Rottinghaus, S.; Tallman, A.; et al. Pharmacokinetic characteristics of tofacitinib in adult patients with moderate to severe chronic plaque psoriasis. Clin. Pharmacol. Drug Dev. 2018, 7, 587–596. [Google Scholar] [CrossRef]
- Vincenti, F.; Tedesco Silva, H.; Busque, S.; O’Connell, P.; Friedewald, J.; Cibrik, D.; Budde, K.; Yoshida, A.; Cohney, S.; Weimar, W.; et al. Randomized phase 2b trial of tofacitinib (CP-690,550) in de novo kidney transplant patients: Efficacy, renal function and safety at 1 year. Am. J. Transplant. 2012, 12, 2446–2456. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.D.; Kang, K.S.; Kwon, R.H.; Yang, J.O.; Lee, J.S.; Ha, B.J. The effect of Rubus coreanum Miquel against lipopolysaccharide-induced oxidative stress and lipid metaobolism. J. Food Hyg. Saf. 2007, 22, 213–217. [Google Scholar]
- Choi, G.H.; Jung, Y.S.; Shin, H.C. The Effects of Haedoksamul-tang on Oxidative Stress and Hyperlipidemia in LPS-induced ICR Mouse. J. Korean Med. 2016, 37, 77–89. [Google Scholar] [CrossRef]
- Kim, K.S.; Ezaki, O.; Ikemoto, S.; Itakura, H. Effects of Platycodon grandiflorum feeding on serum and liver lipid concentrations in rats with diet-induced hyperlipidemia. J. Nutr. Sci. Vitaminol. 1995, 41, 485–491. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, J.L.; Hazzard, W.R.; Schrott, H.G.; Bierman, E.L.; Motulsky, A.G. Hyperlipidemia in coronary heart disease I. Lipid levels in 500 survivors of myocardial infarction. J. Clin. Investig. 1973, 52, 1533–1543. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Oh, J.H.; Lee, Y.J. Effects of experimental hyperlipidemia on the pharmacokinetics of tadalafil in rats. J. Pharm. Pharm. Sci. 2012, 15, 528–537. [Google Scholar] [CrossRef]
- Eliot, L.A.; Foster, R.T.; Jamali, F. Effects of hyperlipidemia on the pharmacokinetics of nifedipine in the rat. Pharm. Res. 1999, 16, 309–313. [Google Scholar] [CrossRef]
- Fukushima, K.; Shibata, M.; Mizuhara, K. Effect of serum lipids on the pharmacokinetics of atazanavir in hyperlipidemic rats. Biomed. Pharmacother. 2009, 63, 635–642. [Google Scholar] [CrossRef]
- Perloff, E.S.; Duan, S.X.; Skolnik, P.R. Atazanavir: Effects on Pglycoprotein transport and CYP3A metabolism in vitro. Drug. Metab. Dispos. 2005, 33, 764–770. [Google Scholar] [CrossRef]
- Lee, J.H.; Oh, J.H.; Lee, Y.J. Effects of experimental hyperlipidaemia on the pharmacokinetics of docetaxel in rats. Xenobiotica 2011, 41, 797–804. [Google Scholar] [CrossRef]
- Kerr, B.M.; Thummel, K.E.; Wurden, C.J. Human liver carbamazepine metabolism. Role of CYP3A4 and CYP2C8 in 10,11-epoxide formation. Biochem. Pharmacol. 1994, 47, 1969–1979. [Google Scholar] [CrossRef] [PubMed]
- Qiu, W.; Jiang, X.H.; Liu, C.X. Effect of berberine on the pharmacokinetics of substrates of CYP3A and P-gp. Phytother. Res. 2009, 23, 1553–1558. [Google Scholar] [CrossRef] [PubMed]
- Hanada, K.; Ikemi, Y.; Kukita, K. Stereoselective first-pass metabolism of verapamil in the small intestine and liver in rats. Drug. Metab. Dispos. 2008, 36, 2037–2042. [Google Scholar] [CrossRef]
- Hu, N.; Xie, S.; Liu, L. Opposite effect of diabetes mellitus induced by streptozotocin on oral and intravenous pharmacokinetics of verapamil in rats. Drug. Metab. Dispos. 2011, 39, 419–425. [Google Scholar] [CrossRef] [PubMed]
- Lee, U.J.; Kwon, M.H.; Kang, H.E. Pharmacokinetic alterations in poloxamer 407-induced hyperlipidemic rats. Xenobiotica 2019, 49, 611–625. [Google Scholar] [CrossRef] [PubMed]
- Bae, S.H.; Choi, H.G.; Park, S.Y.; Chang, S.-Y.; Kim, H.; Kim, S.H. Effects of Isosakuranetin on Pharmacokinetic Changes of Tofacitinib in Rats with N-Dimethylnitrosamine-Induced Liver Cirrhosis. Pharmaceutics 2022, 14, 2684. [Google Scholar] [CrossRef]
- Bae, S.H.; Chang, S.-Y.; Kim, S.H. Slower elimination of tofacitinib in acute renal failure rat models: Contribution of hepatic metabolism and renal excretion. Pharmaceutics 2020, 12, 714. [Google Scholar] [CrossRef]
- Lee, Y.S.; Yoon, J.N.; Yoon, I.S. Pharmacokinetics of verapamil and its metabolite norverapamil in rats with hyperlipidaemia induced by poloxamer 407. Xenobiotica 2012, 42, 766–774. [Google Scholar] [CrossRef]
- Choi, M.R.; Kwon, M.H.; Cho, Y.Y.; Choi, H.D.; Kim, Y.C.; Kang, H.E. Pharmacokinetics of tolbutamide and its metabolite 4-hydroxy tolbutamide in poloxamer 407-induced hyperlipidemic rats. Biopharm Drug Dispos. 2014, 35, 264–274. [Google Scholar] [CrossRef]
- Matos, S.L.; Paula, H.D.; Pedrosa, M.L.; Santos, R.C.D.; Oliveira, E.L.D.; Chianca Júnior, D.A.; Silva, M.E. Dietary models for inducing hypercholesterolemia in rats. Braz. Arch. Biol. Technol. 2005, 48, 203–209. [Google Scholar] [CrossRef]
- Barre, J.; Chamouard, J.M.; Houin, G.; Tillement, J.P. Equilibrium dialysis, ultrafiltration, and ultracentrifugation compared for determining the plasma-protein-binding characteristics of valproic acid. Clin. Chem. 1985, 31, 60–64. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.E.; Park, M.Y.; Kim, S.H. Simple determination and quantification of tofacitinib, a JAK inhibitor, in rat plasma, urine and tissue homogenates by HPLC and its application to a pharmacokinetic study. J. Pharm. Investig. 2020, 50, 603–612. [Google Scholar] [CrossRef]
- Lee, J.S.; Kim, S.H. Dose-Dependent Pharmacokinetics of Tofacitinib in Rats: Influence of Hepatic and Intestinal First-Pass Metabolism. Pharmaceutics 2019, 11, 318. [Google Scholar] [CrossRef]
- Duggleby, R.G. Analysis of enzyme progress curves by nonlinear regression. Methods Enzymol. 1995, 249, 61–90. [Google Scholar] [PubMed]
- Gibaldi, M.; Perrier, D. Pharmacokinetics, 2nd ed.; Marcel-Dekker: New York, NY, USA, 1982. [Google Scholar]
- Chiou, W.L. Critical evaluation of the potential error in pharmacokinetic studies of using the linear trapezoidal rule method for the calculation of the area under the plasma level-time curve. J. Pharmacokinet. Biopharm. 1978, 6, 539–546. [Google Scholar] [CrossRef]
- Johnston, T.P. The P-407-induced murine model of dose-controlled hyperlipidemia and atherosclerosis: A review of findings to date. J. Cardiovasc. Pharmacol. 2004, 43, 595–606. [Google Scholar] [CrossRef]
- Johnston, T.P.; Palmer, W.K. Mechanism of poloxamer 407-induced hypertriglyceridemia in the rat. Biochem. Pharmacol. 1993, 46, 1037–1042. [Google Scholar] [CrossRef] [PubMed]
- Kasim, S.E.; LeBoeuf, R.C.; Khilnani, S.; Tallapaka, L.; Dayananda, D.; Jen, K.L. Mechanisms of triglyceride-lowering effect of an HMG-CoA reductase inhibitor in a hypertriglyceridemic animal model, the Zucker obese rat. J. Lipid Res. 1992, 33, 1–7. [Google Scholar] [CrossRef]
- Wout, Z.G.; Pec, E.A.; Maggiore, J.A.; Williams, R.H.; Palicharla, P.; Johnston, T.P. Poloxamer 407-mediated changes in plasma cholesterol and triglycerides following intraperitoneal injection to rats. J. Parenter. Sci. Technol. 1992, 46, 192–200. [Google Scholar]
- Choi, Y.H.; Yoon, I.; Kim, Y.G.; Lee, M.G. Effects of cysteine on the pharmacokinetics of docetaxel in rats with protein-calorie malnutrition. Xenobiotica 2012, 42, 442–455. [Google Scholar] [CrossRef]
- Shayeganpour, A.; Korashy, H.; Patel, J.P.; El-Kadi, A.O.; Brocks, D.R. The impact of experimental hyperlipidemia on the distribution and metabolism of amiodarone in rat. Int. J. Pharm. 2008, 361, 78–86. [Google Scholar] [CrossRef]
- Sugioka, N.; Haraya, K.; Maeda, Y. Pharmacokinetics of human immunodeficiency virus protease inhibitor, nelfinavir, in poloxamer 407-induced hyperlipidemic model rats. Biol. Pharm. Bull. 2009, 32, 269–275. [Google Scholar] [CrossRef] [PubMed]
- Aliabadi, H.M.; Spencer, T.J.; Mahdipoor, P. Insights into the effects of hyperlipoproteinemia on cyclosporine A biodistribution and relationship to renal function. J. AAPS 2006, 8, 672. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.S.; Kim, Y.W.; Kim, S.G.; Lee, I.; Lee, M.G.; Kang, H.E. Effects of poloxamer 407-induced hyperlipidemia on the pharmacokinetics of carbamazepine and its 10, 11-epoxide metabolite in rats: Impact of decreased expression of both CYP3A1/2 and microsomal epoxide hydrolase. Eur. Neuropsychopharmacol. 2012, 22, 431–440. [Google Scholar] [CrossRef] [PubMed]
- Okabe, H.; Hasunuma, M.; Hashimoto, Y. The hepatic and intestinal metabolic activities of P450 in rats with surgery- and drug-induced renal dysfunction. Pharm. Res. 2003, 20, 1591–1594. [Google Scholar] [CrossRef] [PubMed]
- Hussa, D.A. 2013 new drug update: What do new approvals hold for the elderly? Consult. Pharm. 2014, 29, 224–238. [Google Scholar] [CrossRef] [PubMed]
- Shayeganpour, A.; Jun, A.S.; Brocks, D.R. Pharmacokinetics of Amiodarone in hyperlipidemic and simulated high fat-meal rat models. Biopharm. Drug Dispos. 2005, 26, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Kwong, T.C.; Sparks, J.D.; Sparks, C.E. Lipoprotein and protein binding of the calcium channel blocker diltiazem. Proc. Soc. Exp. Biol. Med. 1985, 178, 313–316. [Google Scholar] [CrossRef]
- Patel, J.P.; Fleischer, J.G.; Wasan, K.M.; Brocks, D.R. The effect of experimental hyperlipidemia on the stereoselective tissue distribution, lipoprotein association and microsomal metabolism of (þ/-)-halofantrine. J. Pharm. Sci. 2009, 98, 2516–2528. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | Intravenous | Oral | ||
|---|---|---|---|---|
| CON (n = 7) | PHL (n = 6) | CON (n = 6) | PHL (n = 7) | |
| Body weight (g) | 296 ± 4.69 | 294 ± 11.4 | 235 ± 4.71 | 250 ± 17.9 |
| Terminal half-life (min) | 29.6 ± 4.55 | 34.5 ± 2.29 | 62.1 ± 16.7 | 84.3 ± 21.2 |
| k (min−1) | 0.0239 ± 0.00337 | 0.0202 ± 0.00131 | 0.0118 ± 0.00301 | 0.00870 ± 0.00233 |
| AUC (µg × min/mL) | 268 ± 34.4 | 465 ± 24.1 *** | 288 ± 38.6 | 477 ± 44.6 *** |
| ka (min−1) | 0.127 ± 0.0389 | 0.156 ± 0.0885 | ||
| Cmax (µg/mL) | 2.96 ± 0.643 | 4.75 ± 1.18 ** | ||
| Tmax (min) | 25.0 ± 14.7 | 30.0 ± 25.4 | ||
| CL (mL/min/kg) | 37.9 ± 4.85 | 21.6 ± 1.13 *** | ||
| CLR (mL/min/kg) | 3.11 ± 1.58 | 2.47 ± 1.10 | 3.52 ± 1.80 | 1.28 ± 0.786 * |
| CLNR (mL/min/kg) | 34.8 ± 4.55 | 18.4 ± 1.25 *** | ||
| MRT (min) | 22.6 ± 5.13 | 22.7 ± 5.88 | ||
| Vss (mL/kg) | 847 ± 191 | 490 ± 136 ** | ||
| Ae0–24 h (% of dose) | 4.06 ± 1.89 | 5.76 ± 2.53 | 4.90 ± 1.95 | 3.02 ± 1.87 |
| F (%) | 53.7 | 51.4 | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Won, J.M.; Choi, H.G.; Park, S.Y.; Kim, J.-H.; Kim, S.H. Effects of Hyperlipidemia on the Pharmacokinetics of Tofacitinib, a JAK 1/3 Inhibitor, in Rats. Pharmaceutics 2023, 15, 2195. https://doi.org/10.3390/pharmaceutics15092195
Won JM, Choi HG, Park SY, Kim J-H, Kim SH. Effects of Hyperlipidemia on the Pharmacokinetics of Tofacitinib, a JAK 1/3 Inhibitor, in Rats. Pharmaceutics. 2023; 15(9):2195. https://doi.org/10.3390/pharmaceutics15092195
Chicago/Turabian StyleWon, Jong Mun, Hyeon Gyeom Choi, So Yeon Park, Jang-Hee Kim, and So Hee Kim. 2023. "Effects of Hyperlipidemia on the Pharmacokinetics of Tofacitinib, a JAK 1/3 Inhibitor, in Rats" Pharmaceutics 15, no. 9: 2195. https://doi.org/10.3390/pharmaceutics15092195
APA StyleWon, J. M., Choi, H. G., Park, S. Y., Kim, J.-H., & Kim, S. H. (2023). Effects of Hyperlipidemia on the Pharmacokinetics of Tofacitinib, a JAK 1/3 Inhibitor, in Rats. Pharmaceutics, 15(9), 2195. https://doi.org/10.3390/pharmaceutics15092195