
Formulation and Evaluation of a Self-Microemulsifying Drug Delivery System of Raloxifene with Improved Solubility and Oral Bioavailability

 ,
,  , , ,
, , ,  , and
, and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Determination of RLX Solubility in Different Oils, Surfactants, and Cosurfactants
2.3. HPLC Analysis
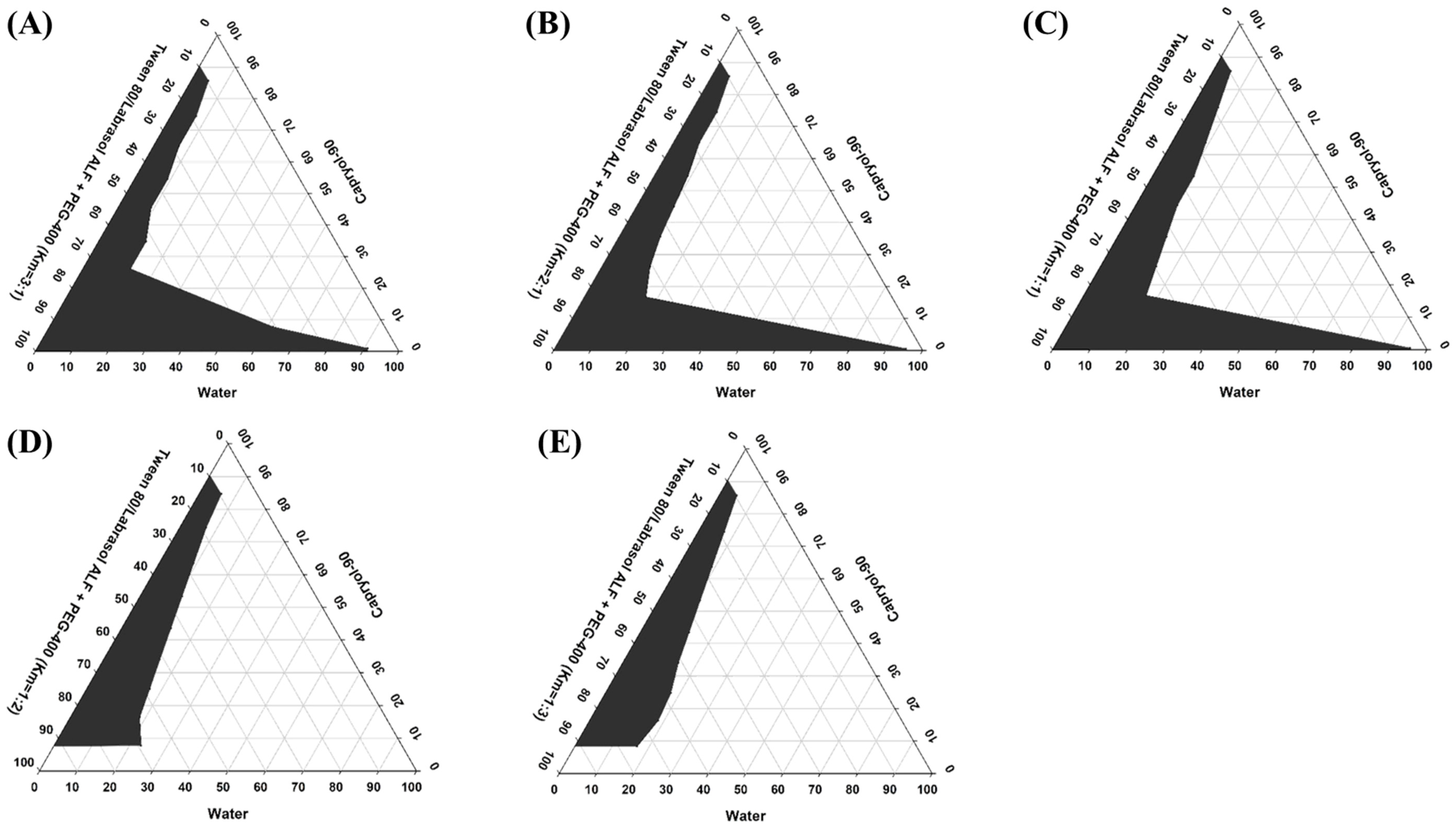
2.4. Construction of Pseudo-Ternary Phase Diagrams
2.5. Preparation of Liquid and Solid RLX-SMEDDS
2.6. Droplet Size Analysis and Self-Microemulsifying Behavior of Liquid RLX-SMEDDS
2.7. Characterization of Lyophilized RLX-SMEDDS
2.7.1. Morphological Analysis
2.7.2. Differential Scanning Calorimetry (DSC)
2.7.3. Powder X-ray Diffraction (PXRD)
2.7.4. Fourier-Transform Infrared (FTIR) Spectroscopy
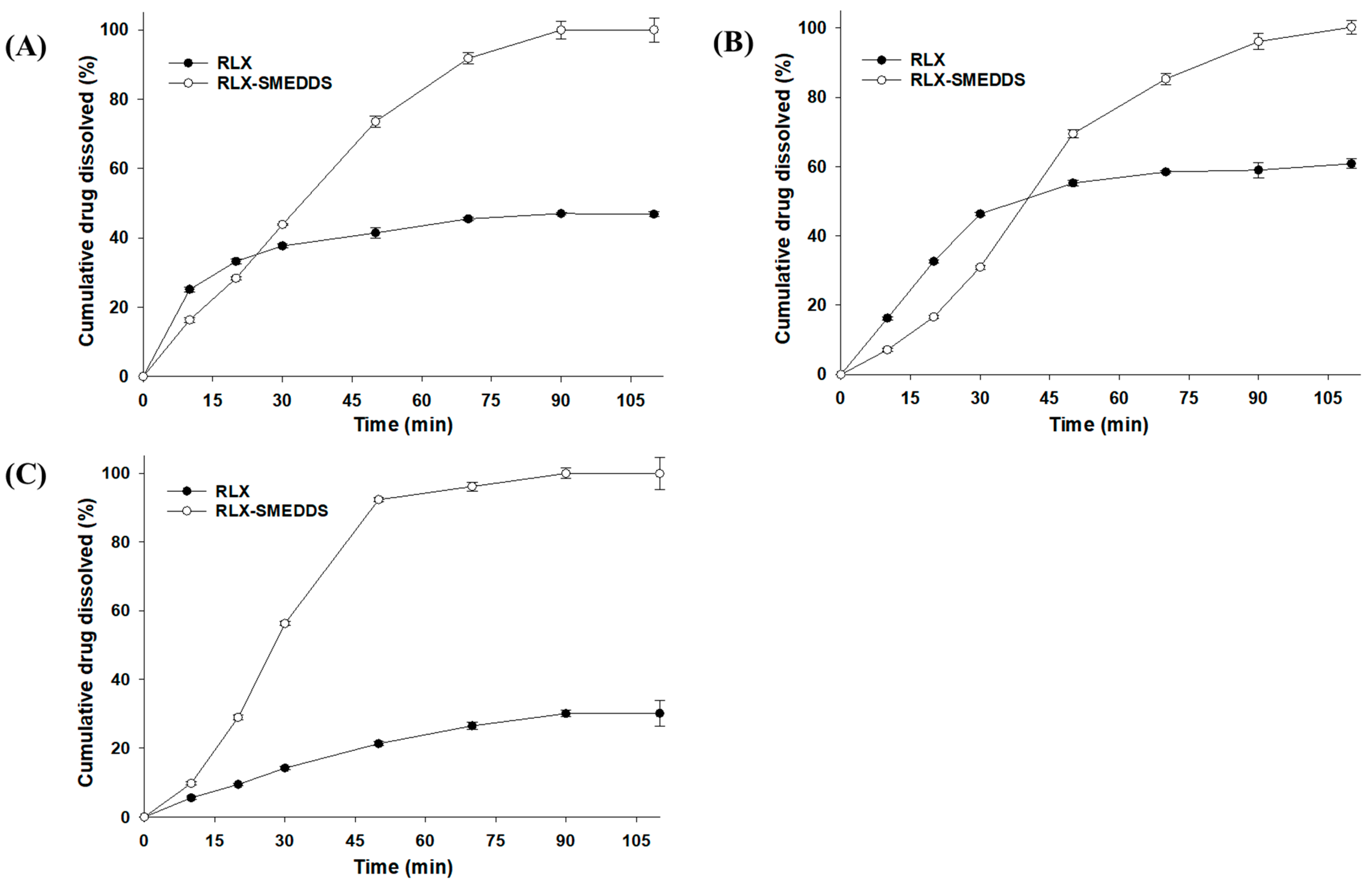
2.8. Saturation Solubility and In Vitro Dissolution Study of RLX-SMEDDS
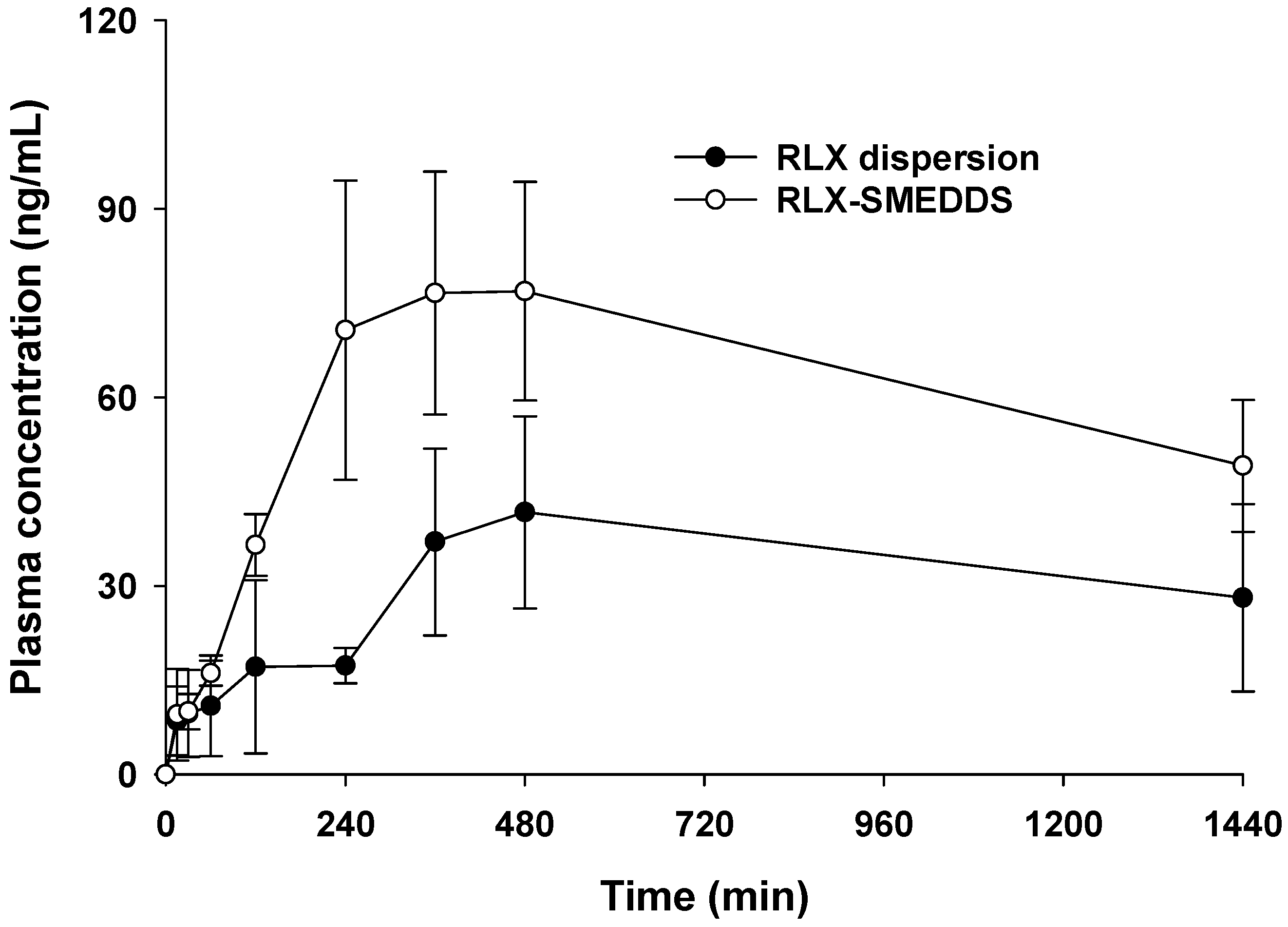
2.9. In Vivo Pharmacokinetic Study
2.9.1. Animals
2.9.2. Oral Dosing, Blood Sampling, and Plasma Collection
2.9.3. Sample Preparation, Liquid Chromatography-Tandem Mass Spectrometry (LC-MS/MS) Analysis and Pharmacokinetic Parameters
2.10. Statistical Analysis
3. Results and Discussion
3.1. Selection of Oil, Surfactants, and Cosurfactants
3.2. Pseudo-Ternary Phase Diagrams
3.3. Optimization of RLX-SMEDDS
3.4. Solid State Characteristics of RLX-SMEDDS
3.5. Saturation Solubility and Dissolution Profile of RLX-SMEDDS
3.6. Pharmacokinetics Profile
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Moen, M.D.; Keating, G.M. Raloxifene: A review of its use in the prevention of invasive breast cancer. Drugs 2008, 68, 2059–2083. [Google Scholar] [CrossRef]
- Muhammad, A.; Mada, S.B.; Malami, I.; Forcados, G.E.; Erukainure, O.L.; Sani, H.; Abubakar, I.B. Postmenopausal osteoporosis and breast cancer: The biochemical links and beneficial effects of functional foods. Biomed. Pharmacother. 2018, 107, 571–582. [Google Scholar] [CrossRef]
- Grady, D.; Rubin, S.M.; Petitti, D.B.; Fox, C.S.; Black, D.; Ettinger, B.; Ernster, V.L.; Cummings, S.R. Hormone therapy to prevent disease and prolong life in postmenopausal women. Ann. Intern. Med. 1992, 117, 1016–1037. [Google Scholar] [CrossRef]
- Sacco, S.M.; Ward, W.E. Revisiting estrogen: Efficacy and safety for postmenopausal bone health. J. Osteoporos. 2010, 2010, 708931. [Google Scholar] [CrossRef]
- Snyder, K.R.; Sparano, N.; Malinowski, J.M. Raloxifene hydrochloride. Am. J. Health Syst. Pharm. 2000, 57, 1669–1675. [Google Scholar] [CrossRef]
- Heringa, M. Review on raloxifene: Profile of a selective estrogen receptor modulator. Int. J. Clin. Pharmacol. Ther. 2003, 41, 331–345. [Google Scholar] [CrossRef]
- Lindstrom, T.D.; Whitaker, N.G.; Whitaker, G.W. Disposition and metabolism of a new benzothiophene antiestrogen in rats, dogs and monkeys. Xenobiotica 1984, 14, 841–847. [Google Scholar] [CrossRef]
- Jeong, E.J.; Liu, Y.; Lin, H.; Hu, M. Species- and disposition model-dependent metabolism of raloxifene in gut and liver: Role of UGT1A10. Drug Metab. Dispos. 2005, 33, 785–794. [Google Scholar] [CrossRef]
- Dodge, J.A.; Lugar, C.W.; Cho, S.; Short, L.L.; Sato, M.; Yang, N.N.; Spangle, L.A.; Martin, M.J.; Phillips, D.L.; Glasebrook, A.L.; et al. Evaluation of the major metabolites of raloxifene as modulators of tissue selectivity. J. Steroid Biochem. Mol. Biol. 1997, 61, 97–106. [Google Scholar] [CrossRef]
- Fontana, F.; Figueiredo, P.; Zhang, P.; Hirvonen, J.T.; Liu, D.; Santos, H.A. Production of pure drug nanocrystals and nano co-crystals by confinement methods. Adv. Drug Deliv. Rev. 2018, 131, 3–21. [Google Scholar] [CrossRef]
- Peltonen, L.; Hirvonen, J. Drug nanocrystals—Versatile option for formulation of poorly soluble materials. Int. J. Pharm. 2018, 537, 73–83. [Google Scholar] [CrossRef]
- Khan, M.A.; Ansari, M.M.; Arif, S.T.; Raza, A.; Choi, H.I.; Lim, C.W.; Noh, H.Y.; Noh, J.S.; Akram, S.; Nawaz, H.A.; et al. Eplerenone nanocrystals engineered by controlled crystallization for enhanced oral bioavailability. Drug Deliv. 2021, 28, 2510–2524. [Google Scholar] [CrossRef]
- Mizuma, T. Intestinal glucuronidation metabolism may have a greater impact on oral bioavailability than hepatic glucuronidation metabolism in humans: A study with raloxifene, substrate for UGT1A1, 1A8, 1A9, and 1A10. Int. J. Pharm. 2009, 378, 140–141. [Google Scholar] [CrossRef]
- Ye, J.Y.; Chen, Z.Y.; Huang, C.L.; Huang, B.; Zheng, Y.R.; Zhang, Y.F.; Lu, B.Y.; He, L.; Liu, C.S.; Long, X.Y. A Non-Lipolysis Nanoemulsion Improved Oral Bioavailability by Reducing the First-Pass Metabolism of Raloxifene, and Related Absorption Mechanisms Being Studied. Int. J. Nanomed. 2020, 15, 6503–6518. [Google Scholar] [CrossRef]
- Murthy, A.; Ravi, P.R.; Kathuria, H.; Vats, R. Self-assembled lecithin-chitosan nanoparticles improve the oral bioavailability and alter the pharmacokinetics of raloxifene. Int. J. Pharm. 2020, 588, 119731. [Google Scholar] [CrossRef]
- Tran, T.H.; Poudel, B.K.; Marasini, N.; Chi, S.-C.; Choi, H.-G.; Yong, C.S.; Kim, J.O. Preparation and evaluation of raloxifene-loaded solid dispersion nanoparticle by spray-drying technique without an organic solvent. Int. J. Pharm. 2013, 443, 50–57. [Google Scholar] [CrossRef]
- Tran, T.H.; Poudel, B.K.; Marasini, N.; Woo, J.S.; Choi, H.G.; Yong, C.S.; Kim, J.O. Development of raloxifene-solid dispersion with improved oral bioavailability via spray-drying technique. Arch. Pharmacal Res. 2013, 36, 86–93. [Google Scholar] [CrossRef]
- Wempe, M.F.; Wacher, V.J.; Ruble, K.M.; Ramsey, M.G.; Edgar, K.J.; Buchanan, N.L.; Buchanan, C.M. Pharmacokinetics of raloxifene in male Wistar–Hannover rats: Influence of complexation with hydroxybutenyl-beta-cyclodextrin. Int. J. Pharm. 2008, 346, 25–37. [Google Scholar] [CrossRef]
- Shah, N.; Seth, A.; Balaraman, R.; Sailor, G.; Javia, A.; Gohil, D. Oral bioavailability enhancement of raloxifene by developing microemulsion using D-optimal mixture design: Optimization and in-vivo pharmacokinetic study. Drug Dev. Ind. Pharm. 2018, 44, 687–696. [Google Scholar] [CrossRef]
- Murthy, A.; Ravi, P.R.; Kathuria, H.; Malekar, S. Oral Bioavailability Enhancement of Raloxifene with Nanostructured Lipid Carriers. Nanomaterials 2020, 10, 1085. [Google Scholar] [CrossRef]
- Shah, N.V.; Seth, A.K.; Balaraman, R.; Aundhia, C.J.; Maheshwari, R.A.; Parmar, G.R. Nanostructured lipid carriers for oral bioavailability enhancement of raloxifene: Design and in vivo study. J. Adv. Res. 2016, 7, 423–434. [Google Scholar] [CrossRef]
- Lu, R.; Liu, S.; Wang, Q.; Li, X. Enhanced bioavailability of raloxifene hydrochloride via dry suspensions prepared from drug/HP-β-cyclodextrin inclusion complexes. Pharmazie 2015, 70, 791–797. [Google Scholar]
- Varshosaz, J.; Dayani, L.; Chegini, S.P.; Minaiyan, M. Production of a new platform based on fumed and mesoporous silica nanoparticles for enhanced solubility and oral bioavailability of raloxifene HCl. IET Nanobiotechnol. 2019, 13, 392–399. [Google Scholar] [CrossRef]
- Jain, A.; Saini, S.; Kumar, R.; Sharma, T.; Swami, R.; Katare, O.P.; Singh, B. Phospholipid-based complex of raloxifene with enhanced biopharmaceutical potential: Synthesis, characterization and preclinical assessment. Int. J. Pharm. 2019, 571, 118698. [Google Scholar] [CrossRef]
- Aditya, N.; Ravi, P.R.; Avula, U.S.; Vats, R. Poly (ε-caprolactone) nanocapsules for oral delivery of raloxifene: Process optimization by hybrid design approach, in vitro and in vivo evaluation. J. Microencapsul. 2014, 31, 508–518. [Google Scholar] [CrossRef]
- Varshosaz, J.; Minaiyan, M.; Dayyani, L. Poly(methyl vinyl ether-co-maleic acid) for enhancement of solubility, oral bioavailability and anti-osteoporotic effects of raloxifene hydrochloride. Eur. J. Pharm. Sci. 2018, 112, 195–206. [Google Scholar] [CrossRef]
- Cho, W.; Kim, M.S.; Kim, J.S.; Park, J.; Park, H.J.; Cha, K.H.; Park, J.S.; Hwang, S.J. Optimized formulation of solid self-microemulsifying sirolimus delivery systems. Int. J. Nanomed. 2013, 8, 1673–1682. [Google Scholar]
- Dokania, S.; Joshi, A.K. Self-microemulsifying drug delivery system (SMEDDS)—Challenges and road ahead. Drug Deliv. 2015, 22, 675–690. [Google Scholar] [CrossRef]
- Kim, D.S.; Cho, J.H.; Park, J.H.; Kim, J.S.; Song, E.S.; Kwon, J.; Giri, B.R.; Jin, S.G.; Kim, K.S.; Choi, H.G.; et al. Self-microemulsifying drug delivery system (SMEDDS) for improved oral delivery and photostability of methotrexate. Int. J. Nanomed. 2019, 14, 4949–4960. [Google Scholar] [CrossRef]
- Marasini, N.; Yan, Y.D.; Poudel, B.K.; Choi, H.G.; Yong, C.S.; Kim, J.O. Development and optimization of self-nanoemulsifying drug delivery system with enhanced bioavailability by Box-Behnken design and desirability function. J. Pharm. Sci. 2012, 101, 4584–4596. [Google Scholar] [CrossRef]
- USP. Raloxifene hydrochloride tablets. In United States Pharmacopeia (USP), 43rd ed.; United States Pharmacopeial Convention; United States Pharmacopeia (USP): Rockville, MD, USA, 2020; p. 3830. [Google Scholar]
- Zhuang, X.; Tian, X.; Zheng, Y.; Lan, N.; Liu, L.; Zhang, R.; Liu, Y. Formulation and physicochemical characterisation of a novel self-microemulsifying delivery system as hydrotropic and solubilising agent for penfluridol. Procedia Eng. 2011, 18, 59–65. [Google Scholar] [CrossRef][Green Version]
- Khoo, S.-M.; Humberstone, A.J.; Porter, C.J.H.; Edwards, G.A.; Charman, W.N. Formulation design and bioavailability assessment of lipidic self-emulsifying formulations of halofantrine. Int. J. Pharm. 1998, 167, 155–164. [Google Scholar] [CrossRef]
- Nasr, A.; Gardouh, A.; Ghorab, M. Novel Solid Self-Nanoemulsifying Drug Delivery System (S-SNEDDS) for Oral Delivery of Olmesartan Medoxomil: Design, Formulation, Pharmacokinetic and Bioavailability Evaluation. Pharmaceutics 2016, 8, 20. [Google Scholar] [CrossRef]
- Thakkar, H.; Nangesh, J.; Parmar, M.; Patel, D. Formulation and characterization of lipid-based drug delivery system of raloxifene-microemulsion and self-microemulsifying drug delivery system. J. Pharm. Bioallied Sci. 2011, 3, 442–448. [Google Scholar] [CrossRef]
- Liu, P.; De Wulf, O.; Laru, J.; Heikkilä, T.; van Veen, B.; Kiesvaara, J.; Hirvonen, J.; Peltonen, L.; Laaksonen, T. Dissolution studies of poorly soluble drug nanosuspensions in non-sink conditions. AAPS PharmSciTech 2013, 14, 748–756. [Google Scholar] [CrossRef]
- Doan, T.N.K.; Vo, D.-K.; Kim, H.; Balla, A.; Lee, Y.; Yoon, I.-S.; Maeng, H.-J. Differential Effects of 1α,25-Dihydroxyvitamin D3 on the Expressions and Functions of Hepatic CYP and UGT Enzymes and Its Pharmacokinetic Consequences In Vivo. Pharmaceutics 2020, 12, 1129. [Google Scholar] [CrossRef]
- Balla, A.; Jeong, Y.-S.; Kim, H.-J.; Lee, Y.-J.; Chung, S.-J.; Chae, Y.-J.; Maeng, H.-J. Effects of 1α,25-Dihydroxyvitamin D3 on the Pharmacokinetics of Procainamide and Its Metabolite N-Acetylprocainamide, Organic Cation Transporter Substrates, in Rats with PBPK Modeling Approach. Pharmaceutics 2021, 13, 1133. [Google Scholar] [CrossRef]
- Le, Q.-D.; Duong, V.-A.; Lee, S.-H.; Nguyen, T.-T.-L.; Maeng, H.-J. Bioanalytical method validation, biopharmaceutical and pharmacokinetic evaluation of GSK-650394, a serum- and glucocorticoid-regulated kinase 1 inhibitor. Arab. J. Chem. 2023, 16, 104462. [Google Scholar] [CrossRef]
- Nguyen, T.-T.-L.; Duong, V.-A.; Vo, D.-K.; Jo, J.; Maeng, H.-J. Development and Validation of a Bioanalytical LC-MS/MS Method for Simultaneous Determination of Sirolimus in Porcine Whole Blood and Lung Tissue and Pharmacokinetic Application with Coronary Stents. Molecules 2021, 26, 425. [Google Scholar] [CrossRef]
- Nguyen, T.-T.-L.; Kim, J.W.; Choi, H.-I.; Maeng, H.-J.; Koo, T.-S. Development of an LC-MS/MS Method for ARV-110, a PROTAC Molecule, and Applications to Pharmacokinetic Studies. Molecules 2022, 27, 1977. [Google Scholar] [CrossRef]
- Kommuru, T.R.; Gurley, B.; Khan, M.A.; Reddy, I.K. Self-emulsifying drug delivery systems (SEDDS) of coenzyme Q10: Formulation development and bioavailability assessment. Int. J. Pharm. 2001, 212, 233–246. [Google Scholar] [CrossRef] [PubMed]
- Pouton, C.W.; Porter, C.J. Formulation of lipid-based delivery systems for oral administration: Materials, methods and strategies. Adv. Drug Deliv. Rev. 2008, 60, 625–637. [Google Scholar] [CrossRef] [PubMed]
- Constantinides, P.P.; Scalart, J.P.; Lancaster, C.; Marcello, J.; Marks, G.; Ellens, H.; Smith, P.L. Formulation and intestinal absorption enhancement evaluation of water-in-oil microemulsions incorporating medium-chain glycerides. Pharm. Res. 1994, 11, 1385–1390. [Google Scholar] [CrossRef] [PubMed]
- Prajapati, H.N.; Dalrymple, D.M.; Serajuddin, A.T.M. A Comparative Evaluation of Mono-, Di- and Triglyceride of Medium Chain Fatty Acids by Lipid/Surfactant/Water Phase Diagram, Solubility Determination and Dispersion Testing for Application in Pharmaceutical Dosage Form Development. Pharm. Res. 2012, 29, 285–305. [Google Scholar] [CrossRef]
- Patel, D.; Sawant, K.K. Oral bioavailability enhancement of acyclovir by self-microemulsifying drug delivery systems (SMEDDS). Drug Dev. Ind. Pharm. 2007, 33, 1318–1326. [Google Scholar] [CrossRef]
- Constantinides, P.P. Lipid microemulsions for improving drug dissolution and oral absorption: Physical and biopharmaceutical aspects. Pharm. Res. 1995, 12, 1561–1572. [Google Scholar] [CrossRef]
- Yin, H.F.; Yin, C.M.; Ouyang, T.; Sun, S.D.; Chen, W.G.; Yang, X.L.; He, X.; Zhang, C.F. Self-Nanoemulsifying Drug Delivery System of Genkwanin: A Novel Approach for Anti-Colitis-Associated Colorectal Cancer. Drug Des. Dev. Ther. 2021, 15, 557–576. [Google Scholar] [CrossRef]
- Kadu, P.J.; Kushare, S.S.; Thacker, D.D.; Gattani, S.G. Enhancement of oral bioavailability of atorvastatin calcium by self-emulsifying drug delivery systems (SEDDS). Pharm. Dev. Technol. 2011, 16, 65–74. [Google Scholar] [CrossRef]
- Li, P.; Ghosh, A.; Wagner, R.F.; Krill, S.; Joshi, Y.M.; Serajuddin, A.T.M. Effect of combined use of nonionic surfactant on formation of oil-in-water microemulsions. Int. J. Pharm. 2005, 288, 27–34. [Google Scholar] [CrossRef]
- Neervannan, S. Preclinical formulations for discovery and toxicology: Physicochemical challenges. Expert Opin. Drug Metab. Toxicol. 2006, 2, 715–731. [Google Scholar] [CrossRef]
- Delongeas, J.L.; de Conchard, G.V.; Beamonte, A.; Bertheux, H.; Spire, C.; Maisonneuve, C.; Becourt-Lhote, N.; Goldfain-Blanc, F.; Claude, N. Assessment of Labrasol®/Labrafil®/Transcutol® (4/4/2, v/v/v) as a non-clinical vehicle for poorly water-soluble compounds after 4-week oral toxicity study in Wistar rats. Regul. Toxicol. Pharmacol. 2010, 57, 284–290. [Google Scholar] [CrossRef] [PubMed]
- Kang, B.K.; Lee, J.S.; Chon, S.K.; Jeong, S.Y.; Yuk, S.H.; Khang, G.; Lee, H.B.; Cho, S.H. Development of self-microemulsifying drug delivery systems (SMEDDS) for oral bioavailability enhancement of simvastatin in beagle dogs. Int. J. Pharm. 2004, 274, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Balakrishnan, P.; Lee, B.-J.; Oh, D.H.; Kim, J.O.; Hong, M.J.; Jee, J.-P.; Kim, J.A.; Yoo, B.K.; Woo, J.S.; Yong, C.S.; et al. Enhanced oral bioavailability of dexibuprofen by a novel solid Self-emulsifying drug delivery system (SEDDS). Eur. J. Pharm. Biopharm. 2009, 72, 539–545. [Google Scholar] [CrossRef] [PubMed]
- Pouton, C.W. Formulation of self-emulsifying drug delivery systems. Adv. Drug Deliv. Rev. 1997, 25, 47–58. [Google Scholar] [CrossRef]
- Narang, A.S.; Delmarre, D.; Gao, D. Stable drug encapsulation in micelles and microemulsions. Int. J. Pharm. 2007, 345, 9–25. [Google Scholar] [CrossRef]
- Tarr, B.D.; Yalkowsky, S. Enhanced intestinal absorption of cyclosporine in rats through the reduction of emulsion droplet size. Pharm. Res. 1989, 6, 40–43. [Google Scholar] [CrossRef]
- Gershanik, T.; Benita, S. Self-dispersing lipid formulations for improving oral absorption of lipophilic drugs. Eur. J. Pharm. Biopharm. 2000, 50, 179–188. [Google Scholar] [CrossRef]
- Burguera, J.L.; Burguera, M. Analytical applications of emulsions and microemulsions. Talanta 2012, 96, 11–20. [Google Scholar] [CrossRef]
- Maji, I.; Mahajan, S.; Sriram, A.; Medtiya, P.; Vasave, R.; Khatri, D.K.; Kumar, R.; Singh, S.B.; Madan, J.; Singh, P.K. Solid self emulsifying drug delivery system: Superior mode for oral delivery of hydrophobic cargos. J. Control. Release 2021, 337, 646–660. [Google Scholar] [CrossRef]
- Cerpnjak, K.; Zvonar, A.; Gašperlin, M.; Vrečer, F. Lipid-based systems as a promising approach for enhancing the bioavailability of poorly water-soluble drugs. Acta Pharm. 2013, 63, 427–445. [Google Scholar] [CrossRef]
- Hsieh, C.-M.; Yang, T.-L.; Putri, A.D.; Chen, C.-T. Application of Design of Experiments in the Development of Self-Microemulsifying Drug Delivery Systems. Pharmaceuticals 2023, 16, 283. [Google Scholar] [CrossRef]
- Meirinho, S.; Rodrigues, M.; Santos, A.O.; Falcão, A.; Alves, G. Self-Emulsifying Drug Delivery Systems: An Alternative Approach to Improve Brain Bioavailability of Poorly Water-Soluble Drugs through Intranasal Administration. Pharmaceutics 2022, 14, 1487. [Google Scholar] [CrossRef] [PubMed]
- McClements, D.J. Nanoemulsions versus microemulsions: Terminology, differences, and similarities. Soft Matter 2012, 8, 1719–1729. [Google Scholar] [CrossRef]
- Burra, M.; Jukanti, R.; Janga, K.Y.; Sunkavalli, S.; Velpula, A.; Ampati, S.; Jayaveera, K. Enhanced intestinal absorption and bioavailability of raloxifene hydrochloride via lyophilized solid lipid nanoparticles. Adv. Powder Technol. 2013, 24, 393–402. [Google Scholar] [CrossRef]
- Akula, S.; Gurram, A.K.; Devireddy, S.R. Self-Microemulsifying Drug Delivery Systems: An Attractive Strategy for Enhanced Therapeutic Profile. Int. Sch. Res. Not. 2014, 2014, 964051. [Google Scholar] [CrossRef]
- Oh, M.J.; Shim, J.B.; Yoo, H.; Lee, G.Y.; Jo, H.; Jeong, S.M.; Yuk, S.H.; Lee, D.; Khang, G. The dissolution property of raloxifene HCl solid dispersion using hydroxypropyl methylcellulose. Macromol. Res. 2012, 20, 835–841. [Google Scholar] [CrossRef]
- US-FDA. FDA Guidance for Industry: Bioanalytical Method Validation; Center for Drug Evaluation and Research: Rockville, MD, USA, 2018. Available online: https://www.fda.gov/downloads/drugs/guidances/ucm070107.Pdf (accessed on 25 June 2023).
- Nair, A.B.; Jacob, S. A simple practice guide for dose conversion between animals and human. J. Basic Clin. Pharm. 2016, 7, 27–31. [Google Scholar] [CrossRef]
- US-FDA. Guidance for Industry: Estimating the Maximum Safe Starting Dose in Adult Healthy Volunteer; Center for Drug Evaluation and Research: Rockville, MD, USA, 2005. [Google Scholar]
- Li, F.; Song, S.; Guo, Y.; Zhao, Q.; Zhang, X.; Pan, W.; Yang, X. Preparation and pharmacokinetics evaluation of oral self-emulsifying system for poorly water-soluble drug Lornoxicam. Drug Deliv. 2015, 22, 487–498. [Google Scholar] [CrossRef]
- Scott, J.A.; Da Camara, C.C.; Early, J.E. Raloxifene: A selective estrogen receptor modulator. Am. Fam. Physician 1999, 60, 1131–1139. [Google Scholar]
- Shanmugam, S.; Baskaran, R.; Balakrishnan, P.; Thapa, P.; Yong, C.S.; Yoo, B.K. Solid self-nanoemulsifying drug delivery system (S-SNEDDS) containing phosphatidylcholine for enhanced bioavailability of highly lipophilic bioactive carotenoid lutein. Eur. J. Pharm. Biopharm. 2011, 79, 250–257. [Google Scholar] [CrossRef]
- Sato, Y.; Joumura, T.; Nashimoto, S.; Yokoyama, S.; Takekuma, Y.; Yoshida, H.; Sugawara, M. Enhancement of lymphatic transport of lutein by oral administration of a solid dispersion and a self-microemulsifying drug delivery system. Eur. J. Pharm. Biopharm. 2018, 127, 171–176. [Google Scholar] [CrossRef] [PubMed]
- McConnell, E.L.; Basit, A.W.; Murdan, S. Measurements of rat and mouse gastrointestinal pH, fluid and lymphoid tissue, and implications for in-vivo experiments. J. Pharm. Pharmacol. 2008, 60, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Teeter, J.S.; Meyerhoff, R.D. Environmental fate and chemistry of raloxifene hydrochloride. Environ. Toxicol. Chem. 2002, 21, 729–736. [Google Scholar] [CrossRef] [PubMed]
- ICH. ICH Guideline M10 on Bioanalytical Method Validation ang Study Samples Analysis; European Medicines Agency, International Council for Harmonization: Amsterdam, The Netherlands, 2019. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/ich-guideline-m10-bioanalytical-method-validation-step-5_en.pdf (accessed on 25 June 2023).









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Vehicle | Description and Composition | RLX Solubility (µg/mL) |
|---|---|---|
| Oils | ||
| Capryol 90 | Propylene glycol caprylate | 259.9 ± 37.5 |
| Labrafac lipophile WL 1349 | Medium-chain triglycerides of caprylic (C8) and capric (C10) acids | 156.1 ± 22.3 |
| Linseed oil | Long chain fatty acid | 148.1 ± 8.3 |
| Isopropyl myristate | Isopropyl tetradecanoate | 111.4 ± 33.0 |
| Oleic acid | Long chain fatty acid | 41.3 ± 16.6 |
| Surfactants | ||
| Tween 80 | Polyoxyethylene sorbitan monooleate | 3195.1 ± 86.8 |
| Labrasol ALF | Caprylocaproyl macrogol-8 glycerides | 246.4 ± 22.4 |
| Maisine CC | Glyceryl monolinoleate | 105.6 ± 22.1 |
| Triton X-100 | Polyoxyethylene octyl phenyl ether | 41.5 ± 18.7 |
| Cosurfactants | ||
| Polyethylene glycol 400 | Polyethylene glycol | 2950.3 ± 73.5 |
| Transcutol P | Diethylene glycol monoethyl ether | 2079.5 ± 62.7 |
| Formulation | Capryol 90 (%) | Tween 80/Labrasol ALF/PEG-400 Mixture (%) | Droplet Size (nm) | PDI | Microemulsion Formed (Visual Inspection) | % Transmittance |
|---|---|---|---|---|---|---|
| F1 | 10 | 90 | 18.4 ± 0.1 | 0.208 ± 0.01 | No | 99.3 ± 0.57 |
| F2 | 15 | 85 | 147.1 ± 1.0 | 0.227 ± 0.01 | Yes | 96.8 ± 0.03 |
| F3 | 20 | 80 | 258.2 ± 8.2 | 0.555 ± 0.03 | Yes | 75.4 ± 1.19 |
| F4 | 30 | 70 | 470.9 ± 22.3 | 0.494 ± 0.23 | Yes | 61.2 ± 0.2 |
| F5 | 40 | 60 | 610.6 ± 51.4 | 0.526 ± 0.29 | Yes | 46.4 ± 0.07 |
| F6 | 50 | 50 | 669.9 ± 56.9 | 0.499 ± 0.21 | No | 13.5 ± 0.07 |
| F7 | 60 | 40 | 297.6 ± 2.8 | 0.419 ± 0.05 | No | 6.3 ± 0.09 |
| F8 | 70 | 30 | 452.9 ± 20.2 | 0.298 ± 0.01 | No | 7.8 ± 0.07 |
| F9 | 80 | 20 | 633.9 ± 38.7 | 0.331 ± 0.17 | No | 4.7 ± 0.13 |
| F10 | 90 | 10 | 2792.0 ± 58.1 | 0.567 ± 0.43 | No | 6.1 ± 0.23 |
| Parameters | RLX Dispersion | RLX-SMEDDS |
|---|---|---|
| AUC24h (ng × min/mL) | 44,907.5 ± 15,657.7 | 87,144.5 ± 13,815.1 ** |
| Cmax (ng/mL) | 45.6 ± 16.1 | 81.6 ± 17.0 * |
| Tmax (min) | 420.0 ± 69.3 | 390.0 ± 114.9 |
| MRT (min) | 712.4 ± 79.6 | 688.51 ± 71.5 |
| BArel (%) | - | 194.0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ansari, M.M.; Vo, D.-K.; Choi, H.-I.; Ryu, J.-S.; Bae, Y.; Bukhari, N.I.; Zeb, A.; Kim, J.-K.; Maeng, H.-J. Formulation and Evaluation of a Self-Microemulsifying Drug Delivery System of Raloxifene with Improved Solubility and Oral Bioavailability. Pharmaceutics 2023, 15, 2073. https://doi.org/10.3390/pharmaceutics15082073
Ansari MM, Vo D-K, Choi H-I, Ryu J-S, Bae Y, Bukhari NI, Zeb A, Kim J-K, Maeng H-J. Formulation and Evaluation of a Self-Microemulsifying Drug Delivery System of Raloxifene with Improved Solubility and Oral Bioavailability. Pharmaceutics. 2023; 15(8):2073. https://doi.org/10.3390/pharmaceutics15082073
Chicago/Turabian StyleAnsari, Muhammad Mohsin, Dang-Khoa Vo, Ho-Ik Choi, Jeong-Su Ryu, Yumi Bae, Nadeem Irfan Bukhari, Alam Zeb, Jin-Ki Kim, and Han-Joo Maeng. 2023. "Formulation and Evaluation of a Self-Microemulsifying Drug Delivery System of Raloxifene with Improved Solubility and Oral Bioavailability" Pharmaceutics 15, no. 8: 2073. https://doi.org/10.3390/pharmaceutics15082073
APA StyleAnsari, M. M., Vo, D.-K., Choi, H.-I., Ryu, J.-S., Bae, Y., Bukhari, N. I., Zeb, A., Kim, J.-K., & Maeng, H.-J. (2023). Formulation and Evaluation of a Self-Microemulsifying Drug Delivery System of Raloxifene with Improved Solubility and Oral Bioavailability. Pharmaceutics, 15(8), 2073. https://doi.org/10.3390/pharmaceutics15082073