
Hydrophilic Copolymers with Hydroxamic Acid Groups as a Protective Biocompatible Coating of Maghemite Nanoparticles: Synthesis, Physico-Chemical Characterization and MRI Biodistribution Study
Abstract
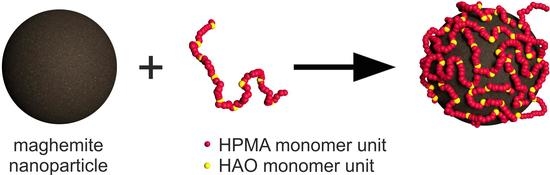
1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Synthesis of Maghemite Nanoparticles
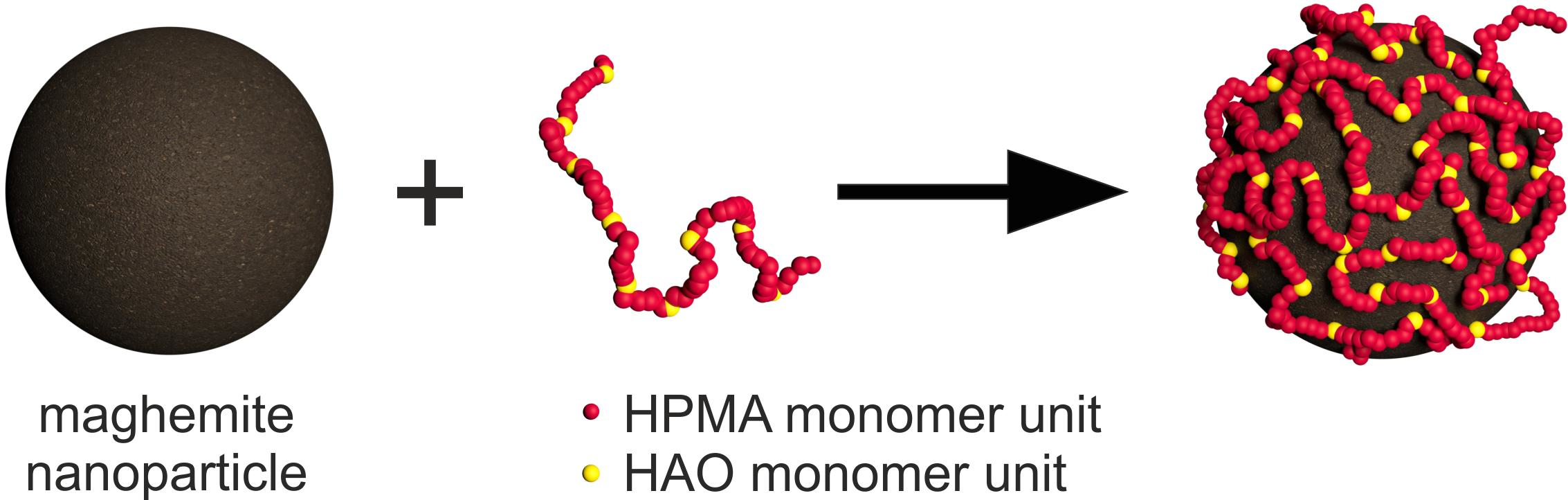
2.3. Synthesis of Methyl 2-(2-Methylprop-2-enoylamino)acetate (MMA)
2.4. Synthesis of N-[2-(Hydroxyamino)-2-oxo-ethyl]-2-methyl-prop-2-enamide (HAO)
2.5. RAFT Polymerization of HPMA and Its Copolymerization with MMA or HAO
2.6. Functional Modifications of P(HPMA-co-MMA)
2.6.1. Hydrolysis: Synthesis of N-(2-Methyl-1-oxo-1,2-propandiyl)glycine Comonomer Unit (GLM)
2.6.2. Aminolysis: Synthesis of N-[2-[(2-Aminoethyl)amino]-2-oxoethyl]-2-methyl-1,2-propandiyl-2-amide Comonomer Unit (AEM)
2.6.3. Derivatization by Hydroxamic Acid: Synthesis of N-[2-(Hydroxyamino)-2-oxo-ethyl]-2-methyl-1,2-propandiyl-2-amide (HAM)
2.7. Nuclear Magnetic Resonance Spectroscopy (NMR)
2.8. Elemental Analysis
2.9. Size Exclusion Chromatography Analysis (SEC)
2.10. Dynamic Light Scattering (DLS)
2.11. Transmission Electron Microscopy (TEM)
2.12. Thermogravimetric Analysis (TGA)
2.13. Biodistribution of the Nanoparticles In Vivo
3. Results and Discussion
3.1. Synthesis and Coating of the Nanoparticles
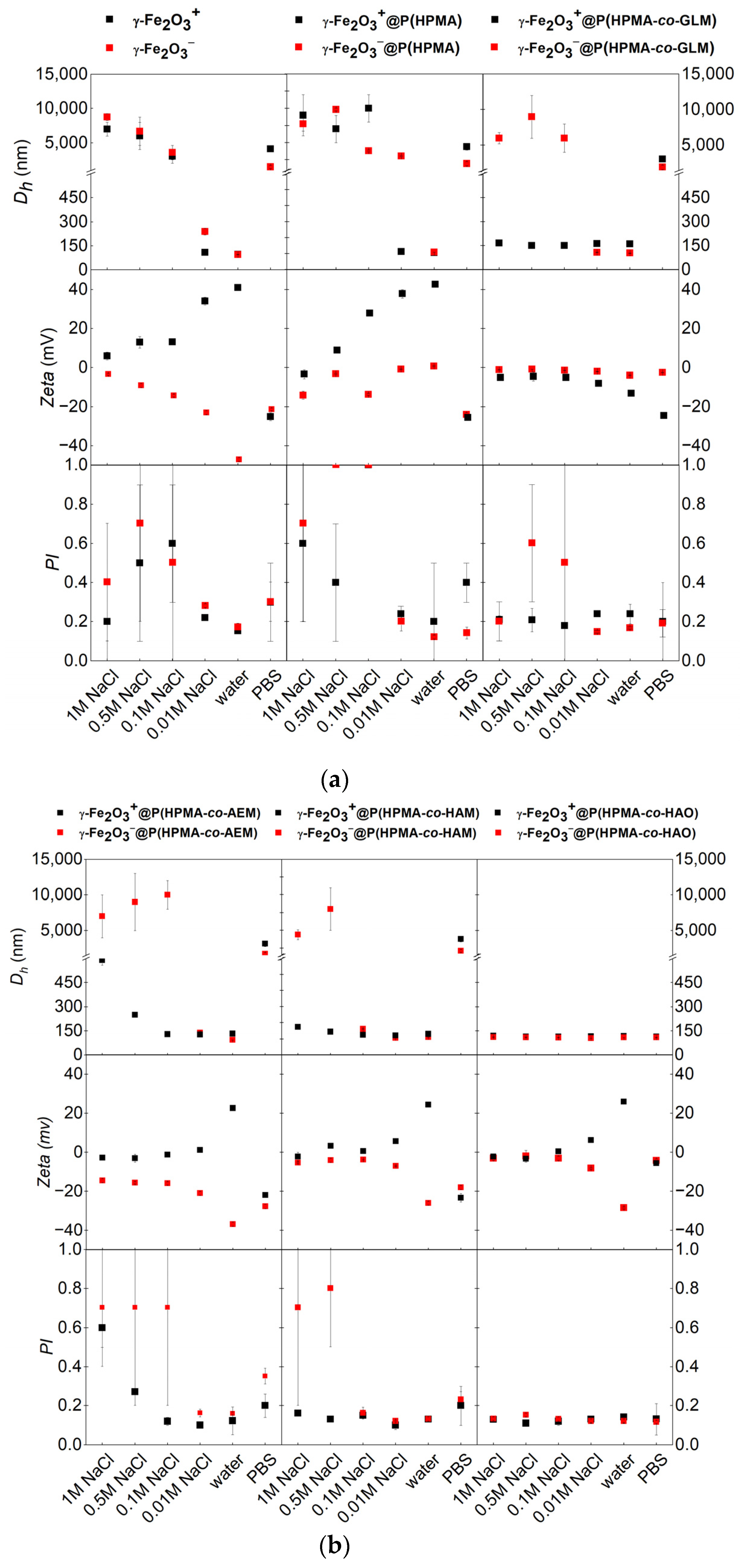
3.2. Effect of Coating on Hydrodynamic Stability in Different Liquid Media
3.3. Evaluation of the Size and Shape Changes of the (Un)Coated Nanoparticles by TEM Measurement
3.4. TGA Analysis
3.5. Study of the Influence of pH Changes on the Colloidal Behavior of the Selected (Un)Coated Particles
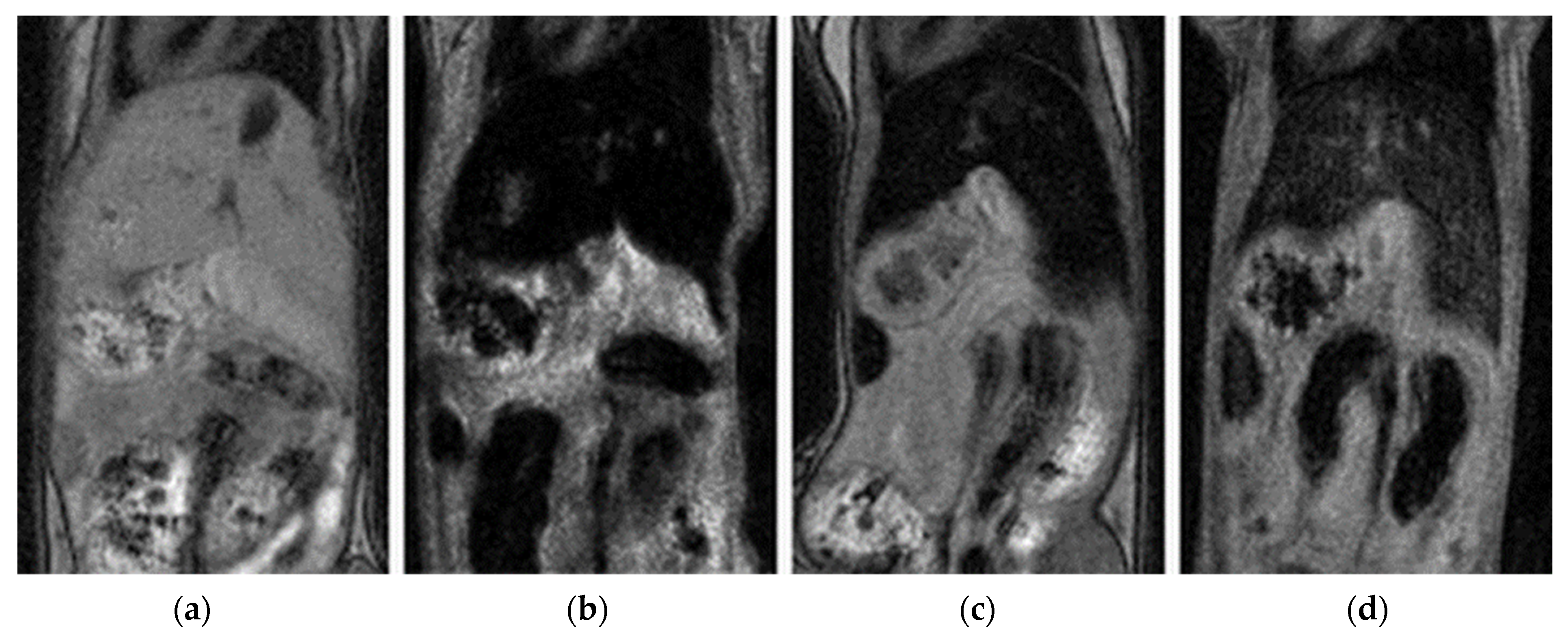
3.6. In Vivo Study
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Shabatina, T.I.; Vernaya, O.I.; Shabatin, V.P.; Melnikov, M.Y. Magnetic Nanoparticles for Biomedical Purposes: Modern Trends and Prospects. Magnetochemistry 2020, 6, 30. [Google Scholar] [CrossRef]
- Nelson, N.; Port, J.; Pandey, M. Use of Superparamagnetic Iron Oxide Nanoparticles (SPIONs) via Multiple Imaging Modalities and Modifications to Reduce Cytotoxicity: An Educational Review. J. Nanotheranostics 2020, 1, 105–135. [Google Scholar] [CrossRef]
- Shubayev, V.I.; Pisanic, T.R.; Jin, S. Magnetic Nanoparticles for Theragnostics. Adv. Drug Deliv. Rev. 2009, 61, 467–477. [Google Scholar] [CrossRef]
- Uskoković, V.; Tang, S.; Wu, V.M. Targeted Magnetic Separation of Biomolecules and Cells Using Earthicle-Based Ferrofluids. Nanoscale 2019, 11, 11236–11253. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Yu, B.; Wang, S.; Shen, Y.; Cong, H. Preparation, Surface Functionalization and Application of Fe3O4 Magnetic Nanoparticles. Adv. Colloid Interface Sci. 2020, 281, 102165. [Google Scholar] [CrossRef]
- Guo, S.; Dong, S. Biomolecule-Nanoparticle Hybrids for Electrochemical Biosensors. TrAC Trends Anal. Chem. 2009, 28, 96–109. [Google Scholar] [CrossRef]
- Bilgic, A.; Cimen, A. A Highly Sensitive and Selective ON-OFF Fluorescent Sensor Based on Functionalized Magnetite Nanoparticles for Detection of Cr(VI) Metal Ions in the Aqueous Medium. J. Mol. Liq. 2020, 312, 113398. [Google Scholar] [CrossRef]
- Xue, T.; Wang, S.; Ou, G.; Li, Y.; Ruan, H.; Li, Z.; Ma, Y.; Zou, R.; Qiu, J.; Shen, Z.; et al. Detection of Circulating Tumor Cells Based on Improved SERS-Active Magnetic Nanoparticles. Anal. Methods 2019, 11, 2918–2928. [Google Scholar] [CrossRef]
- Jordan, A.; Scholz, R.; Maier-Hauff, K.; Johannsen, M.; Wust, P.; Nadobny, J.; Schirra, H.; Schmidt, H.; Deger, S.; Loening, S.; et al. Presentation of a New Magnetic Field Therapy System for the Treatment of Human Solid Tumors with Magnetic Fluid Hyperthermia. J. Magn. Magn. Mater. 2001, 225, 118–126. [Google Scholar] [CrossRef]
- Piazza, R.D.; Viali, W.R.; dos Santos, C.C.; Nunes, E.S.; Marques, R.F.C.; Morais, P.C.; da Silva, S.W.; Coaquira, J.A.H.; Jafelicci, M. PEGlatyon-SPION Surface Functionalization with Folic Acid for Magnetic Hyperthermia Applications. Mater. Res. Express 2020, 7, 015078. [Google Scholar] [CrossRef]
- Gharibkandi, N.A.; Żuk, M.; Muftuler, F.Z.B.; Wawrowicz, K.; Żelechowska-Matysiak, K.; Bilewicz, A. 198Au-Coated Superparamagnetic Iron Oxide Nanoparticles for Dual Magnetic Hyperthermia and Radionuclide Therapy of Hepatocellular Carcinoma. Int. J. Mol. Sci. 2023, 24, 5282. [Google Scholar] [CrossRef]
- Zhi, D.; Yang, T.; Yang, J.; Fu, S.; Zhang, S. Targeting Strategies for Superparamagnetic Iron Oxide Nanoparticles in Cancer Therapy. Acta Biomater. 2020, 102, 13–34. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhao, J.; Jiang, J.; Chen, F.; Fang, X. Doxorubicin Delivered Using Nanoparticles Camouflaged with Mesenchymal Stem Cell Membranes to Treat Colon Cancer. Int. J. Nanomed. 2020, 15, 2873–2884. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Deng, L.; Liu, H.; Mai, S.; Cheng, Z.; Shi, G.; Zeng, H.; Wu, Z. Enhanced Fluorescence/Magnetic Resonance Dual Imaging and Gene Therapy of Liver Cancer Using Cationized Amylose Nanoprobe. Mater. Today Bio 2022, 13, 100220. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.-Y.; Liu, Z.-H.; Weng, W.-H.; Chang, C.-W. Magnetic Nanocomplexes for Gene Delivery Applications. J. Mater. Chem. B 2021, 9, 4267–4286. [Google Scholar] [CrossRef]
- Antonelli, A.; Magnani, M. SPIO Nanoparticles and Magnetic Erythrocytes as Contrast Agents for Biomedical and Diagnostic Applications. J. Magn. Magn. Mater. 2022, 541, 168520. [Google Scholar] [CrossRef]
- Chen, C.; Ge, J.; Gao, Y.; Chen, L.; Cui, J.; Zeng, J.; Gao, M. Ultrasmall Superparamagnetic Iron Oxide Nanoparticles: A next Generation Contrast Agent for Magnetic Resonance Imaging. WIREs Nanomed. Nanobiotechnol. 2022, 14, e1740. [Google Scholar] [CrossRef]
- Bulte, J.W.M. Superparamagnetic Iron Oxides as MPI Tracers: A Primer and Review of Early Applications. Adv. Drug Deliv. Rev. 2019, 138, 293–301. [Google Scholar] [CrossRef]
- Canese, R.; Vurro, F.; Marzola, P. Iron Oxide Nanoparticles as Theranostic Agents in Cancer Immunotherapy. Nanomaterials 2021, 11, 1950. [Google Scholar] [CrossRef]
- Antonelli, A.; Szwargulski, P.; Scarpa, E.; Thieben, F.; Cordula, G.; Ambrosi, G.; Guidi, L.; Ludewig, P.; Knopp, T.; Magnani, M. Development of Long Circulating Magnetic Particle Imaging Tracers: Use of Novel Magnetic Nanoparticles and Entrapment into Human Erythrocytes. Nanomedicine 2020, 15, 739–753. [Google Scholar] [CrossRef]
- Tay, Z.W.; Savliwala, S.; Hensley, D.W.; Fung, K.L.B.; Colson, C.; Fellows, B.D.; Zhou, X.; Huynh, Q.; Lu, Y.; Zheng, B.; et al. Superferromagnetic Nanoparticles Enable Order-of-Magnitude Resolution & Sensitivity Gain in Magnetic Particle Imaging. Small Methods 2021, 5, 2100796. [Google Scholar] [CrossRef]
- Avasthi, A.; Caro, C.; Pozo-Torres, E.; Leal, M.P.; García-Martín, M.L. Magnetic Nanoparticles as MRI Contrast Agents. Top. Curr. Chem. 2020, 378, 40. [Google Scholar] [CrossRef] [PubMed]
- Laurent, S.; Elst, L.V.; Muller, R.N. Comparative Study of the Physicochemical Properties of Six Clinical Low Molecular Weight Gadolinium Contrast Agents. Contrast Media Mol. Imaging 2006, 1, 128–137. [Google Scholar] [CrossRef] [PubMed]
- Ramalho, J.; Ramalho, M.; Jay, M.; Burke, L.M.; Semelka, R.C. Gadolinium Toxicity and Treatment. Magn. Reson. Imaging 2016, 34, 1394–1398. [Google Scholar] [CrossRef]
- Rogosnitzky, M.; Branch, S. Gadolinium-Based Contrast Agent Toxicity: A Review of Known and Proposed Mechanisms. BioMetals 2016, 29, 365–376. [Google Scholar] [CrossRef] [PubMed]
- Buhaescu, I.; Izzedine, H. Gadolinium-Induced Nephrotoxicity. Int. J. Clin. Pract. 2008, 62, 1113–1118. [Google Scholar] [CrossRef]
- Liu, Y.; Li, Y.; Huang, J.; Zhang, Y.; Ruan, Z.; Hu, T. Science of the Total Environment An Advanced Sol—Gel Strategy for Enhancing Interfacial Reactivity of Iron Oxide Nanoparticles on Rosin Biochar Substrate to Remove Cr (VI). Sci. Total Environ. 2019, 690, 438–446. [Google Scholar] [CrossRef]
- Soleymani, M.; Velashjerdi, M.; Shaterabadi, Z.; Barati, A. One-Pot Preparation of Hyaluronic Acid-coated Iron Oxide Nanoparticles for Magnetic Hyperthermia Therapy and Targeting CD44-Overexpressing Cancer Cells. Carbohydr. Polym. 2020, 237, 116130. [Google Scholar] [CrossRef]
- Salazar-Alvarez, G.; Muhammed, M.; Zagorodni, A.A. Novel Flow Injection Synthesis of Iron Oxide Nanoparticles with Narrow Size Distribution. Chem. Eng. Sci. 2006, 61, 4625–4633. [Google Scholar] [CrossRef]
- Cabrera, L.; Gutierrez, S.; Menendez, N.; Morales, M.P.; Herrasti, P. Magnetite Nanoparticles: Electrochemical Synthesis and Characterization. Electrochim. Acta 2008, 53, 3436–3441. [Google Scholar] [CrossRef]
- Aghazadeh, M.; Karimzadeh, I.; Doroudi, T.; Ganjali, M.R.; Kolivand, P.H.; Gharailou, D. Facile Electrosynthesis and Characterization of Superparamagnetic Nanoparticles Coated with Cysteine, Glycine and Glutamine. Appl. Phys. A 2017, 123, 529. [Google Scholar] [CrossRef]
- Ansari, S.R.; Hempel, N.-J.; Asad, S.; Svedlindh, P.; Bergström, C.A.S.; Löbmann, K.; Teleki, A. Hyperthermia-Induced In Situ Drug Amorphization by Superparamagnetic Nanoparticles in Oral Dosage Forms. ACS Appl. Mater. Interfaces 2022, 14, 21978–21988. [Google Scholar] [CrossRef] [PubMed]
- Fuentes-García, J.A.; Carvalho Alavarse, A.; Moreno Maldonado, A.C.; Toro-Córdova, A.; Ibarra, M.R.; Goya, G.F. Simple Sonochemical Method to Optimize the Heating Efficiency of Magnetic Nanoparticles for Magnetic Fluid Hyperthermia. ACS Omega 2020, 5, 26357–26364. [Google Scholar] [CrossRef] [PubMed]
- Dheyab, M.A.; Aziz, A.A.; Jameel, M.S.; Noqta, O.A.; Khaniabadi, P.M.; Mehrdel, B. Excellent Relaxivity and X-Ray Attenuation Combo Properties of Fe3O4@Au CSNPs Produced via Rapid Sonochemical Synthesis for MRI and CT Imaging. Mater. Today Commun. 2020, 25, 101368. [Google Scholar] [CrossRef]
- Chamorro, E.; Tenorio, M.J.; Calvo, L.; Torralvo, M.J.; Sáez-Puche, R.; Cabañas, A. One-Step Sustainable Preparation of Superparamagnetic Iron Oxide Nanoparticles Supported on Mesoporous SiO2. J. Supercrit. Fluids 2020, 159, 104775. [Google Scholar] [CrossRef]
- Abu Bakar, M.; Tan, W.L.; Abu Bakar, N.H.H. A Simple Synthesis of Size-Reduce Magnetite Nano-Crystals via Aqueous to Toluene Phase-Transfer Method. J. Magn. Magn. Mater. 2007, 314, 1–6. [Google Scholar] [CrossRef]
- El-Gendy, N.S.; Nassar, H.N. Biosynthesized Magnetite Nanoparticles as an Environmental Opulence and Sustainable Wastewater Treatment. Sci. Total Environ. 2021, 774, 145610. [Google Scholar] [CrossRef]
- Kianpour, S.; Ebrahiminezhad, A.; Deyhimi, M.; Negahdaripour, M.; Raee, M.J.; Mohkam, M.; Rezaee, H.; Irajie, C.; Berenjian, A.; Ghasemi, Y. Structural Characterization of Polysaccharide-Coated Iron Oxide Nanoparticles Produced by Staphylococcus warneri, Isolated from a Thermal Spring. J. Basic Microbiol. 2019, 59, 569–578. [Google Scholar] [CrossRef]
- Fokina, V.; Wilke, M.; Dulle, M.; Ehlert, S.; Förster, S. Size Control of Iron Oxide Nanoparticles Synthesized by Thermal Decomposition Methods. J. Phys. Chem. C 2022, 126, 21356–21367. [Google Scholar] [CrossRef]
- Mieloch, A.A.; Żurawek, M.; Giersig, M.; Rozwadowska, N.; Rybka, J.D. Bioevaluation of Superparamagnetic Iron Oxide Nanoparticles (SPIONs) Functionalized with Dihexadecyl Phosphate (DHP). Sci. Rep. 2020, 10, 2725. [Google Scholar] [CrossRef]
- Babič, M.; Horák, D.; Trchová, M.; Jendelová, P.; Glogarová, K.; Lesný, P.; Herynek, V.; Hájek, M.; Syková, E. Poly(L-Lysine)-Modified Iron Oxide Nanoparticles for Stem Cell Labeling. Bioconjug. Chem. 2008, 19, 740–750. [Google Scholar] [CrossRef] [PubMed]
- Sodipo, B.K.; Noqta, O.A.; Aziz, A.A.; Katsikini, M.; Pinakidou, F.; Paloura, E.C. Influence of Capping Agents on Fraction of Fe Atoms Occupying Octahedral Site and Magnetic Property of Magnetite (Fe3O4) Nanoparticles by One-Pot Co-Precipitation Method. J. Alloys Compd. 2023, 938, 168558. [Google Scholar] [CrossRef]
- LaGrow, A.P.; Besenhard, M.O.; Hodzic, A.; Sergides, A.; Bogart, L.K.; Gavriilidis, A.; Thanh, N.T.K. Unravelling the Growth Mechanism of the Co-Precipitation of Iron Oxide Nanoparticles with the Aid of Synchrotron X-ray Diffraction in Solution. Nanoscale 2019, 11, 6620–6628. [Google Scholar] [CrossRef]
- Novotna, B.; Jendelova, P.; Kapcalova, M.; Rossner, P.; Turnovcova, K.; Bagryantseva, Y.; Babic, M.; Horak, D.; Sykova, E. Oxidative Damage to Biological Macromolecules in Human Bone Marrow Mesenchymal Stromal Cells Labeled with Various Types of Iron Oxide Nanoparticles. Toxicol. Lett. 2012, 210, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Arbab, A.S.; Bashaw, L.A.; Miller, B.R.; Jordan, E.K.; Lewis, B.K.; Kalish, H.; Frank, J.A. Characterization of Biophysical and Metabolic Properties of Cells Labeled with Superparamagnetic Iron Oxide Nanoparticles and Transfection Agent for Cellular MR Imaging. Radiology 2003, 229, 838–846. [Google Scholar] [CrossRef]
- Gershon, H.; Ghirlando, R.; Guttman, S.B.; Minsky, A. Mode of Formation and Structural Features of DNA-Cationic Liposome Complexes Used for Transfection. Biochemistry 1993, 32, 7143–7151. [Google Scholar] [CrossRef]
- Babič, M.; Horák, D.; Jendelová, P.; Glogarová, K.; Herynek, V.; Trchová, M.; Likavčanová, K.; Lesný, P.; Pollert, E.; Hájek, M.; et al. Poly(N,N-dimethylacrylamide)-Coated Maghemite Nanoparticles for Stem Cell Labeling. Bioconjug. Chem. 2009, 20, 283–294. [Google Scholar] [CrossRef]
- Mukhopadhyay, A.; Joshi, N.; Chattopadhyay, K.; De, G. A Facile Synthesis of PEG-Coated Magnetite (Fe3O4) Nanoparticles and Their Prevention of the Reduction of Cytochrome C. ACS Appl. Mater. Interfaces 2012, 4, 142–149. [Google Scholar] [CrossRef]
- Valdiglesias, V.; Fernández-Bertólez, N.; Kiliç, G.; Costa, C.; Costa, S.; Fraga, S.; Bessa, M.J.; Pásaro, E.; Teixeira, J.P.; Laffon, B. Are Iron Oxide Nanoparticles Safe? Current Knowledge and Future Perspectives. J. Trace Elem. Med. Biol. 2016, 38, 53–63. [Google Scholar] [CrossRef]
- Reddy, L.H.; Arias, J.L.; Nicolas, J.; Couvreur, P. Magnetic Nanoparticles: Design and Characterization, Toxicity and Biocompatibility, Pharmaceutical and Biomedical Applications. Chem. Rev. 2012, 112, 5818–5878. [Google Scholar] [CrossRef]
- Cotin, G.; Blanco-Andujar, C.; Perton, F.; Asín, L.; de la Fuente, J.M.; Reichardt, W.; Schaffner, D.; Ngyen, D.-V.; Mertz, D.; Kiefer, C.; et al. Unveiling the Role of Surface, Size, Shape and Defects of Iron Oxide Nanoparticles for Theranostic Applications. Nanoscale 2021, 13, 14552–14571. [Google Scholar] [CrossRef] [PubMed]
- Aisida, S.O.; Akpa, P.A.; Ahmad, I.; Zhao, T.; Maaza, M.; Ezema, F.I. Bio-Inspired Encapsulation and Functionalization of Iron Oxide Nanoparticles for Biomedical Applications. Eur. Polym. J. 2020, 122, 109371. [Google Scholar] [CrossRef]
- Naha, P.C.; Liu, Y.; Hwang, G.; Huang, Y.; Gubara, S.; Jonnakuti, V.; Simon-Soro, A.; Kim, D.; Gao, L.; Koo, H.; et al. Dextran-Coated Iron Oxide Nanoparticles as Biomimetic Catalysts for Localized and PH-Activated Biofilm Disruption. ACS Nano 2019, 13, 4960–4971. [Google Scholar] [CrossRef] [PubMed]
- Chircov, C.; Ștefan, R.-E.; Dolete, G.; Andrei, A.; Holban, A.M.; Oprea, O.-C.; Vasile, B.S.; Neacșu, I.A.; Tihăuan, B. Dextran-Coated Iron Oxide Nanoparticles Loaded with Curcumin for Antimicrobial Therapies. Pharmaceutics 2022, 14, 1057. [Google Scholar] [CrossRef]
- Badawy, M.M.M.; Abdel-Hamid, G.R.; Mohamed, H.E. Antitumor Activity of Chitosan-Coated Iron Oxide Nanocomposite Against Hepatocellular Carcinoma in Animal Models. Biol. Trace Elem. Res. 2023, 201, 1274–1285. [Google Scholar] [CrossRef]
- Yu, S.; Xu, X.; Feng, J.; Liu, M.; Hu, K. Chitosan and Chitosan Coating Nanoparticles for the Treatment of Brain Disease. Int. J. Pharm. 2019, 560, 282–293. [Google Scholar] [CrossRef]
- Lazaro-Carrillo, A.; Filice, M.; Guillén, M.J.; Amaro, R.; Viñambres, M.; Tabero, A.; Paredes, K.O.; Villanueva, A.; Calvo, P.; del Puerto Morales, M.; et al. Tailor-Made PEG Coated Iron Oxide Nanoparticles as Contrast Agents for Long Lasting Magnetic Resonance Molecular Imaging of Solid Cancers. Mater. Sci. Eng. C 2020, 107, 110262. [Google Scholar] [CrossRef]
- Salehipour, M.; Rezaei, S.; Mosafer, J.; Pakdin-Parizi, Z.; Motaharian, A.; Mogharabi-Manzari, M. Recent Advances in Polymer-Coated Iron Oxide Nanoparticles as Magnetic Resonance Imaging Contrast Agents. J. Nanopart. Res. 2021, 23, 48. [Google Scholar] [CrossRef]
- Pongrac, I.M.; Dobrivojević, M.; Ahmed, L.B.; Babič, M.; Šlouf, M.; Horák, D.; Gajović, S. Improved Biocompatibility and Efficient Labeling of Neural Stem Cells with Poly(L-Lysine)-Coated Maghemite Nanoparticles. Beilstein J. Nanotechnol. 2016, 7, 926–936. [Google Scholar] [CrossRef]
- Plichta, Z.; Kozak, Y.; Panchuk, R.; Sokolova, V.; Epple, M.; Kobylinska, L.; Jendelová, P.; Horák, D. Cytotoxicity of Doxorubicin-Conjugated Poly[N-(2-Hydroxypropyl)Methacrylamide]-Modified γ-Fe2O3 Nanoparticles towards Human Tumor Cells. Beilstein J. Nanotechnol. 2018, 9, 2533–2545. [Google Scholar] [CrossRef]
- Gupta, S.P. Hydroxamic Acids a Unique Family of Chemicals with Multiple Biological Activities; Springer Science & Business Media: Meerut, India, 2013. [Google Scholar]
- Al Shaer, D.; Al Musaimi, O.; de la Torre, B.G.; Albericio, F. Hydroxamate Siderophores: Natural Occurrence, Chemical Synthesis, Iron Binding Affinity and Use as Trojan Horses against Pathogens. Eur. J. Med. Chem. 2020, 208, 112791. [Google Scholar] [CrossRef] [PubMed]
- Winston, A.; Varaprasad, D.V.P.R. Polymeric Iron Chelators. WO 86/00891, 1986. [Google Scholar]
- Timofeeva, A.M.; Galyamova, M.R.; Sedykh, S.E. Bacterial Siderophores: Classification, Biosynthesis, Perspectives of Use in Agriculture. Plants 2022, 11, 3065. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.K.; Bera, T.; Chakrabarty, A.M. Microbial Siderophore—A Boon to Agricultural Sciences. Biol. Control 2020, 144, 104214. [Google Scholar] [CrossRef]
- Herynek, V.; Babič, M.; Kaman, O.; Charvátová, H.; Veselá, M.; Buchholz, O.; Vosmanská, M.; Kubániová, D.; Kohout, J.; Hofmann, U.G.; et al. Maghemite Nanoparticles Coated by Methacrylamide-Based Polymer for Magnetic Particle Imaging. J. Nanopart. Res. 2021, 23, 52. [Google Scholar] [CrossRef]
- Chytil, P.; Etrych, T.; Kříž, J.; Šubr, V.; Ulbrich, K. N-(2-Hydroxypropyl)Methacrylamide-Based Polymer Conjugates with PH-Controlled Activation of Doxorubicin for Cell-Specific or Passive Tumour Targeting. Synthesis by RAFT Polymerisation and Physicochemical Characterisation. Eur. J. Pharm. Sci. 2010, 41, 473–482. [Google Scholar] [CrossRef]
- Reimer, P.; Balzer, T. Ferucarbotran (Resovist): A New Clinically Approved RES-Specific Contrast Agent for Contrast-Enhanced MRI of the Liver: Properties, Clinical Development, and Applications. Eur. Radiol. 2003, 13, 1266–1276. [Google Scholar] [CrossRef]
- Ulbrich, K.; Šubr, V.; Strohalm, J.; Plocová, D.; Jelínková, M.; Říhová, B. Polymeric Drugs Based on Conjugates of Synthetic and Natural Macromolecules. J. Control. Release 2000, 64, 63–79. [Google Scholar] [CrossRef]
- Rasband, W.S. ImageJ National institutes of Health, Bethesda, Maryland, USA. Available online: http://imagej.nih.gov/ij (accessed on 10 June 2023).
- Gilbert, R.G.; Hess, M.; Jenkins, A.D.; Jones, R.G.; Kratochvíl, P.; Stepto, R.F.T. Dispersity in Polymer Science (IUPAC Recommendations 2009). Pure Appl. Chem. 2009, 81, 351–353. [Google Scholar] [CrossRef]
- Nair, A.; Jacob, S. A Simple Practice Guide for Dose Conversion between Animals and Human. J. Basic Clin. Pharm. 2016, 7, 27. [Google Scholar] [CrossRef]
- FDA; U.S.F.; D.A. Feraheme/Ferumoxytol FDA Label—AMAG Pharmaceuticals. 2018; pp. 1–17. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/022180s009lbl.pdf (accessed on 18 February 2018).
- Horák, D.; Babič, M.; Jendelová, P.; Herynek, V.; Trchová, M.; Pientka, Z.; Pollert, E.; Hájek, M.; Syková, E. D-Mannose-Modified Iron Oxide Nanoparticles for Stem Cell Labeling. Bioconjug. Chem. 2007, 18, 635–644. [Google Scholar] [CrossRef]
- Lucas, I.T.; Durand-Vidal, S.; Dubois, E.; Chevalet, J.; Turq, P. Surface Charge Density of Maghemite Nanoparticles: Role of Electrostatics in the Proton Exchange. J. Phys. Chem. C 2007, 111, 18568–18576. [Google Scholar] [CrossRef]
- Luengo, Y.; Nardecchia, S.; Morales, M.P.; Serrano, M.C. Different Cell Responses Induced by Exposure to Maghemite Nanoparticles. Nanoscale 2013, 5, 11428. [Google Scholar] [CrossRef] [PubMed]
- Calatayud, M.P.; Sanz, B.; Raffa, V.; Riggio, C.; Ibarra, M.R.; Goya, G.F. The Effect of Surface Charge of Functionalized Fe3O4 Nanoparticles on Protein Adsorption and Cell Uptake. Biomaterials 2014, 35, 6389–6399. [Google Scholar] [CrossRef] [PubMed]
- Oddsson, Á.; Patrakka, J.; Tryggvason, K. Glomerular Filtration Barrier. In Reference Module in Biomedical Sciences; Elsevier: Amsterdam, The Netherlands, 2014; pp. 1–11. ISBN 9780128012383. [Google Scholar]
- Ruggiero, A.; Villa, C.H.; Bander, E.; Rey, D.A.; Bergkvist, M.; Batt, C.A.; Manova-Todorova, K.; Deen, W.M.; Scheinberg, D.A.; McDevitt, M.R. Paradoxical Glomerular Filtration of Carbon Nanotubes. Proc. Natl. Acad. Sci. USA 2010, 107, 12369–12374. [Google Scholar] [CrossRef] [PubMed]
- Mayadunne, R.T.A.; Rizzardo, E.; Chiefari, J.; Krstina, J.; Moad, G.; Postma, A.; Thang, S.H. Living Polymers by the Use of Trithiocarbonates as Reversible Addition−Fragmentation Chain Transfer (RAFT) Agents: ABA Triblock Copolymers by Radical Polymerization in Two Steps. Macromolecules 2000, 33, 243–245. [Google Scholar] [CrossRef]
- Spitzer, J.J. Colloidal Interactions: Contact Limiting Laws, Double-Layer Dissociation, and “Non-DLVO” (Derjaguin–Landau–Verwey–Overbeek) Forces. Colloid Polym. Sci. 2003, 281, 589–592. [Google Scholar] [CrossRef]
- Spitzer, J.J. Maxwellian Double Layer Forces: From Infinity to Contact. Langmuir 2003, 19, 7099–7111. [Google Scholar] [CrossRef]
- Spitzer, J.J. Theory of Dissociative Electrical Double Layers: The Limit of Close Separations and “Hydration” Forces. Langmuir 1992, 8, 1659–1662. [Google Scholar] [CrossRef]
- Debayle, M.; Balloul, E.; Dembele, F.; Xu, X.; Hanafi, M.; Ribot, F.; Monzel, C.; Coppey, M.; Fragola, A.; Dahan, M.; et al. Zwitterionic Polymer Ligands: An Ideal Surface Coating to Totally Suppress Protein-Nanoparticle Corona Formation? Biomaterials 2019, 219, 119357. [Google Scholar] [CrossRef]
- Gandhi, S.N.; Brown, M.A.; Wong, J.G.; Aguirre, D.A.; Sirlin, C.B. MR Contrast Agents for Liver Imaging: What, When, How. RadioGraphics 2006, 26, 1621–1636. [Google Scholar] [CrossRef]
- Ferrucci, J.T.; Stark, D.D. Iron Oxide-Enhanced MR Imaging of the Liver and Spleen: Review of the First 5 Years. Am. J. Roentgenol. 1990, 155, 943–950. [Google Scholar] [CrossRef] [PubMed]
- Keselman, P.; Yu, E.Y.; Zhou, X.Y.; Goodwill, P.W.; Chandrasekharan, P.; Ferguson, R.M.; Khandhar, A.P.; Kemp, S.J.; Krishnan, K.M.; Zheng, B.; et al. Tracking Short-Term Biodistribution and Long-Term Clearance of SPIO Tracers in Magnetic Particle Imaging. Phys. Med. Biol. 2017, 62, 3440–3453. [Google Scholar] [CrossRef] [PubMed]


















{kind=link}
{kind=link}
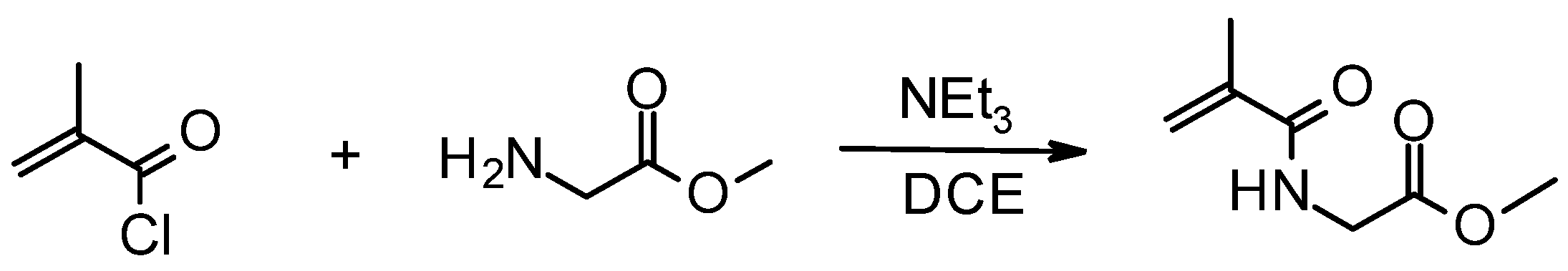{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | DLS | TEM | pH | ||||
|---|---|---|---|---|---|---|---|
| Dh (nm) | PI | Zeta (mV) | Dn (nm) | Dw (nm) | PDI | ||
| γ-Fe2O3⨁ | 96.9 ± 0.9 | 0.153 ± 0.01 | +41 | 8.0 | 10.0 | 1.3 | 3.9 |
| γ-Fe2O3⊖ | 96 ± 1 | 0.17 ± 0.02 | −51 | 8.7 | 11.1 | 1.3 | 7.4 |
| Compound | Mn (kg·mol−1) | Mw (kg·mol−1) | Ð |
|---|---|---|---|
| P(HPMA) | 37.6 | 44.7 | 1.19 |
| P(HPMA-co-MMA) | 40.6 | 48.7 | 1.20 |
| P(HPMA-co-HAO) | 32.3 | 40.6 | 1.26 |
| Compound | Mn (kg·mol−1) | Mw (kg·mol−1) | Ð |
|---|---|---|---|
| P(HPMA-co-GLM) | 32.2 | 39.0 | 1.21 |
| P(HPMA-co-AEM) | 47.9 | 58.3 | 1.22 |
| P(HPMA-co-HAM) | 39.9 | 48.1 | 1.21 |
| Compound | Burned Mass (%) | Nanoparticles (%) | Polymer Bound to the Particles (%) | Portion of Bound Polymer Related to the Mass Used for Coating |
|---|---|---|---|---|
| γ-Fe2O3⨁ | 9.64 | – | – | – |
| γ-Fe2O3⊖ | 7.21 | – | – | – |
| P(HPMA-co-HAO) | 97.75 | – | – | – |
| γ-Fe2O3⨁@P(HPMA-co-HAO) | 17.2 | 91 | 9 | 26 |
| γ-Fe2O3⊖@P(HPMA-co-HAO) | 20.50 | 88 | 12 | 38 |
| Nanoparticles | Clearance Half-Time (Days) |
|---|---|
| γ-Fe2O3⨁ | 38 ± 8 |
| γ-Fe2O3⊖ | 59 ± 36 |
| γ-Fe2O3⨁@P(HPMA-co-HAO) | 28 ± 13 |
| γ-Fe2O3⊖@P(HPMA-co-HAO) | 47 ± 43 |
| Resovist® | 272 ± 150 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Charvátová, H.; Plichta, Z.; Hromádková, J.; Herynek, V.; Babič, M. Hydrophilic Copolymers with Hydroxamic Acid Groups as a Protective Biocompatible Coating of Maghemite Nanoparticles: Synthesis, Physico-Chemical Characterization and MRI Biodistribution Study. Pharmaceutics 2023, 15, 1982. https://doi.org/10.3390/pharmaceutics15071982
Charvátová H, Plichta Z, Hromádková J, Herynek V, Babič M. Hydrophilic Copolymers with Hydroxamic Acid Groups as a Protective Biocompatible Coating of Maghemite Nanoparticles: Synthesis, Physico-Chemical Characterization and MRI Biodistribution Study. Pharmaceutics. 2023; 15(7):1982. https://doi.org/10.3390/pharmaceutics15071982
Chicago/Turabian StyleCharvátová, Hana, Zdeněk Plichta, Jiřina Hromádková, Vít Herynek, and Michal Babič. 2023. "Hydrophilic Copolymers with Hydroxamic Acid Groups as a Protective Biocompatible Coating of Maghemite Nanoparticles: Synthesis, Physico-Chemical Characterization and MRI Biodistribution Study" Pharmaceutics 15, no. 7: 1982. https://doi.org/10.3390/pharmaceutics15071982
APA StyleCharvátová, H., Plichta, Z., Hromádková, J., Herynek, V., & Babič, M. (2023). Hydrophilic Copolymers with Hydroxamic Acid Groups as a Protective Biocompatible Coating of Maghemite Nanoparticles: Synthesis, Physico-Chemical Characterization and MRI Biodistribution Study. Pharmaceutics, 15(7), 1982. https://doi.org/10.3390/pharmaceutics15071982