
Fluoxetine Protects Retinal Ischemic Damage in Mice

 ,
,  ,
, 
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Animals
2.3. Retinal Ischemia
2.4. Pattern Electroretinogram
2.5. ddPCR Analysis
2.6. Statistical Analysis
3. Results
3.1. Retinal Function
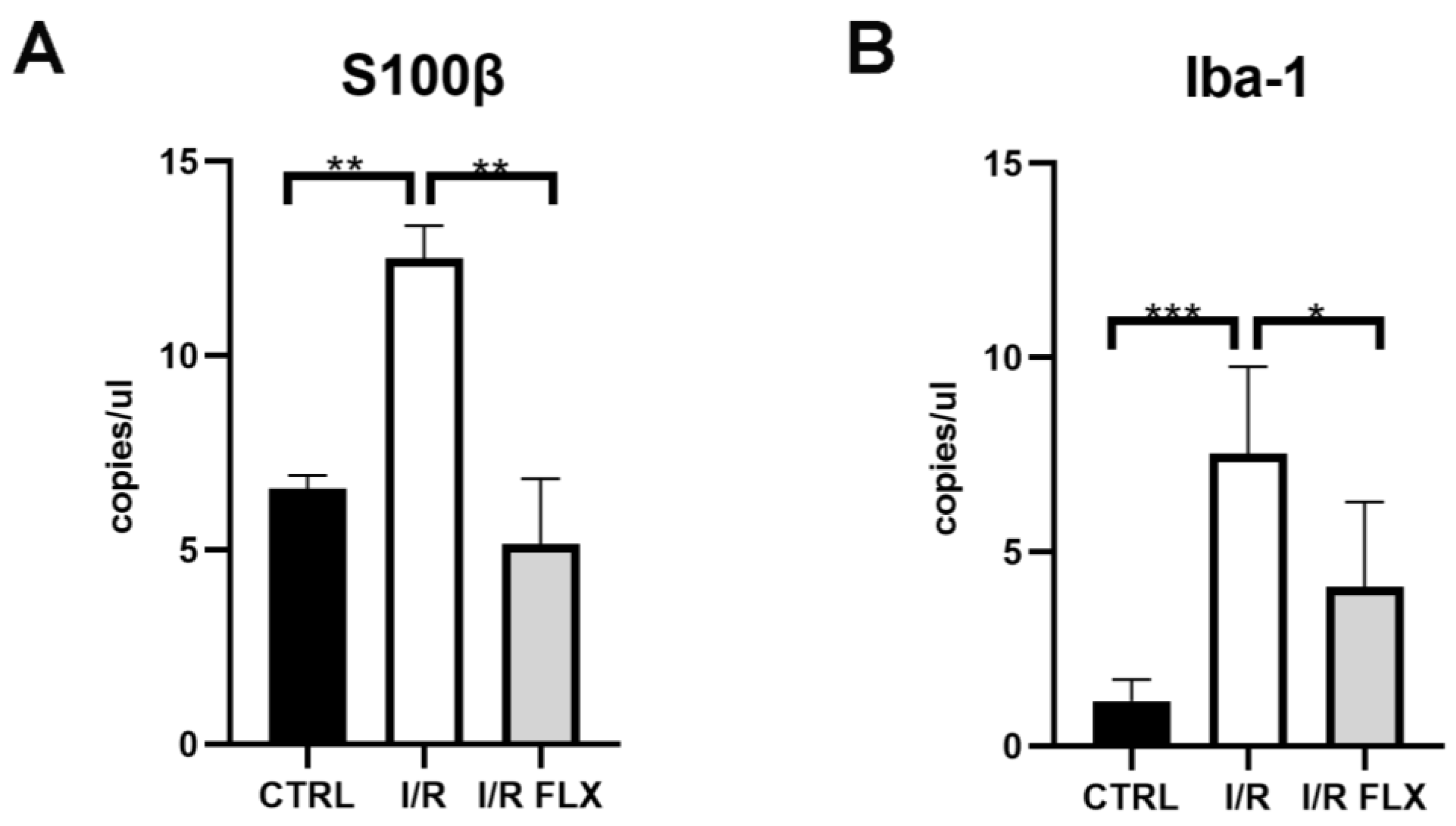
3.2. Biomarkers
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bucolo, C.; Drago, F. Carbon monoxide and the eye: Implications for glaucoma therapy. Pharmacol. Ther. 2011, 130, 191–201. [Google Scholar] [CrossRef] [PubMed]
- La Morgia, C.; Di Vito, L.; Carelli, V.; Carbonelli, M. Patterns of Retinal Ganglion Cell Damage in Neurodegenerative Disorders: Parvocellular vs Magnocellular Degeneration in Optical Coherence Tomography Studies. Front. Neurol. 2017, 8, 710. [Google Scholar] [CrossRef]
- Banitt, M.R.; Ventura, L.M.; Feuer, W.J.; Savatovsky, E.; Luna, G.; Shif, O.; Bosse, B.; Porciatti, V. Progressive loss of retinal ganglion cell function precedes structural loss by several years in glaucoma suspects. Investig. Ophthalmol. Vis. Sci. 2013, 54, 2346–2352. [Google Scholar] [CrossRef] [PubMed]
- Gagliano, C.; Caruso, S.; Napolitano, G.; Malaguarnera, G.; Cicinelli, M.V.; Amato, R.; Reibaldi, M.; Incarbone, G.; Bucolo, C.; Drago, F.; et al. Low levels of 17-beta-oestradiol, oestrone and testosterone correlate with severe evaporative dysfunctional tear syndrome in postmenopausal women: A case-control study. Br. J. Ophthalmol. 2014, 98, 371–376. [Google Scholar] [CrossRef] [PubMed]
- Vernazza, S.; Oddone, F.; Tirendi, S.; Bassi, A.M. Risk Factors for Retinal Ganglion Cell Distress in Glaucoma and Neuroprotective Potential Intervention. Int. J. Mol. Sci. 2021, 22, 7994. [Google Scholar] [CrossRef]
- Casson, R.J. Medical therapy for glaucoma: A review. Clin. Exp. Ophthalmol. 2022, 50, 198–212. [Google Scholar] [CrossRef]
- Shalaby, W.S.; Ahmed, O.M.; Waisbourd, M.; Katz, L.J. A review of potential novel glaucoma therapeutic options independent of intraocular pressure. Surv. Ophthalmol. 2022, 67, 1062–1080. [Google Scholar] [CrossRef]
- Wubben, T.J.; Zacks, D.N.; Besirli, C.G. Retinal neuroprotection: Current strategies and future directions. Curr. Opin. Ophthalmol. 2019, 30, 199–205. [Google Scholar] [CrossRef]
- Ng, K.L.; Gibson, E.M.; Hubbard, R.; Yang, J.; Caffo, B.; O’Brien, R.J.; Krakauer, J.W.; Zeiler, S.R. Fluoxetine Maintains a State of Heightened Responsiveness to Motor Training Early After Stroke in a Mouse Model. Stroke 2015, 46, 2951–2960. [Google Scholar] [CrossRef]
- O’Sullivan, J.B.; Ryan, K.M.; Curtin, N.M.; Harkin, A.; Connor, T.J. Noradrenaline reuptake inhibitors limit neuroinflammation in rat cortex following a systemic inflammatory challenge: Implications for depression and neurodegeneration. Int. J. Neuropsychopharmacol. 2009, 12, 687–699. [Google Scholar] [CrossRef]
- Sheline, Y.I.; West, T.; Yarasheski, K.; Jasielec, M.S.; Hettinger, J.C.; Tripoli, D.L.; Xiong, C.; Frederiksen, C.; Grzelak, M.V.; Bateman, R.J.; et al. Reply to comment on “An antidepressant decreases CSF Abeta production in healthy individuals and in transgenic AD mice”. Sci. Transl. Med. 2014, 6, 268lr4. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Kim, W.; Kong, S.Y. Antidepressants for neuro-regeneration: From depression to Alzheimer’s disease. Arch. Pharm. Res. 2013, 36, 1279–1290. [Google Scholar] [CrossRef] [PubMed]
- Imoto, Y.; Kira, T.; Sukeno, M.; Nishitani, N.; Nagayasu, K.; Nakagawa, T.; Kaneko, S.; Kobayashi, K.; Segi-Nishida, E. Role of the 5-HT4 receptor in chronic fluoxetine treatment-induced neurogenic activity and granule cell dematuration in the dentate gyrus. Mol. Brain 2015, 8, 29. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Sun, X.; Liu, T.; Zhao, M.; Zhao, S.; Xiao, T.; Jolkkonen, J.; Zhao, C. Fluoxetine enhanced neurogenesis is not translated to functional outcome in stroke rats. Neurosci. Lett. 2015, 603, 31–36. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Sun, X.; Qu, H.; Zhao, S.; Xiao, T.; Zhao, C. Neuroplasticity and behavioral effects of fluoxetine after experimental stroke. Restor. Neurol. Neurosci. 2017, 35, 457–468. [Google Scholar] [CrossRef] [PubMed]
- Khodanovich, M.; Kisel, A.; Kudabaeva, M.; Chernysheva, G.; Smolyakova, V.; Krutenkova, E.; Wasserlauf, I.; Plotnikov, M.; Yarnykh, V. Effects of Fluoxetine on Hippocampal Neurogenesis and Neuroprotection in the Model of Global Cerebral Ischemia in Rats. Int. J. Mol. Sci. 2018, 19, 162. [Google Scholar] [CrossRef]
- Tizabi, Y. Duality of Antidepressants and Neuroprotectants. Neurotox. Res. 2016, 30, 1–13. [Google Scholar] [CrossRef]
- Corbett, A.M.; Sieber, S.; Wyatt, N.; Lizzi, J.; Flannery, T.; Sibbit, B.; Sanghvi, S. Increasing neurogenesis with fluoxetine, simvastatin and ascorbic Acid leads to functional recovery in ischemic stroke. Recent Pat. Drug Deliv. Formul. 2015, 9, 158–166. [Google Scholar] [CrossRef]
- Correia, A.S.; Vale, N. Antidepressants in Alzheimer’s Disease: A Focus on the Role of Mirtazapine. Pharmaceuticals 2021, 14, 930. [Google Scholar] [CrossRef]
- Dafsari, F.S.; Jessen, F. Depression-an underrecognized target for prevention of dementia in Alzheimer’s disease. Transl. Psychiatry 2020, 10, 160. [Google Scholar] [CrossRef]
- Bartels, C.; Wagner, M.; Wolfsgruber, S.; Ehrenreich, H.; Schneider, A.; Alzheimer’s Disease Neuroimaging, I. Impact of SSRI Therapy on Risk of Conversion From Mild Cognitive Impairment to Alzheimer’s Dementia in Individuals With Previous Depression. Am. J. Psychiatry 2018, 175, 232–241. [Google Scholar] [CrossRef] [PubMed]
- Cesareo, M.; Martucci, A.; Ciuffoletti, E.; Mancino, R.; Cerulli, A.; Sorge, R.P.; Martorana, A.; Sancesario, G.; Nucci, C. Association Between Alzheimer’s Disease and Glaucoma: A Study Based on Heidelberg Retinal Tomography and Frequency Doubling Technology Perimetry. Front. Neurosci. 2015, 9, 479. [Google Scholar] [CrossRef] [PubMed]
- Guidoboni, G.; Sacco, R.; Szopos, M.; Sala, L.; Verticchio Vercellin, A.C.; Siesky, B.; Harris, A. Neurodegenerative Disorders of the Eye and of the Brain: A Perspective on Their Fluid-Dynamical Connections and the Potential of Mechanism-Driven Modeling. Front. Neurosci. 2020, 14, 566428. [Google Scholar] [CrossRef] [PubMed]
- Jain, S.; Aref, A.A. Senile Dementia and Glaucoma: Evidence for a Common Link. J. Ophthalmic. Vis. Res. 2015, 10, 178–183. [Google Scholar] [CrossRef] [PubMed]
- Bayer, A.U.; Keller, O.N.; Ferrari, F.; Maag, K.P. Association of glaucoma with neurodegenerative diseases with apoptotic cell death: Alzheimer’s disease and Parkinson’s disease. Am. J. Ophthalmol. 2002, 133, 135–137. [Google Scholar] [CrossRef] [PubMed]
- Bayer, A.U.; Ferrari, F.; Erb, C. High occurrence rate of glaucoma among patients with Alzheimer’s disease. Eur. Neurol. 2002, 47, 165–168. [Google Scholar] [CrossRef]
- Yochim, B.P.; Mueller, A.E.; Kane, K.D.; Kahook, M.Y. Prevalence of cognitive impairment, depression, and anxiety symptoms among older adults with glaucoma. J. Glaucoma 2012, 21, 250–254. [Google Scholar] [CrossRef]
- Osborne, N.N.; Casson, R.J.; Wood, J.P.; Chidlow, G.; Graham, M.; Melena, J. Retinal ischemia: Mechanisms of damage and potential therapeutic strategies. Prog. Retin. Eye Res. 2004, 23, 91–147. [Google Scholar] [CrossRef]
- Fortmann, S.D.; Grant, M.B. Molecular mechanisms of retinal ischemia. Curr. Opin. Physiol. 2019, 7, 41–48. [Google Scholar] [CrossRef]
- Minhas, G.; Morishita, R.; Anand, A. Preclinical models to investigate retinal ischemia: Advances and drawbacks. Front. Neurol. 2012, 3, 75. [Google Scholar] [CrossRef]
- Wareham, L.K.; Liddelow, S.A.; Temple, S.; Benowitz, L.I.; Di Polo, A.; Wellington, C.; Goldberg, J.L.; He, Z.; Duan, X.; Bu, G.; et al. Solving neurodegeneration: Common mechanisms and strategies for new treatments. Mol. Neurodegener. 2022, 17, 23. [Google Scholar] [CrossRef] [PubMed]
- Marchesi, N.; Fahmideh, F.; Boschi, F.; Pascale, A.; Barbieri, A. Ocular Neurodegenerative Diseases: Interconnection between Retina and Cortical Areas. Cells 2021, 10, 2394. [Google Scholar] [CrossRef] [PubMed]
- Dhande, O.S.; Huberman, A.D. Retinal ganglion cell maps in the brain: Implications for visual processing. Curr. Opin. Neurobiol. 2014, 24, 133–142. [Google Scholar] [CrossRef] [PubMed]
- Kim, U.S.; Mahroo, O.A.; Mollon, J.D.; Yu-Wai-Man, P. Retinal Ganglion Cells-Diversity of Cell Types and Clinical Relevance. Front. Neurol. 2021, 12, 661938. [Google Scholar] [CrossRef]
- Pardue, M.T.; Allen, R.S. Neuroprotective strategies for retinal disease. Prog. Retin. Eye Res. 2018, 65, 50–76. [Google Scholar] [CrossRef]
- Chang, E.E.; Goldberg, J.L. Glaucoma 2.0: Neuroprotection, neuroregeneration, neuroenhancement. Ophthalmology 2012, 119, 979–986. [Google Scholar] [CrossRef]
- European Glaucoma Society Terminology and Guidelines for Glaucoma, 4th Edition—Chapter 3: Treatment principles and options Supported by the EGS Foundation: Part 1: Foreword; Introduction; Glossary; Chapter 3 Treatment principles and options. Br. J. Ophthalmol. 2017, 101, 130–195. [CrossRef]
- Kaufman, P.L.; Rasmussen, C.A. Advances in glaucoma treatment and management: Outflow drugs. Investig. Ophthalmol. Vis. Sci. 2012, 53, 2495–2500. [Google Scholar] [CrossRef]
- Nucci, C.; Martucci, A.; Giannini, C.; Morrone, L.A.; Bagetta, G.; Mancino, R. Neuroprotective agents in the management of glaucoma. Eye 2018, 32, 938–945. [Google Scholar] [CrossRef]
- Vohra, R.; Tsai, J.C.; Kolko, M. The role of inflammation in the pathogenesis of glaucoma. Surv. Ophthalmol. 2013, 58, 311–320. [Google Scholar] [CrossRef]
- Baudouin, C.; Kolko, M.; Melik-Parsadaniantz, S.; Messmer, E.M. Inflammation in Glaucoma: From the back to the front of the eye, and beyond. Prog. Retin. Eye Res. 2021, 83, 100916. [Google Scholar] [CrossRef] [PubMed]
- Bell, K.; Und Hohenstein-Blaul, N.V.T.; Teister, J.; Grus, F. Modulation of the Immune System for the Treatment of Glaucoma. Curr. Neuropharmacol. 2018, 16, 942–958. [Google Scholar] [CrossRef] [PubMed]
- Caruso, G.; Grasso, M.; Fidilio, A.; Torrisi, S.A.; Musso, N.; Geraci, F.; Tropea, M.R.; Privitera, A.; Tascedda, F.; Puzzo, D.; et al. Antioxidant Activity of Fluoxetine and Vortioxetine in a Non-Transgenic Animal Model of Alzheimer’s Disease. Front. Pharmacol. 2021, 12, 809541. [Google Scholar] [CrossRef] [PubMed]
- Caraci, F.; Tascedda, F.; Merlo, S.; Benatti, C.; Spampinato, S.F.; Munafo, A.; Leggio, G.M.; Nicoletti, F.; Brunello, N.; Drago, F.; et al. Fluoxetine Prevents Abeta1-42-Induced Toxicity via a Paracrine Signaling Mediated by Transforming-Growth-Factor-beta1. Front. Pharmacol. 2016, 7, 389. [Google Scholar] [CrossRef]
- Behr, G.A.; Moreira, J.C.; Frey, B.N. Preclinical and clinical evidence of antioxidant effects of antidepressant agents: Implications for the pathophysiology of major depressive disorder. Oxid. Med. Cell Longev. 2012, 2012, 609421. [Google Scholar] [CrossRef]
- Szalach, L.P.; Lisowska, K.A.; Cubala, W.J. The Influence of Antidepressants on the Immune System. Arch. Immunol. Ther. Exp. 2019, 67, 143–151. [Google Scholar] [CrossRef]
- Torrisi, S.A.; Geraci, F.; Tropea, M.R.; Grasso, M.; Caruso, G.; Fidilio, A.; Musso, N.; Sanfilippo, G.; Tascedda, F.; Palmeri, A.; et al. Fluoxetine and Vortioxetine Reverse Depressive-Like Phenotype and Memory Deficits Induced by Abeta1-42 Oligomers in Mice: A Key Role of Transforming Growth Factor-beta1. Front. Pharmacol. 2019, 10, 693. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, Y.; Xu, H.; Zhu, S.; Wang, H.; He, J.; Zhang, H.; Guo, H.; Kong, J.; Huang, Q.; et al. Fluoxetine improves behavioral performance by suppressing the production of soluble beta-amyloid in APP/PS1 mice. Curr. Alzheimer. Res. 2014, 11, 672–680. [Google Scholar] [CrossRef]
- Wu, J.Q.; Kosten, T.R.; Zhang, X.Y. Free radicals, antioxidant defense systems, and schizophrenia. Prog. Neuropsychopharmacol. Biol. Psychiatry 2013, 46, 200–206. [Google Scholar] [CrossRef]
- Jin, L.; Gao, L.F.; Sun, D.S.; Wu, H.; Wang, Q.; Ke, D.; Lei, H.; Wang, J.Z.; Liu, G.P. Long-term Ameliorative Effects of the Antidepressant Fluoxetine Exposure on Cognitive Deficits in 3 × TgAD Mice. Mol. Neurobiol. 2017, 54, 4160–4171. [Google Scholar] [CrossRef]
- Mariani, N.; Everson, J.; Pariante, C.M.; Borsini, A. Modulation of microglial activation by antidepressants. J. Psychopharmacol. 2022, 36, 131–150. [Google Scholar] [CrossRef] [PubMed]
- Stefan, M.G.; Kiss, B.; Gutleb, A.C.; Loghin, F. Redox metabolism modulation as a mechanism in SSRI toxicity and pharmacological effects. Arch. Toxicol. 2020, 94, 1417–1441. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.I.; Chung, Y.C.; Jin, B.K. Norfluoxetine Prevents Degeneration of Dopamine Neurons by Inhibiting Microglia-Derived Oxidative Stress in an MPTP Mouse Model of Parkinson’s Disease. Mediators. Inflamm. 2018, 2018, 4591289. [Google Scholar] [CrossRef] [PubMed]
- Lauterbach, E.C. Repurposing psychiatric medicines to target activated microglia in anxious mild cognitive impairment and early Parkinson’s disease. Am. J. Neurodegener. Dis. 2016, 5, 29–51. [Google Scholar] [PubMed]
- Xu, H.; Steven Richardson, J.; Li, X.M. Dose-related effects of chronic antidepressants on neuroprotective proteins BDNF, Bcl-2 and Cu/Zn-SOD in rat hippocampus. Neuropsychopharmacology 2003, 28, 53–62. [Google Scholar] [CrossRef]
- Peric, I.; Stanisavljevic, A.; Gass, P.; Filipovic, D. Fluoxetine reverses behavior changes in socially isolated rats: Role of the hippocampal GSH-dependent defense system and proinflammatory cytokines. Eur. Arch. Psychiatry Clin. Neurosci. 2017, 267, 737–749. [Google Scholar] [CrossRef] [PubMed]
- Ludka, F.K.; Dal-Cim, T.; Binder, L.B.; Constantino, L.C.; Massari, C.; Tasca, C.I. Atorvastatin and Fluoxetine Prevent Oxidative Stress and Mitochondrial Dysfunction Evoked by Glutamate Toxicity in Hippocampal Slices. Mol. Neurobiol. 2017, 54, 3149–3161. [Google Scholar] [CrossRef]
- Lee, J.Y.; Kang, S.R.; Yune, T.Y. Fluoxetine prevents oligodendrocyte cell death by inhibiting microglia activation after spinal cord injury. J. Neurotrauma. 2015, 32, 633–644. [Google Scholar] [CrossRef]
- Ubhi, K.; Inglis, C.; Mante, M.; Patrick, C.; Adame, A.; Spencer, B.; Rockenstein, E.; May, V.; Winkler, J.; Masliah, E. Fluoxetine ameliorates behavioral and neuropathological deficits in a transgenic model mouse of alpha-synucleinopathy. Exp. Neurol. 2012, 234, 405–416. [Google Scholar] [CrossRef]
- Moretti, M.; Colla, A.; de Oliveira Balen, G.; dos Santos, D.B.; Budni, J.; de Freitas, A.E.; Farina, M.; Severo Rodrigues, A.L. Ascorbic acid treatment, similarly to fluoxetine, reverses depressive-like behavior and brain oxidative damage induced by chronic unpredictable stress. J. Psychiatr. Res. 2012, 46, 331–340. [Google Scholar] [CrossRef]
- Chung, E.S.; Chung, Y.C.; Bok, E.; Baik, H.H.; Park, E.S.; Park, J.Y.; Yoon, S.H.; Jin, B.K. Fluoxetine prevents LPS-induced degeneration of nigral dopaminergic neurons by inhibiting microglia-mediated oxidative stress. Brain Res. 2010, 1363, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Kalogiannis, M.; Delikatny, E.J.; Jeitner, T.M. Serotonin as a putative scavenger of hypohalous acid in the brain. Biochim. Biophys. Acta 2016, 1862, 651–661. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Zhang, R.; Zhang, S.; Wu, J.; Sun, X. Activation of 5-HT1A Receptors Promotes Retinal Ganglion Cell Function by Inhibiting the cAMP-PKA Pathway to Modulate Presynaptic GABA Release in Chronic Glaucoma. J. Neurosci. 2019, 39, 1484–1504. [Google Scholar] [CrossRef] [PubMed]
- Sharif, N.A.; Senchyna, M. Serotonin receptor subtype mRNA expression in human ocular tissues, determined by RT-PCR. Mol. Vis. 2006, 12, 1040–1047. [Google Scholar] [PubMed]
- Trakhtenberg, E.F.; Pita-Thomas, W.; Fernandez, S.G.; Patel, K.H.; Venugopalan, P.; Shechter, J.M.; Morkin, M.I.; Galvao, J.; Liu, X.; Dombrowski, S.M.; et al. Serotonin receptor 2C regulates neurite growth and is necessary for normal retinal processing of visual information. Dev. Neurobiol. 2017, 77, 419–437. [Google Scholar] [CrossRef]
- Collier, R.J.; Patel, Y.; Martin, E.A.; Dembinska, O.; Hellberg, M.; Krueger, D.S.; Kapin, M.A.; Romano, C. Agonists at the serotonin receptor (5-HT(1A)) protect the retina from severe photo-oxidative stress. Investig. Ophthalmol. Vis. Sci. 2011, 52, 2118–2126. [Google Scholar] [CrossRef]
- Gustavsson, C.; Agardh, C.D.; Hagert, P.; Agardh, E. Inflammatory markers in nondiabetic and diabetic rat retinas exposed to ischemia followed by reperfusion. Retina 2008, 28, 645–652. [Google Scholar] [CrossRef]
- Stankowska, D.L.; Dibas, A.; Li, L.; Zhang, W.; Krishnamoorthy, V.R.; Chavala, S.H.; Nguyen, T.P.; Yorio, T.; Ellis, D.Z.; Acharya, S. Hybrid Compound SA-2 is Neuroprotective in Animal Models of Retinal Ganglion Cell Death. Investig. Ophthalmol. Vis. Sci. 2019, 60, 3064–3073. [Google Scholar] [CrossRef]
- Hartsock, M.J.; Cho, H.; Wu, L.; Chen, W.J.; Gong, J.; Duh, E.J. A Mouse Model of Retinal Ischemia-Reperfusion Injury Through Elevation of Intraocular Pressure. J. Vis. Exp. 2016, 113, 54065. [Google Scholar] [CrossRef]
- Conti, F.; Romano, G.L.; Eandi, C.M.; Toro, M.D.; Rejdak, R.; Di Benedetto, G.; Lazzara, F.; Bernardini, R.; Drago, F.; Cantarella, G.; et al. Brimonidine is Neuroprotective in Animal Paradigm of Retinal Ganglion Cell Damage. Front. Pharmacol. 2021, 12, 705405. [Google Scholar] [CrossRef]
- Rovere, G.; Nadal-Nicolas, F.M.; Wang, J.; Bernal-Garro, J.M.; Garcia-Carrillo, N.; Villegas-Perez, M.P.; Agudo-Barriuso, M.; Vidal-Sanz, M. Melanopsin-Containing or Non-Melanopsin-Containing Retinal Ganglion Cells Response to Acute Ocular Hypertension With or Without Brain-Derived Neurotrophic Factor Neuroprotection. Investig. Ophthalmol. Vis. Sci. 2016, 57, 6652–6661. [Google Scholar] [CrossRef]
- Wang, J.; Valiente-Soriano, F.J.; Nadal-Nicolas, F.M.; Rovere, G.; Chen, S.; Huang, W.; Agudo-Barriuso, M.; Jonas, J.B.; Vidal-Sanz, M.; Zhang, X. MicroRNA regulation in an animal model of acute ocular hypertension. Acta Ophthalmol. 2017, 95, e10–e21. [Google Scholar] [CrossRef] [PubMed]
- Chou, T.H.; Musada, G.R.; Romano, G.L.; Bolton, E.; Porciatti, V. Anesthetic Preconditioning as Endogenous Neuroprotection in Glaucoma. Int. J. Mol. Sci. 2018, 19, 237. [Google Scholar] [CrossRef] [PubMed]
- Porciatti, V. Electrophysiological assessment of retinal ganglion cell function. Exp. Eye Res. 2015, 141, 164–170. [Google Scholar] [CrossRef] [PubMed]
- Almasieh, M.; Wilson, A.M.; Morquette, B.; Cueva Vargas, J.L.; Di Polo, A. The molecular basis of retinal ganglion cell death in glaucoma. Prog. Retin. Eye Res. 2012, 31, 152–181. [Google Scholar] [CrossRef]
- Almasieh, M.; Levin, L.A. Neuroprotection in Glaucoma: Animal Models and Clinical Trials. Annu. Rev. Vis. Sci. 2017, 3, 91–120. [Google Scholar] [CrossRef]
- Teleanu, D.M.; Niculescu, A.G.; Lungu, I.I.; Radu, C.I.; Vladacenco, O.; Roza, E.; Costachescu, B.; Grumezescu, A.M.; Teleanu, R.I. An Overview of Oxidative Stress, Neuroinflammation, and Neurodegenerative Diseases. Int. J. Mol. Sci. 2022, 23, 5938. [Google Scholar] [CrossRef]
- Dreyer, E.B.; Zurakowski, D.; Schumer, R.A.; Podos, S.M.; Lipton, S.A. Elevated glutamate levels in the vitreous body of humans and monkeys with glaucoma. Arch. Ophthalmol. 1996, 114, 299–305. [Google Scholar] [CrossRef]
- Kawasaki, A.; Otori, Y.; Barnstable, C.J. Muller cell protection of rat retinal ganglion cells from glutamate and nitric oxide neurotoxicity. Investig. Ophthalmol. Vis. Sci. 2000, 41, 3444–3450. [Google Scholar]
- Tezel, G.; Wax, M.B. Increased production of tumor necrosis factor-alpha by glial cells exposed to simulated ischemia or elevated hydrostatic pressure induces apoptosis in cocultured retinal ganglion cells. J. Neurosci. 2000, 20, 8693–8700. [Google Scholar] [CrossRef]
- Gozzo, L.; Romano, G.L.; Romano, F.; Brancati, S.; Longo, L.; Vitale, D.C.; Drago, F. Health Technology Assessment of Advanced Therapy Medicinal Products: Comparison Among 3 European Countries. Front. Pharmacol. 2021, 12, 755052. [Google Scholar] [CrossRef] [PubMed]
- Bonfiglio, V.; Rejdak, R.; Nowomiejska, K.; Zweifel, S.A.; Justus Wiest, M.R.; Romano, G.L.; Bucolo, C.; Gozzo, L.; Castellino, N.; Patane, C.; et al. Efficacy and Safety of Subthreshold Micropulse Yellow Laser for Persistent Diabetic Macular Edema After Vitrectomy: A Pilot Study. Front. Pharmacol. 2022, 13, 832448. [Google Scholar] [CrossRef] [PubMed]
- Williams, P.A.; Marsh-Armstrong, N.; Howell, G.R.; Lasker, I.I.o.A.; Glaucomatous Neurodegeneration, P. Neuroinflammation in glaucoma: A new opportunity. Exp. Eye Res. 2017, 157, 20–27. [Google Scholar] [CrossRef]
- Tezel, G. Immune regulation toward immunomodulation for neuroprotection in glaucoma. Curr. Opin. Pharmacol. 2013, 13, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Mac Nair, C.E.; Nickells, R.W. Neuroinflammation in Glaucoma and Optic Nerve Damage. Prog. Mol. Biol. Transl. Sci. 2015, 134, 343–363. [Google Scholar] [CrossRef]
- Yoo, H.S.; Shanmugalingam, U.; Smith, P.D. Harnessing Astrocytes and Muller Glial Cells in the Retina for Survival and Regeneration of Retinal Ganglion Cells. Cells 2021, 10, 1339. [Google Scholar] [CrossRef]
- Puglia, C.; Santonocito, D.; Romeo, G.; Intagliata, S.; Romano, G.L.; Strettoi, E.; Novelli, E.; Ostacolo, C.; Campiglia, P.; Sommella, E.M.; et al. Lipid Nanoparticles Traverse Non-Corneal Path to Reach the Posterior Eye Segment: In Vivo Evidence. Molecules 2021, 26, 4673. [Google Scholar] [CrossRef]
- Kaminska, A.; Romano, G.L.; Rejdak, R.; Zweifel, S.; Fiedorowicz, M.; Rejdak, M.; Bajka, A.; Amato, R.; Bucolo, C.; Avitabile, T.; et al. Influence of Trace Elements on Neurodegenerative Diseases of The Eye-The Glaucoma Model. Int. J. Mol. Sci. 2021, 22, 4323. [Google Scholar] [CrossRef]
- Boulkrane, M.S.; Ilina, V.; Melchakov, R.; Fedotova, J.; Drago, F.; Gozzo, L.; Das, U.N.; Abd El-Aty, A.M.; Baranenko, D. COVID-19 Disease and Vitamin D: A Mini-Review. Front. Pharmacol. 2020, 11, 604579. [Google Scholar] [CrossRef]
- Rolle, T.; Ponzetto, A.; Malinverni, L. The Role of Neuroinflammation in Glaucoma: An Update on Molecular Mechanisms and New Therapeutic Options. Front. Neurol. 2020, 11, 612422. [Google Scholar] [CrossRef]
- Lambuk, L.; Ahmad, S.; Sadikan, M.Z.; Nordin, N.A.; Kadir, R.; Nasir, N.A.A.; Chen, X.; Boer, J.; Plebanski, M.; Mohamud, R. Targeting Differential Roles of Tumor Necrosis Factor Receptors as a Therapeutic Strategy for Glaucoma. Front. Immunol. 2022, 13, 857812. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, B.B. Signalling pathways of the TNF superfamily: A double-edged sword. Nat. Rev. Immunol. 2003, 3, 745–756. [Google Scholar] [CrossRef] [PubMed]
- Clark, J.; Vagenas, P.; Panesar, M.; Cope, A.P. What does tumour necrosis factor excess do to the immune system long term? Ann. Rheum. Dis. 2005, 64 (Suppl. S4), 70–76. [Google Scholar] [CrossRef] [PubMed]
- McCoy, M.K.; Ruhn, K.A.; Blesch, A.; Tansey, M.G. TNF: A key neuroinflammatory mediator of neurotoxicity and neurodegeneration in models of Parkinson’s disease. Adv. Exp. Med. Biol. 2011, 691, 539–540. [Google Scholar] [CrossRef] [PubMed]
- Zelova, H.; Hosek, J. TNF-alpha signalling and inflammation: Interactions between old acquaintances. Inflamm. Res. 2013, 62, 641–651. [Google Scholar] [CrossRef]
- Shi, K.; Zhang, J.; Dong, J.F.; Shi, F.D. Dissemination of brain inflammation in traumatic brain injury. Cell Mol. Immunol. 2019, 16, 523–530. [Google Scholar] [CrossRef]
- Jung, Y.J.; Tweedie, D.; Scerba, M.T.; Greig, N.H. Neuroinflammation as a Factor of Neurodegenerative Disease: Thalidomide Analogs as Treatments. Front. Cell Dev. Biol. 2019, 7, 313. [Google Scholar] [CrossRef]
- von Bernhardi, R.; Cornejo, F.; Parada, G.E.; Eugenin, J. Role of TGFbeta signaling in the pathogenesis of Alzheimer’s disease. Front. Cell Neurosci. 2015, 9, 426. [Google Scholar] [CrossRef]
- Cai, Y.; Liu, J.; Wang, B.; Sun, M.; Yang, H. Microglia in the Neuroinflammatory Pathogenesis of Alzheimer’s Disease and Related Therapeutic Targets. Front. Immunol. 2022, 13, 856376. [Google Scholar] [CrossRef]
- Xu, S.; Lu, J.; Shao, A.; Zhang, J.H.; Zhang, J. Glial Cells: Role of the Immune Response in Ischemic Stroke. Front. Immunol. 2020, 11, 294. [Google Scholar] [CrossRef]
- Tezel, G. TNF-alpha signaling in glaucomatous neurodegeneration. Prog. Brain Res. 2008, 173, 409–421. [Google Scholar] [CrossRef] [PubMed]
- Tezel, G.; Li, L.Y.; Patil, R.V.; Wax, M.B. TNF-alpha and TNF-alpha receptor-1 in the retina of normal and glaucomatous eyes. Investig. Ophthalmol. Vis. Sci. 2001, 42, 1787–1794. [Google Scholar]
- Desai, D.; He, S.; Yorio, T.; Krishnamoorthy, R.R.; Prasanna, G. Hypoxia augments TNF-alpha-mediated endothelin-1 release and cell proliferation in human optic nerve head astrocytes. Biochem. Biophys. Res. Commun. 2004, 318, 642–648. [Google Scholar] [CrossRef]
- Fontaine, V.; Mohand-Said, S.; Hanoteau, N.; Fuchs, C.; Pfizenmaier, K.; Eisel, U. Neurodegenerative and neuroprotective effects of tumor Necrosis factor (TNF) in retinal ischemia: Opposite roles of TNF receptor 1 and TNF receptor 2. J. Neurosci. 2002, 22, RC216. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Goeddel, D.V. TNF-R1 signaling: A beautiful pathway. Science 2002, 296, 1634–1635. [Google Scholar] [CrossRef]
- Probert, L. TNF and its receptors in the CNS: The essential, the desirable and the deleterious effects. Neuroscience 2015, 302, 2–22. [Google Scholar] [CrossRef] [PubMed]
- Chou, T.H.; Tomarev, S.; Porciatti, V. Transgenic mice expressing mutated Tyr437His human myocilin develop progressive loss of retinal ganglion cell electrical responsiveness and axonopathy with normal iop. Investig. Ophthalmol. Vis. Sci. 2014, 55, 5602–5609. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Migallon, M.C.; Nadal-Nicolas, F.M.; Jimenez-Lopez, M.; Sobrado-Calvo, P.; Vidal-Sanz, M.; Agudo-Barriuso, M. Brain derived neurotrophic factor maintains Brn3a expression in axotomized rat retinal ganglion cells. Exp. Eye Res. 2011, 92, 260–267. [Google Scholar] [CrossRef]
- Valiente-Soriano, F.J.; Nadal-Nicolas, F.M.; Salinas-Navarro, M.; Jimenez-Lopez, M.; Bernal-Garro, J.M.; Villegas-Perez, M.P.; Agudo-Barriuso, M.; Vidal-Sanz, M. BDNF Rescues RGCs But Not Intrinsically Photosensitive RGCs in Ocular Hypertensive Albino Rat Retinas. Investig. Ophthalmol. Vis. Sci. 2015, 56, 1924–1936. [Google Scholar] [CrossRef]
- Boia, R.; Ruzafa, N.; Aires, I.D.; Pereiro, X.; Ambrosio, A.F.; Vecino, E.; Santiago, A.R. Neuroprotective Strategies for Retinal Ganglion Cell Degeneration: Current Status and Challenges Ahead. Int. J. Mol. Sci. 2020, 21, 2262. [Google Scholar] [CrossRef]
- Buccarello, L.; Dragotto, J.; Hassanzadeh, K.; Maccarone, R.; Corbo, M.; Feligioni, M. Retinal ganglion cell loss in an ex vivo mouse model of optic nerve cut is prevented by curcumin treatment. Cell Death Discov. 2021, 7, 394. [Google Scholar] [CrossRef] [PubMed]
- Lambuk, L.; Mohd Lazaldin, M.A.; Ahmad, S.; Iezhitsa, I.; Agarwal, R.; Uskokovic, V.; Mohamud, R. Brain-Derived Neurotrophic Factor-Mediated Neuroprotection in Glaucoma: A Review of Current State of the Art. Front. Pharmacol. 2022, 13, 875662. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Shang, P.; Wang, M.; Xie, M.; Liu, J. Neuroprotective Effects of Fluoxetine Against Chronic Stress-Induced Neural Inflammation and Apoptosis: Involvement of the p38 Activity. Front. Physiol. 2020, 11, 351. [Google Scholar] [CrossRef] [PubMed]
- Bastos, A.G.; Guimaraes, L.S.; Trentini, C.M. The efficacy of long-term psychodynamic psychotherapy, fluoxetine and their combination in the outpatient treatment of depression. Psychother. Res. 2015, 25, 612–624. [Google Scholar] [CrossRef] [PubMed]
- Dooley, L.N.; Kuhlman, K.R.; Robles, T.F.; Eisenberger, N.I.; Craske, M.G.; Bower, J.E. The role of inflammation in core features of depression: Insights from paradigms using exogenously-induced inflammation. Neurosci. Biobehav. Rev. 2018, 94, 219–237. [Google Scholar] [CrossRef] [PubMed]
- Dantzer, R.; O’Connor, J.C.; Freund, G.G.; Johnson, R.W.; Kelley, K.W. From inflammation to sickness and depression: When the immune system subjugates the brain. Nat. Rev. Neurosci. 2008, 9, 46–56. [Google Scholar] [CrossRef]
- Miller, A.H.; Maletic, V.; Raison, C.L. Inflammation and its discontents: The role of cytokines in the pathophysiology of major depression. Biol. Psychiatry 2009, 65, 732–741. [Google Scholar] [CrossRef]
- Garcia-Garcia, M.L.; Tovilla-Zarate, C.A.; Villar-Soto, M.; Juarez-Rojop, I.E.; Gonzalez-Castro, T.B.; Genis-Mendoza, A.D.; Ramos-Mendez, M.A.; Lopez-Narvaez, M.L.; Saucedo-Osti, A.S.; Ruiz-Quinones, J.A.; et al. Fluoxetine modulates the pro-inflammatory process of IL-6, IL-1beta and TNF-alpha levels in individuals with depression: A systematic review and meta-analysis. Psychiatry Res. 2022, 307, 114317. [Google Scholar] [CrossRef]
- Shan, H.; Bian, Y.; Shu, Z.; Zhang, L.; Zhu, J.; Ding, J.; Lu, M.; Xiao, M.; Hu, G. Fluoxetine protects against IL-1beta-induced neuronal apoptosis via downregulation of p53. Neuropharmacology 2016, 107, 68–78. [Google Scholar] [CrossRef]
- Copani, A.; Condorelli, F.; Caruso, A.; Vancheri, C.; Sala, A.; Giuffrida Stella, A.M.; Canonico, P.L.; Nicoletti, F.; Sortino, M.A. Mitotic signaling by beta-amyloid causes neuronal death. FASEB J. 1999, 13, 2225–2234. [Google Scholar] [CrossRef]
- Caraci, F.; Pappalardo, G.; Basile, L.; Giuffrida, A.; Copani, A.; Tosto, R.; Sinopoli, A.; Giuffrida, M.L.; Pirrone, E.; Drago, F.; et al. Neuroprotective effects of the monoamine oxidase inhibitor tranylcypromine and its amide derivatives against Abeta(1-42)-induced toxicity. Eur. J. Pharmacol. 2015, 764, 256–263. [Google Scholar] [CrossRef] [PubMed]
- Kingston, R.; Amin, D.; Misra, S.; Gross, J.M.; Kuwajima, T. Serotonin transporter-mediated molecular axis regulates regional retinal ganglion cell vulnerability and axon regeneration after nerve injury. PLoS Genet. 2021, 17, e1009885. [Google Scholar] [CrossRef] [PubMed]
- Bosco, A.; Steele, M.R.; Vetter, M.L. Early microglia activation in a mouse model of chronic glaucoma. J. Comp. Neurol. 2011, 519, 599–620. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward Primer | Reverse Primer |
|---|---|---|
| IL-6 | 5′-ACAACCACGGCCTTCCCTA-3′ | 5′-TTGCCATTGCACAACTCTTTTCTC-3′ |
| TNF-α | 5′-CAGGCGGTGCCTATGTCTC-3′ | 5′-CCATTTGGGAACTTCTCATCCCTT-3′ |
| Iba-1 | 5′-GCAATTCCTCGATGATCCCAAA-3′ | 5′-GATCAAACTCCATGTACTTCACCTT-3′ |
| IL-1β | 5′-GGGCCTCAAAGGAAAGAATC-3′ | 5′-TACCAGTTGGGGAACTCTGC-3′ |
| S100β | 5′-ACTTCCTGGAGGAAATCAAGGAGC-3′ | 5′-ACACTCCCCATCCCCATCTTC-3′ |
| HPRT | 5′-TCAGTCAACGGGGGACATAAA-3′ | 5′-GGGGCTGTACTGCTTAACCAG-3′ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Romano, G.L.; Gozzo, L.; Maurel, O.M.; Di Martino, S.; Riolo, V.; Micale, V.; Drago, F.; Bucolo, C. Fluoxetine Protects Retinal Ischemic Damage in Mice. Pharmaceutics 2023, 15, 1370. https://doi.org/10.3390/pharmaceutics15051370
Romano GL, Gozzo L, Maurel OM, Di Martino S, Riolo V, Micale V, Drago F, Bucolo C. Fluoxetine Protects Retinal Ischemic Damage in Mice. Pharmaceutics. 2023; 15(5):1370. https://doi.org/10.3390/pharmaceutics15051370
Chicago/Turabian StyleRomano, Giovanni Luca, Lucia Gozzo, Oriana Maria Maurel, Serena Di Martino, Valentina Riolo, Vincenzo Micale, Filippo Drago, and Claudio Bucolo. 2023. "Fluoxetine Protects Retinal Ischemic Damage in Mice" Pharmaceutics 15, no. 5: 1370. https://doi.org/10.3390/pharmaceutics15051370
APA StyleRomano, G. L., Gozzo, L., Maurel, O. M., Di Martino, S., Riolo, V., Micale, V., Drago, F., & Bucolo, C. (2023). Fluoxetine Protects Retinal Ischemic Damage in Mice. Pharmaceutics, 15(5), 1370. https://doi.org/10.3390/pharmaceutics15051370