

Structural Adaptation of the Single-Stranded DNA-Binding Protein C-Terminal to DNA Metabolizing Partners Guides Inhibitor Design

 , , , ,
, , , ,
Abstract
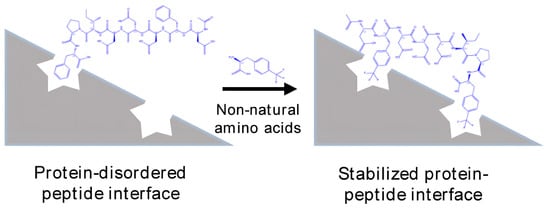
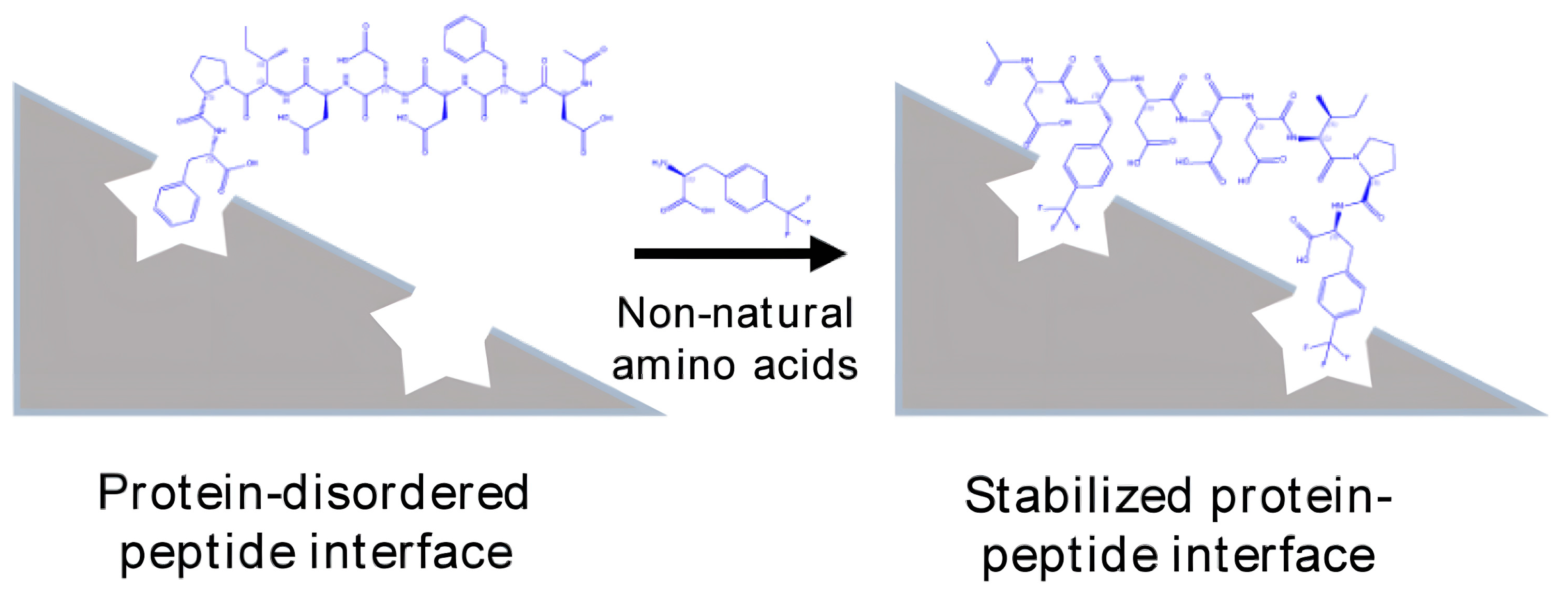
1. Introduction
2. Materials and Methods
2.1. Buffers and Reagents
2.2. Proteins and Peptides
2.3. Fluorescence Anisotropy Assay
2.4. Isothermal Titration Calorimetry
2.5. Pull down Assay
2.6. Modeling
3. Results
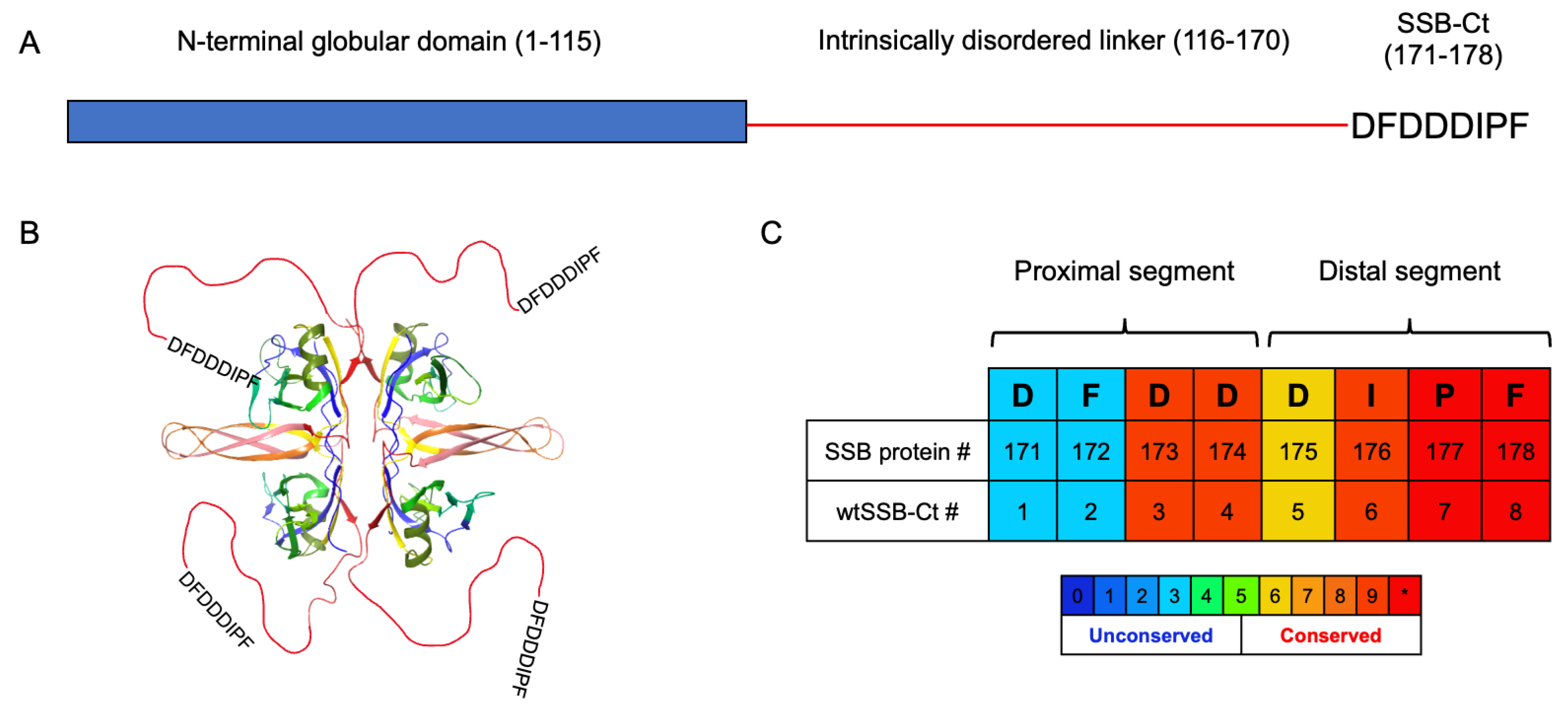
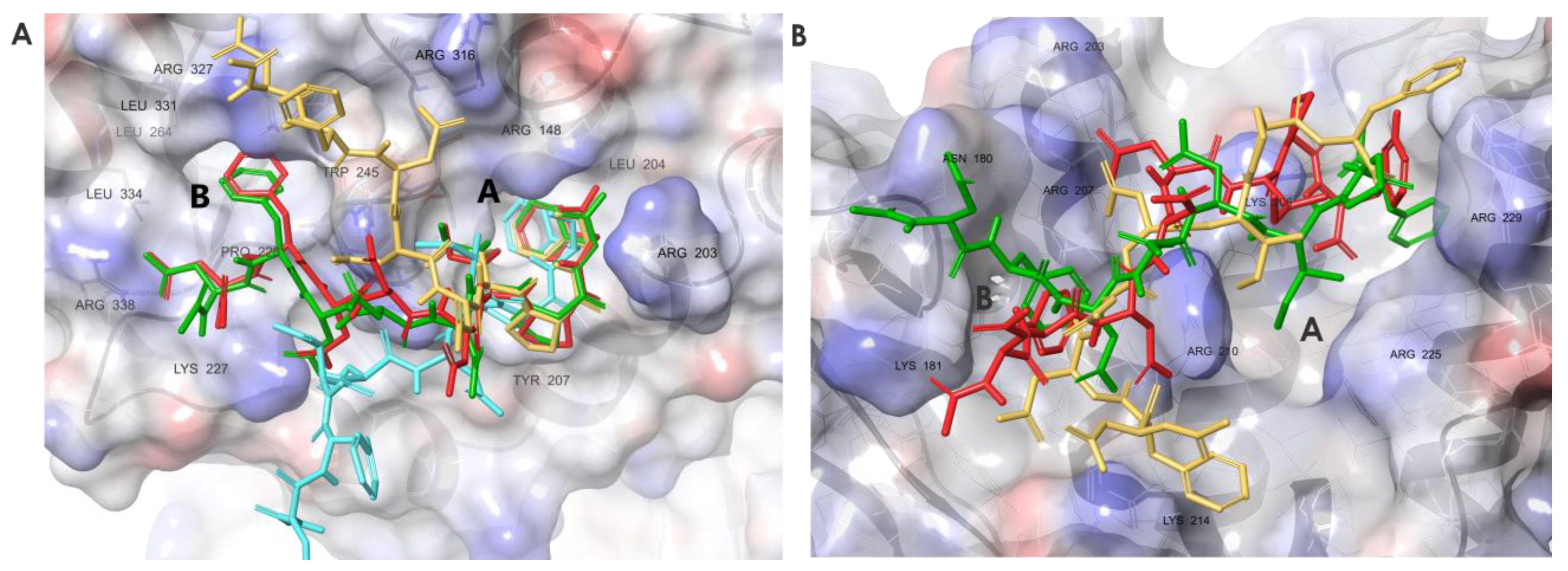
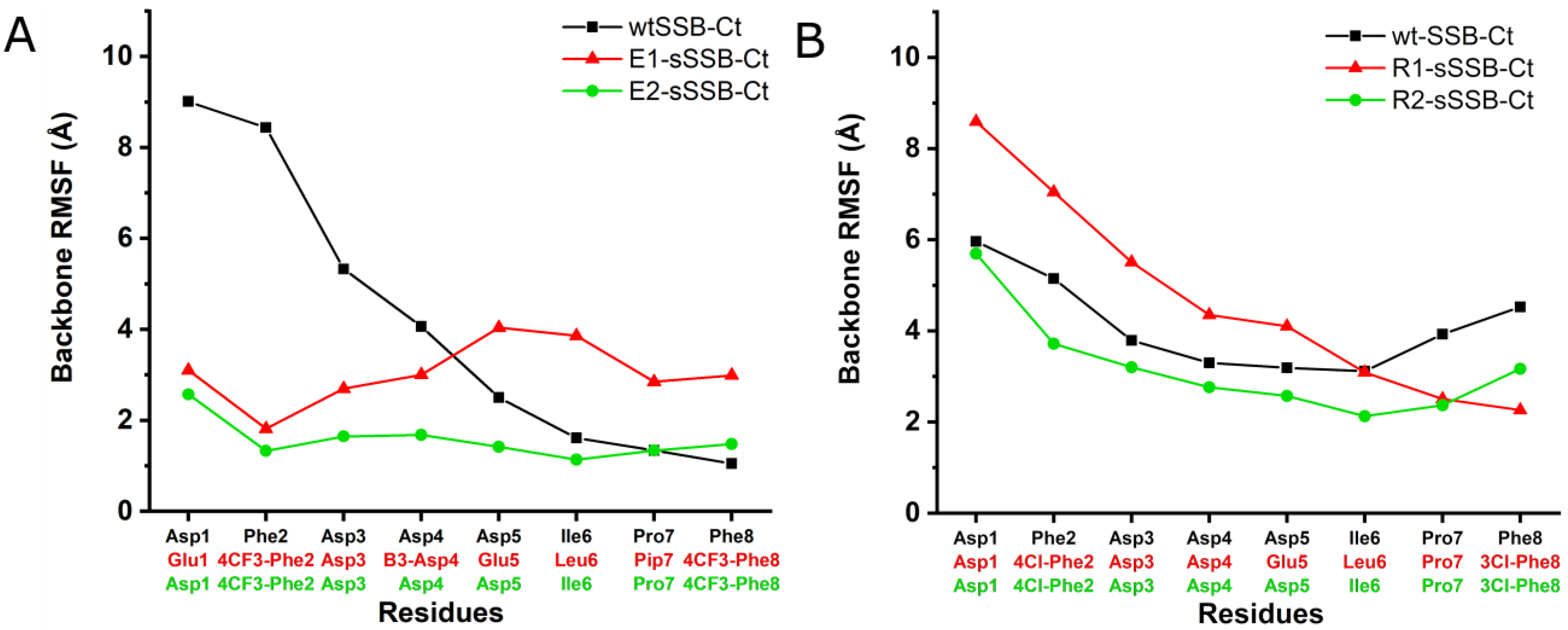
3.1. Molecular Dynamics Simulations of SSB-Ct–SIP Interactions
3.2. Contributions of the Proximal DFDD and the Distal DIPF Segments to the Binding Affinity
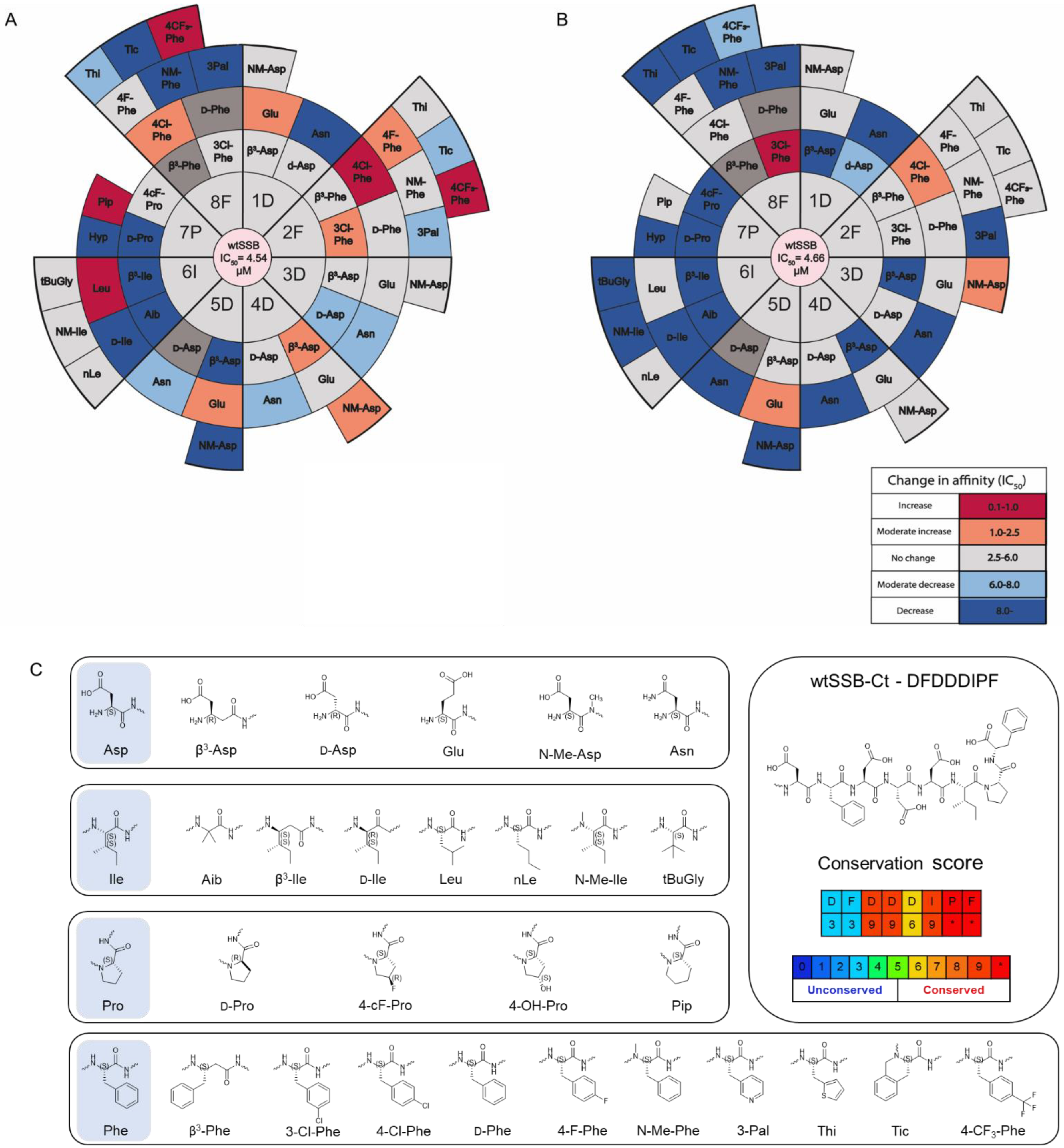
3.3. Synthesis and Screening of the Single Mutant wtSSB-Ct Library
3.4. Modifications in Both the Proximal DFDD and the Distal DIPF Segments Increase Affinity
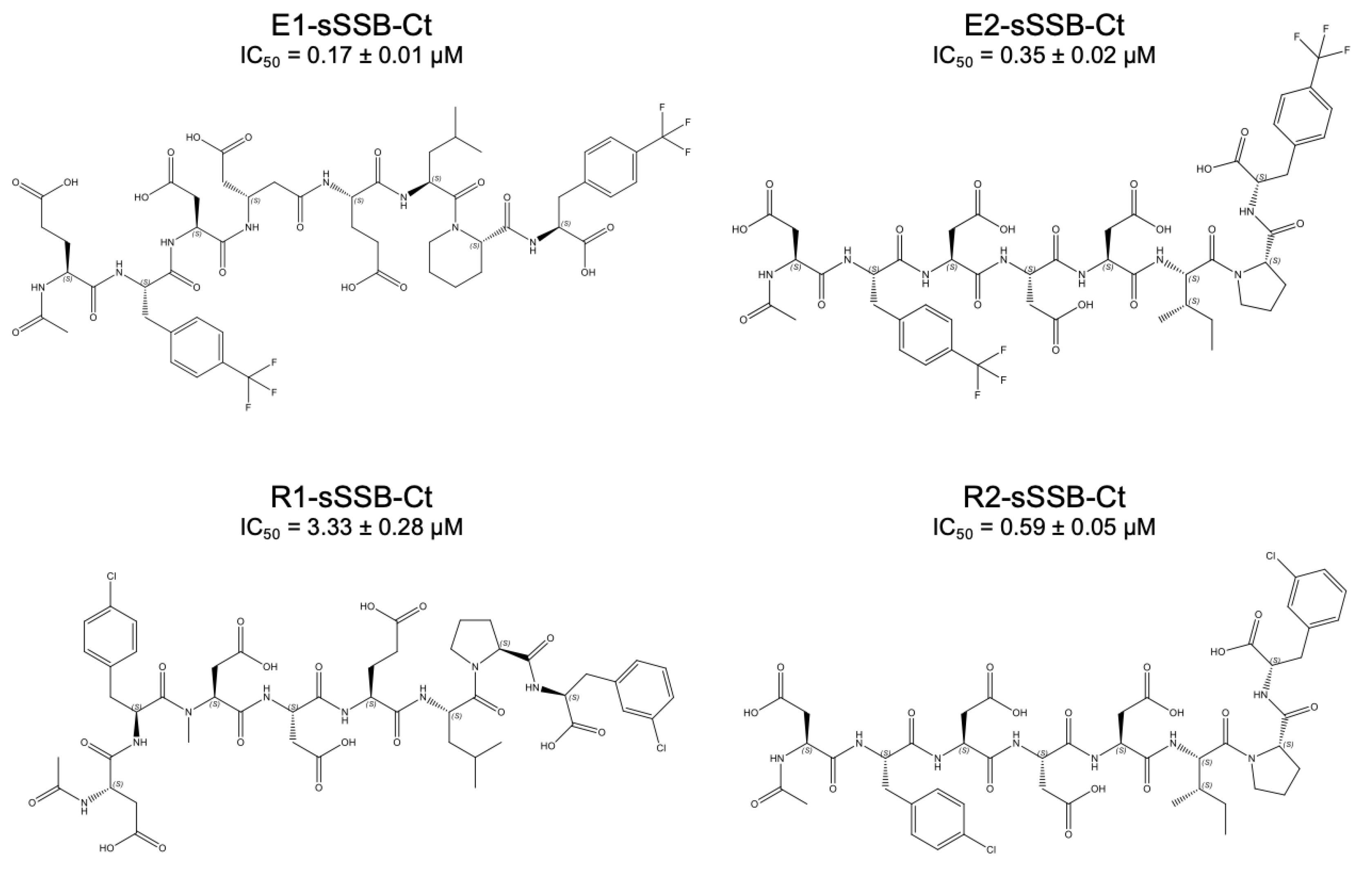
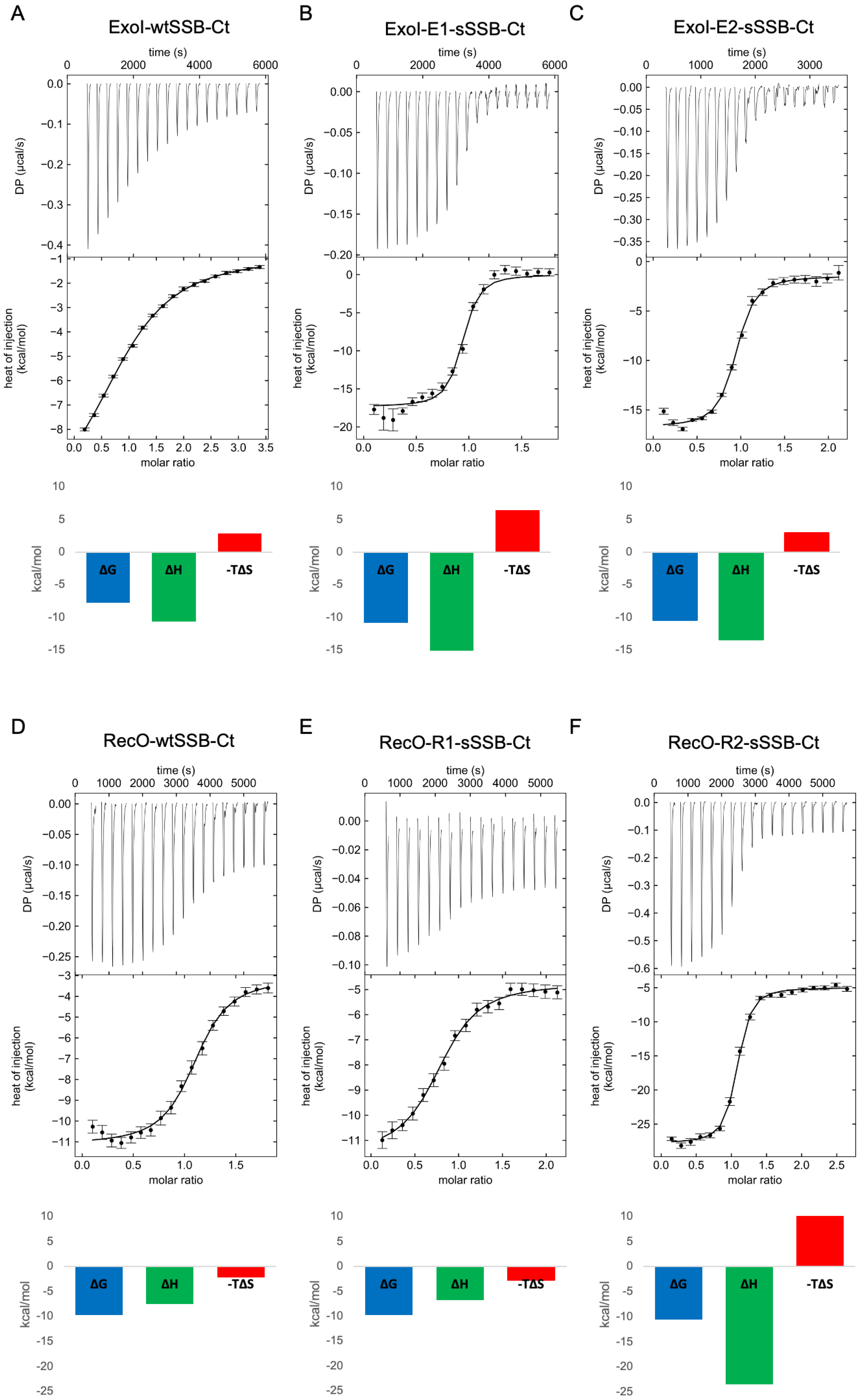
3.5. Combined Modifications Yielded High-Affinity Ligands
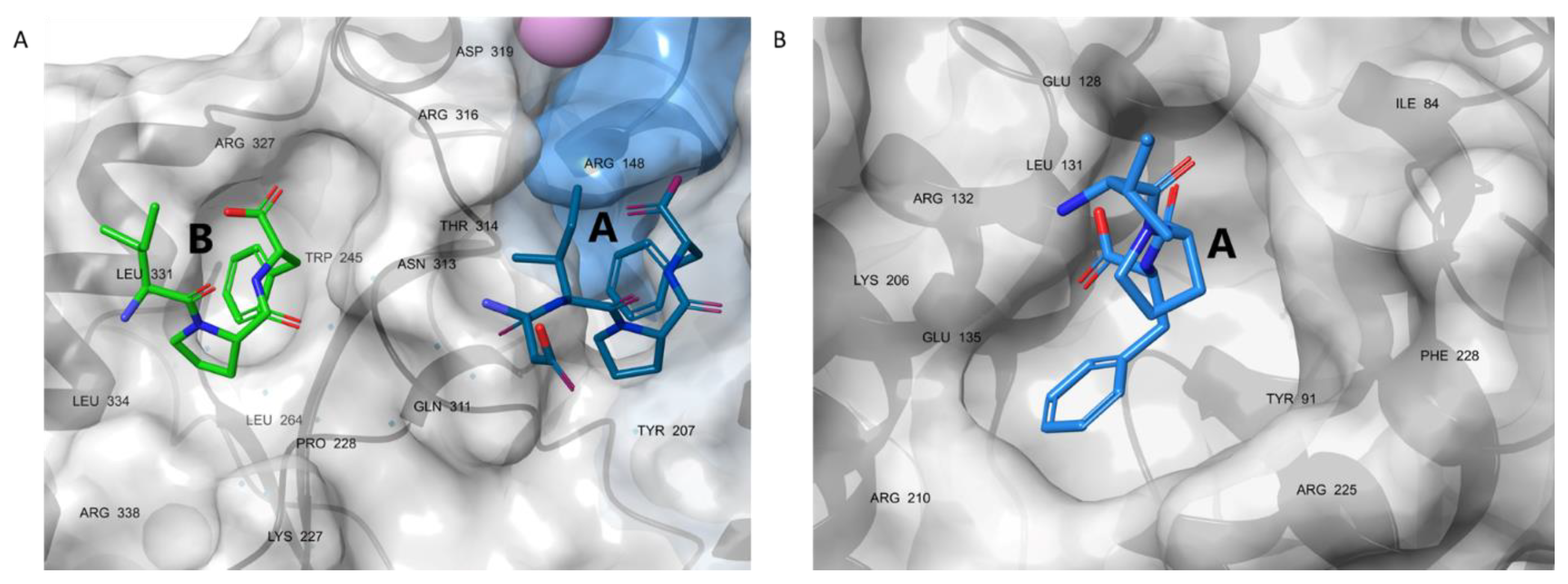
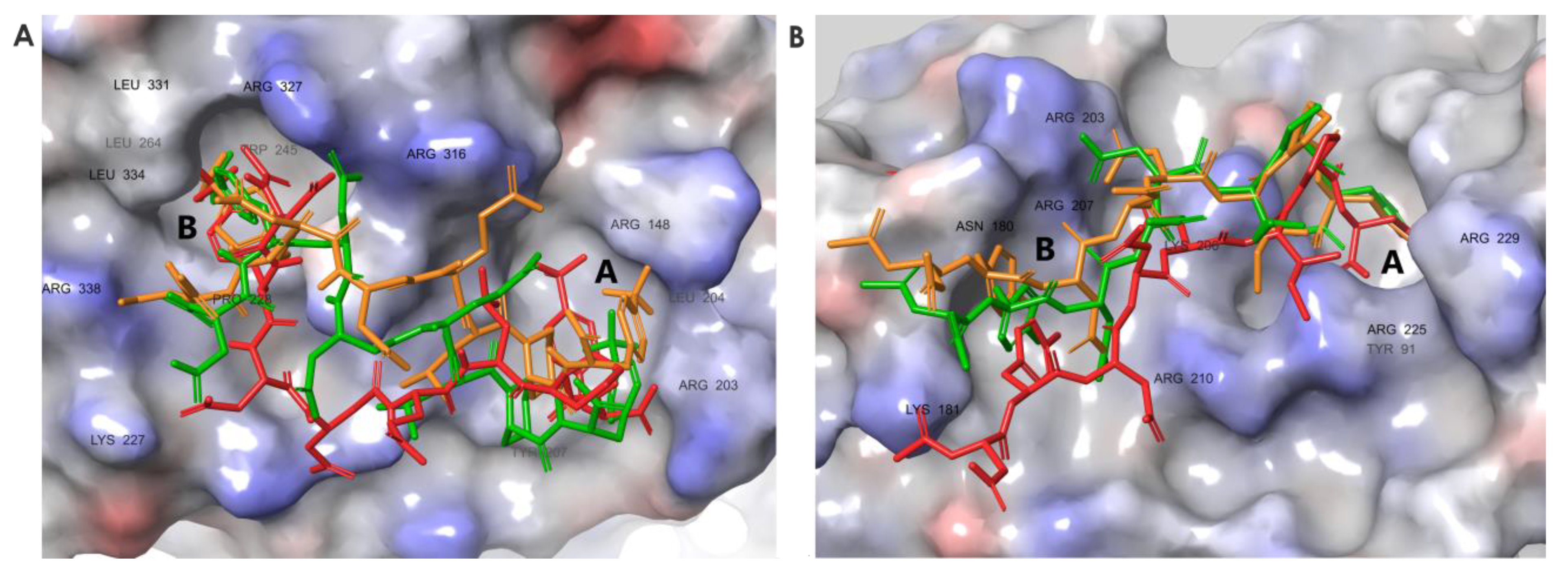
3.6. Molecular Dynamics Simulations Provided Insight into the Binding Modes of E1-sSSB-Ct and R2-sSSB-Ct
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Murray, C.J.; Ikuta, K.S.; Sharara, F.; Swetschinski, L.; Robles Aguilar, G.; Gray, A.; Han, C.; Bisignano, C.; Rao, P.; Wool, E.; et al. Global burden of bacterial antimicrobial resistance in 2019: A systematic analysis. Lancet 2022, 399, 629–655. [Google Scholar] [CrossRef] [PubMed]
- Miethke, M.; Pieroni, M.; Weber, T.; Brönstrup, M.; Hammann, P.; Halby, L.; Arimondo, P.B.; Glaser, P.; Aigle, B.; Bode, H.B.; et al. Towards the sustainable discovery and development of new antibiotics. Nat. Rev. Chem. 2021, 5, 726–749. [Google Scholar] [CrossRef] [PubMed]
- Theuretzbacher, U.; Outterson, K.; Engel, A.; Karlén, A. The global preclinical antibacterial pipeline. Nat. Rev. Microbiol. 2020, 18, 275–285. [Google Scholar] [CrossRef] [PubMed]
- Brown, E.D.; Wright, G.D. Antibacterial drug discovery in the resistance era. Nature 2016, 529, 336–343. [Google Scholar] [CrossRef] [PubMed]
- Tommasi, R.; Brown, D.G.; Walkup, G.K.; Manchester, J.I.; Miller, A.A. ESKAPEing the labyrinth of antibacterial discovery. Nat. Rev. Drug. Discov. 2015, 14, 529–542. [Google Scholar] [CrossRef] [PubMed]
- Lu, D.; Bernstein, D.A.; Satyshur, K.A.; Keck, J.L. Small-molecule tools for dissecting the roles of SSB/protein interactions in genome maintenance. Proc. Natl. Acad. Sci. USA 2010, 107, 633–638. [Google Scholar] [CrossRef]
- Marceau, A.H.; Bernstein, D.A.; Walsh, B.W.; Shapiro, W.; Simmons, L.A.; Keck, J.L. Protein Interactions in Genome Maintenance as Novel Antibacterial Targets. PLoS ONE 2013, 8, e58765. [Google Scholar] [CrossRef]
- van Eijk, E.; Wittekoek, B.; Kuijper, E.J.; Smits, W.K. DNA replication proteins as potential targets for antimicrobials in drug-resistant bacterial pathogens. J. Antimicrob. Chemother. 2017, 72, 1275–1284. [Google Scholar] [CrossRef]
- Rowen, L.; Kornberg, A. Primase, the dnaG protein of Escherichia coli. An enzyme which starts DNA chains. J. Biol. Chem. 1978, 253, 758–764. [Google Scholar] [CrossRef]
- Lu, D.; Keck, J.L. Structural basis of Escherichia coli single-stranded DNA-binding protein stimulation of exonuclease I. Proc. Natl. Acad. Sci. USA 2008, 105, 9169–9174. [Google Scholar] [CrossRef]
- Ryzhikov, M.; Koroleva, O.; Postnov, D.; Tran, A.; Korolev, S. Mechanism of RecO recruitment to DNA by single-stranded DNA binding protein. Nucleic Acids Res. 2011, 39, 6305–6314. [Google Scholar] [CrossRef] [PubMed]
- Cadman, C.J. PriA helicase and SSB interact physically and functionally. Nucleic Acids Res. 2004, 32, 6378–6387. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.-Y.; Lu, C.-H.; Li, H.-W. RecA-SSB Interaction Modulates RecA Nucleoprotein Filament Formation on SSB-Wrapped DNA. Sci. Rep. 2017, 7, 11876. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Godson, G.N. Structure of the Escherichia coli primase/single-strand DNA-binding protein/phage G4ori complex required for primer RNA synthesis. J. Mol. Biol. 1998, 276, 689–703. [Google Scholar] [CrossRef] [PubMed]
- Lehman, I.R.; Nussbaum, A.L. The deoxyribonucleases of escherichia coli. v. on the specificity of exonuclease i (phosphodiesterase). J. Biol. Chem. 1964, 239, 2628–2636. [Google Scholar] [CrossRef]
- Henry, C.; Henrikus, S.S. Elucidating Recombination Mediator Function Using Biophysical Tools. Biology 2021, 10, 288. [Google Scholar] [CrossRef]
- Handa, N.; Morimatsu, K.; Lovett, S.T.; Kowalczykowski, S.C. Reconstitution of initial steps of dsDNA break repair by the RecF pathway of E. coli. Genes Dev. 2009, 23, 1234–1245. [Google Scholar] [CrossRef]
- Morimatsu, K.; Wu, Y.; Kowalczykowski, S.C. RecFOR Proteins Target RecA Protein to a DNA Gap with Either DNA or RNA at the 5′ Terminus. J. Biol. Chem. 2012, 287, 35621–35630. [Google Scholar] [CrossRef]
- Bell, J.C.; Kowalczykowski, S.C. RecA: Regulation and Mechanism of a Molecular Search Engine. Trends Biochem. Sci. 2016, 41, 491–507. [Google Scholar] [CrossRef]
- Maslowska, K.H.; Makiela-Dzbenska, K.; Fijalkowska, I.J. The SOS system: A complex and tightly regulated response to DNA damage. Environ. Mol. Mutagen. 2019, 60, 368–384. [Google Scholar] [CrossRef]
- Bianco, P.R. The tale of SSB. Prog. Biophys. Mol. Biol. 2017, 127, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Kowalczykowski, S.C. DNA strand exchange proteins a biochemical and physical comparison. Front. Biosci. 1998, 3, 570–603. [Google Scholar] [CrossRef] [PubMed]
- Bianco, P.R. OB-fold Families of Genome Guardians: A Universal Theme Constructed From the Small β-barrel Building Block. Front. Mol. Biosci. 2022, 9, 784451. [Google Scholar] [CrossRef] [PubMed]
- Bianco, P.R. The mechanism of action of the SSB interactome reveals it is the first OB-fold family of genome guardians in prokaryotes. Protein Sci. 2021, 30, 1757–1775. [Google Scholar] [CrossRef]
- Ding, W.; Tan, H.Y.; Zhang, J.X.; Wilczek, L.A.; Hsieh, K.R.; Mulkin, J.A.; Bianco, P.R. The mechanism of Single strand binding protein–RecG binding: Implications for SSB interactome function. Protein Sci. 2020, 29, 1211–1227. [Google Scholar] [CrossRef]
- Shereda, R.D.; Kozlov, A.G.; Lohman, T.M.; Cox, M.M.; Keck, J.L. SSB as an Organizer/Mobilizer of Genome Maintenance Complexes. Crit. Rev. Biochem. Mol. Biol. 2008, 43, 289–318. [Google Scholar] [CrossRef]
- Chase, J.W.; L’Italien, J.J.; Murphy, J.B.; Spicer, E.K.; Williams, K.R. Characterization of the Escherichia coli SSB-113 mutant single-stranded DNA-binding protein. Cloning of the gene, DNA and protein sequence analysis, high pressure liquid chromatography peptide mapping, and DNA-binding studies. J. Biol. Chem. 1984, 259, 805–814. [Google Scholar] [CrossRef]
- Genschel, J.; Curth, U.; Urbanke, C. Interaction of E coli Single-Stranded DNA Binding Protein (SSB) with Exonuclease I. The Carboxy-Terminus of SSB Is the Recognition Site for the Nuclease. Biol. Chem. 2000, 381. [Google Scholar] [CrossRef]
- Lu, D.; Windsor, M.A.; Gellman, S.H.; Keck, J.L. Peptide Inhibitors Identify Roles for SSB C-Terminal Residues in SSB/Exonuclease I Complex Formation. Biochemistry 2009, 48, 6764–6771. [Google Scholar] [CrossRef]
- Fedorov, R.; Böhl, M.; Tsiavaliaris, G.; Hartmann, F.K.; Taft, M.H.; Baruch, P.; Brenner, B.; Martin, R.; Knölker, H.-J.; Gutzeit, H.O.; et al. The mechanism of pentabromopseudilin inhibition of myosin motor activity. Nat. Struct. Mol. Biol. 2009, 16, 80–88. [Google Scholar] [CrossRef]
- Bawono, P.; Heringa, J. PRALINE: A Versatile Multiple Sequence Alignment Toolkit. In Multiple Sequence Alignment Methods; Russell, D.J., Ed.; Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2014; Volume 1079, pp. 245–262. ISBN 978-1-62703-645-0. [Google Scholar]
- Chilingaryan, Z.; Headey, S.; Lo, A.; Xu, Z.-Q.; Otting, G.; Dixon, N.; Scanlon, M.; Oakley, A. Fragment-Based Discovery of Inhibitors of the Bacterial DnaG-SSB Interaction. Antibiotics 2018, 7, 14. [Google Scholar] [CrossRef] [PubMed]
- Voter, A.F.; Killoran, M.P.; Ananiev, G.E.; Wildman, S.A.; Hoffmann, F.M.; Keck, J.L. A High-Throughput Screening Strategy to Identify Inhibitors of SSB Protein–Protein Interactions in an Academic Screening Facility. Slas Discov. Adv. Sci. Drug Discov. 2018, 23, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Alnammi, M.; Liu, S.; Ericksen, S.S.; Ananiev, G.E.; Voter, A.F.; Guo, S.; Keck, J.L.; Hoffmann, F.M.; Wildman, S.A.; Gitter, A. Evaluating Scalable Supervised Learning for Synthesize-on-Demand Chemical Libraries. Chemistry 2021. [Google Scholar] [CrossRef]
- Shereda, R.D.; Bernstein, D.A.; Keck, J.L. A Central Role for SSB in Escherichia coli RecQ DNA Helicase Function. J. Biol. Chem. 2007, 282, 19247–19258. [Google Scholar] [CrossRef] [PubMed]
- Korada, S.K.C.; Johns, T.D.; Smith, C.E.; Jones, N.D.; McCabe, K.A.; Bell, C.E. Crystal structures of Escherichia coli exonuclease I in complex with single-stranded DNA provide insights into the mechanism of processive digestion. Nucleic Acids Res. 2013, 41, 5887–5897. [Google Scholar] [CrossRef]
- Ryzhikov, M.; Korolev, S. Structural Studies of SSB Interaction with RecO. In Single-Stranded DNA Binding Proteins; Keck, J.L., Ed.; Humana Press: Totowa, NJ, USA, 2012; pp. 123–131. ISBN 978-1-62703-031-1. [Google Scholar]
- Gasteiger, E.; Hoogland, C.; Gattiker, A.; Duvaud, S.; Wilkins, M.R.; Appel, R.D.; Bairoch, A. Protein Identification and Analysis Tools on the ExPASy Server. In The Proteomics Protocols Handbook; Walker, J.M., Ed.; Humana Press: Totowa, NJ, USA, 2005; pp. 571–607. ISBN 978-1-58829-343-5. [Google Scholar]
- Scheuermann, T.H.; Brautigam, C.A. High-precision, automated integration of multiple isothermal titration calorimetric thermograms: New features of NITPIC. Methods 2015, 76, 87–98. [Google Scholar] [CrossRef]
- Zhao, H.; Piszczek, G.; Schuck, P. SEDPHAT—A platform for global ITC analysis and global multi-method analysis of molecular interactions. Methods 2015, 76, 137–148. [Google Scholar] [CrossRef]
- Wang, L.; Friesner, R.A.; Berne, B.J. Replica Exchange with Solute Scaling: A More Efficient Version of Replica Exchange with Solute Tempering (REST2). J. Phys. Chem. B 2011, 115, 9431–9438. [Google Scholar] [CrossRef]
- Liu, P.; Kim, B.; Friesner, R.A.; Berne, B.J. Replica exchange with solute tempering: A method for sampling biological systems in explicit water. Proc. Natl. Acad. Sci. USA 2005, 102, 13749–13754. [Google Scholar] [CrossRef]
- Schrödinger Release 2021-1; Schrödinger, LLC: New York, NY, USA, 2021.
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; Hermans, J. Interaction Models for Water in Relation to Protein Hydration. In Intermolecular Forces; Pullman, B., Ed.; The Jerusalem Symposia on Quantum Chemistry and Biochemistry; Springer: Dordrecht, The Netherlands, 1981; Volume 14, pp. 331–342. ISBN 978-90-481-8368-5. [Google Scholar]
- Lu, C.; Wu, C.; Ghoreishi, D.; Chen, W.; Wang, L.; Damm, W.; Ross, G.A.; Dahlgren, M.K.; Russell, E.; Von Bargen, C.D.; et al. OPLS4: Improving Force Field Accuracy on Challenging Regimes of Chemical Space. J. Chem. Theory Comput. 2021, 17, 4291–4300. [Google Scholar] [CrossRef]
- Kovács, É.; Ali, H.; Minorics, R.; Traj, P.; Resch, V.; Paragi, G.; Bruszel, B.; Zupkó, I.; Mernyák, E. Synthesis and Antiproliferative Activity of Steroidal Diaryl Ethers. Molecules 2023, 28, 1196. [Google Scholar] [CrossRef] [PubMed]
- Schieferdecker, S.; Vock, E. Development of Pharmacophore Models for the Important Off-Target 5-HT 2B Receptor. J. Med. Chem. 2023, 66, 1509–1521. [Google Scholar] [CrossRef] [PubMed]
- Lihan, M.; Lupyan, D.; Oehme, D. Target-template relationships in protein structure prediction and their effect on the accuracy of thermostability calculations. Protein Sci. 2023, 32, e4557. [Google Scholar] [CrossRef]
- Frey, B.J.; Dueck, D. Clustering by Passing Messages Between Data Points. Science 2007, 315, 972–976. [Google Scholar] [CrossRef] [PubMed]
- Shinn, M.K.; Kozlov, A.G.; Lohman, T.M. Allosteric effects of SSB C-terminal tail on assembly of E. coli RecOR proteins. Nucleic Acids Res. 2021, 49, 1987–2004. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Zhou, Q.; He, J.; Jiang, Z.; Peng, C.; Tong, R.; Shi, J. Recent advances in the development of protein–protein interactions modulators: Mechanisms and clinical trials. Signal Transduct. Target. Ther. 2020, 5, 213. [Google Scholar] [CrossRef]
- Wells, J.A.; McClendon, C.L. Reaching for high-hanging fruit in drug discovery at protein–protein interfaces. Nature 2007, 450, 1001–1009. [Google Scholar] [CrossRef]
- Mabonga, L.; Kappo, A.P. Protein-protein interaction modulators: Advances, successes and remaining challenges. Biophys. Rev. 2019, 11, 559–581. [Google Scholar] [CrossRef]
- Borcherds, W.; Theillet, F.-X.; Katzer, A.; Finzel, A.; Mishall, K.M.; Powell, A.T.; Wu, H.; Manieri, W.; Dieterich, C.; Selenko, P.; et al. Disorder and residual helicity alter p53-Mdm2 binding affinity and signaling in cells. Nat. Chem. Biol. 2014, 10, 1000–1002. [Google Scholar] [CrossRef]
- Neira, J.L.; Bintz, J.; Arruebo, M.; Rizzuti, B.; Bonacci, T.; Vega, S.; Lanas, A.; Velázquez-Campoy, A.; Iovanna, J.L.; Abián, O. Identification of a Drug Targeting an Intrinsically Disordered Protein Involved in Pancreatic Adenocarcinoma. Sci. Rep. 2017, 7, 39732. [Google Scholar] [CrossRef]
- Su, B.G.; Henley, M.J. Drugging Fuzzy Complexes in Transcription. Front. Mol. Biosci. 2021, 8, 795743. [Google Scholar] [CrossRef]
- Oláh, J.; Szénási, T.; Lehotzky, A.; Norris, V.; Ovádi, J. Challenges in Discovering Drugs That Target the Protein–Protein Interactions of Disordered Proteins. IJMS 2022, 23, 1550. [Google Scholar] [CrossRef] [PubMed]
- Blaskovich, M.A.T. Unusual Amino Acids in Medicinal Chemistry. J. Med. Chem. 2016, 59, 10807–10836. [Google Scholar] [CrossRef] [PubMed]
- Kurczab, R.; Canale, V.; Satała, G.; Zajdel, P.; Bojarski, A.J. Amino Acid Hot Spots of Halogen Bonding: A Combined Theoretical and Experimental Case Study of the 5-HT7 Receptor. J. Med. Chem. 2018, 61, 8717–8733. [Google Scholar] [CrossRef] [PubMed]
- Shinada, N.K.; de Brevern, A.G.; Schmidtke, P. Halogens in Protein–Ligand Binding Mechanism: A Structural Perspective. J. Med. Chem. 2019, 62, 9341–9356. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | ExoI | RecO | ||||
|---|---|---|---|---|---|---|
| Peptide | wtSSB-Ct | E1-sSSB-Ct | E2-sSSB-Ct | wtSSB-Ct | R1-sSSB-Ct | R2-sSSB-Ct |
| cell (µM) | 7.5 | 3 | 5 | 4 | 2 | 2 |
| syringe (µM) | 120 | 25 | 50 | 50 | 20 | 50 |
| mSSB | IC50 on ExoI (µM) | EC50 on ExoI (µM) | IC50 on RecO (µM) | EC50 on RecO (µM) |
|---|---|---|---|---|
| F-wtSSB | 4.54 ± 0.34 | 0.35 ± 0.04 | 4.66± 0.24 | 0.37 ± 0.03 |
| F-4Cl-Phe2 | 0.56 ± 0.03 | 0.15 ± 0.02 | 0.41 ± 0.27 | 0.26 ± 0.01 |
| F-4F-Phe2 | 1.93 ± 0.81 | 0.36 ± 0.03 | 0.52 ± 0.28 | 0.23 ± 0.02 |
| F-4CF3-Phe2 | 0.33 ± 0.02 | 0.04 ± 0.002 | 0.56 ± 0.42 | 0.18 ± 0.03 |
| F-NM-Asp3 | 3.88 ± 0.13 | 0.86 ± 0.11 | 1.88 ± 0.09 | 0.27 ± 0.04 |
| F-Glu5 | 1.49 ± 0.10 | 0.11 ± 0.01 | 1.41 ± 0.55 | 0.20 ± 0.02 |
| F-Leu6 | 0.92 ± 0.06 | 0.20 ± 0.03 | 3.23 ± 0.16 | 0.31 ± 0.03 |
| F-nLe6 | 2.68 ± 0.54 | 0.20 ± 0.01 | 9.36 ± 0.08 | 0.20 ± 0.01 |
| F-Pip7 | 0.89 ± 0.14 | 0.09 ± 0.01 | 3.31 ± 0.44 | 0.25 ± 0.03 |
| F-3Cl-Phe8 | 3.40 ± 2.41 | 0.25 ± 0.02 | 0.36 ± 0.04 | 0.22 ± 0.03 |
| F-4CF3-Phe8 | 0.39 ± 0.07 | 0.09 ± 0.01 | 5.97 ± 2.43 | 0.26 ± 0.02 |
| ExoI | RecO | |||||
|---|---|---|---|---|---|---|
| wtSSB-Ct | E1-sSSB-Ct | E2-sSSB-Ct | wtSSB-Ct | R1-sSSB-Ct | R2-sSSB-Ct | |
| N | 0.9593 | 1.0000 | 0.8846 | 1.0032 | 0.9353 | 0.9999 |
| KD (µM) | 3.07 (2.62–3.64) | 0.02 (0.01–0.03) | 0.04 (0.02–0.07) | 0.14 (0.08–0.24) | 0.13 (0.08–0.22) | 0.04 (0.02–0.05) |
| ΔG (kcal/mol) | −7.78 | −10.89 | −10.43 | −9.77 | −9.71 | −10.54 |
| ΔH (kcal/mol) | −10.67 (−11.64–−9.97) | −17.35 (−18.34–−16.43) | −13.45 (−14.46–−12.49) | −7.54 (−8.28–−6.90) | −6.79 (−7.84–−6.05) | −23.48 (−24.34–−22.73) |
| −TΔS (kcal/mol) | 2.90 | 6.46 | 3.02 | −2.28 | −2.91 | 12.32 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tököli, A.; Bodnár, B.; Bogár, F.; Paragi, G.; Hetényi, A.; Bartus, É.; Wéber, E.; Hegedüs, Z.; Szabó, Z.; Kecskeméti, G.; et al. Structural Adaptation of the Single-Stranded DNA-Binding Protein C-Terminal to DNA Metabolizing Partners Guides Inhibitor Design. Pharmaceutics 2023, 15, 1032. https://doi.org/10.3390/pharmaceutics15041032
Tököli A, Bodnár B, Bogár F, Paragi G, Hetényi A, Bartus É, Wéber E, Hegedüs Z, Szabó Z, Kecskeméti G, et al. Structural Adaptation of the Single-Stranded DNA-Binding Protein C-Terminal to DNA Metabolizing Partners Guides Inhibitor Design. Pharmaceutics. 2023; 15(4):1032. https://doi.org/10.3390/pharmaceutics15041032
Chicago/Turabian StyleTököli, Attila, Brigitta Bodnár, Ferenc Bogár, Gábor Paragi, Anasztázia Hetényi, Éva Bartus, Edit Wéber, Zsófia Hegedüs, Zoltán Szabó, Gábor Kecskeméti, and et al. 2023. "Structural Adaptation of the Single-Stranded DNA-Binding Protein C-Terminal to DNA Metabolizing Partners Guides Inhibitor Design" Pharmaceutics 15, no. 4: 1032. https://doi.org/10.3390/pharmaceutics15041032
APA StyleTököli, A., Bodnár, B., Bogár, F., Paragi, G., Hetényi, A., Bartus, É., Wéber, E., Hegedüs, Z., Szabó, Z., Kecskeméti, G., Szakonyi, G., & Martinek, T. A. (2023). Structural Adaptation of the Single-Stranded DNA-Binding Protein C-Terminal to DNA Metabolizing Partners Guides Inhibitor Design. Pharmaceutics, 15(4), 1032. https://doi.org/10.3390/pharmaceutics15041032