
Design, Synthesis, and Antisickling Investigation of a Thiazolidine Prodrug of TD-7 That Prolongs the Duration of Action of Antisickling Aromatic Aldehyde

 , ,
, ,  , ,
, , 
Abstract
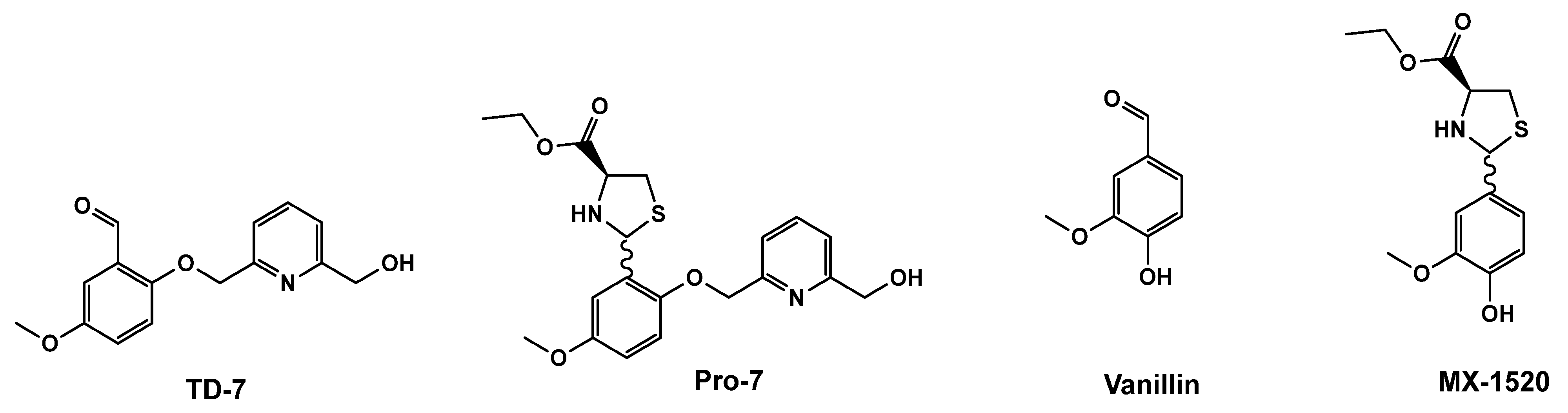
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Study Approvals
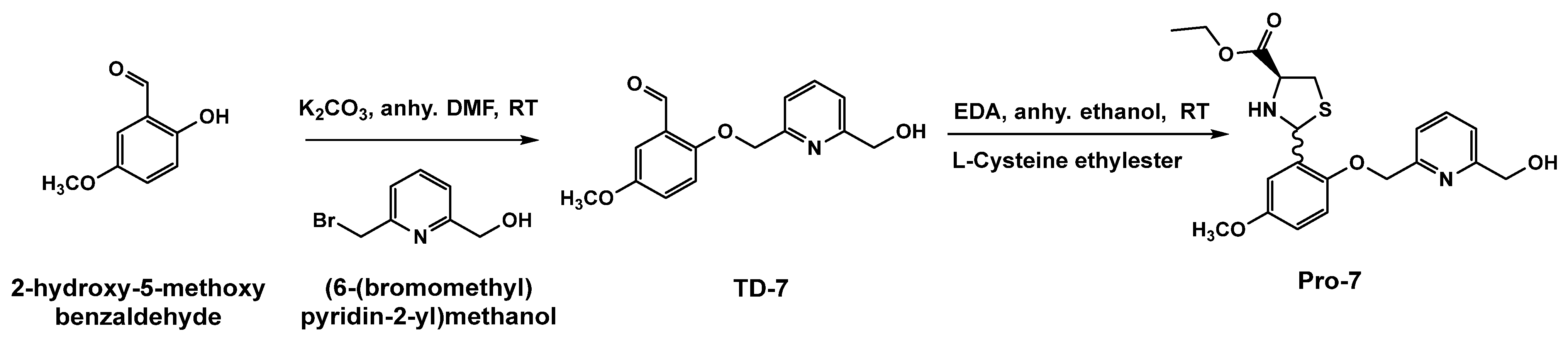
2.3. Chemistry Synthesis of Pro-7, the Prodrug of TD-7
2.4. In-Vitro Stability Studies
2.5. In-Vitro Time-Dependent Hb Modification Studies Using Human Normal Whole Blood
2.6. In-Vitro Time-Dependent Oxygen Equilibrium Studies Using Human Normal Whole Blood
2.7. In-Vitro Time-Dependent Sickling Inhibition Studies Using Homozygous Sickle Cell (SS) Blood
2.8. Development of Enteric-Coated Beads and Intravenous (IV) Formulations
2.8.1. Solubility Study
2.8.2. Preparation of Enteric-Coated Microspheres
2.8.3. Stability of the Prepared Microspheres in Buffer pH 1.2 and 7.4
2.8.4. Preparation of IV Formulation
2.9. In-Vivo Pharmacokinetic Studies in Rat
2.9.1. Animal Housing
2.9.2. Chromatographic Conditions
2.9.3. Pharmacokinetic Analysis
2.9.4. Statistical Analysis
3. Results and Discussion
3.1. Protection of TD-7 by L-Cysteine Ethyl Ester
3.2. The Stability of Pro-7 Is pH Dependent
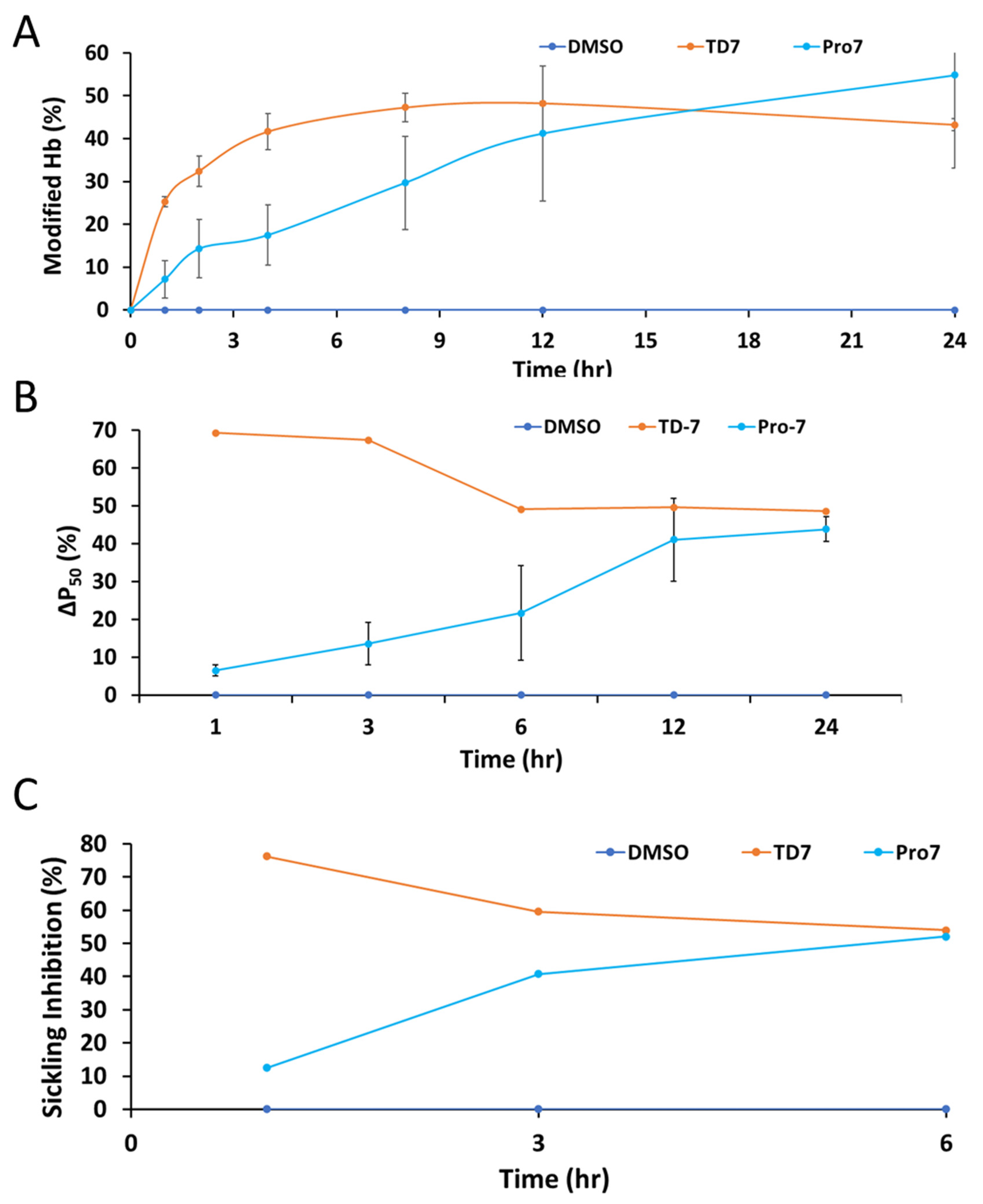
3.3. Pro-7 Modifies Hb to Increase the Protein Affinity for Oxygen and Prevents Hypoxia Induced RBC Sickling in a Sustained Manner
3.4. Development of Enteric Coated Beads
3.5. Development of Intravenous (IV) Formulation
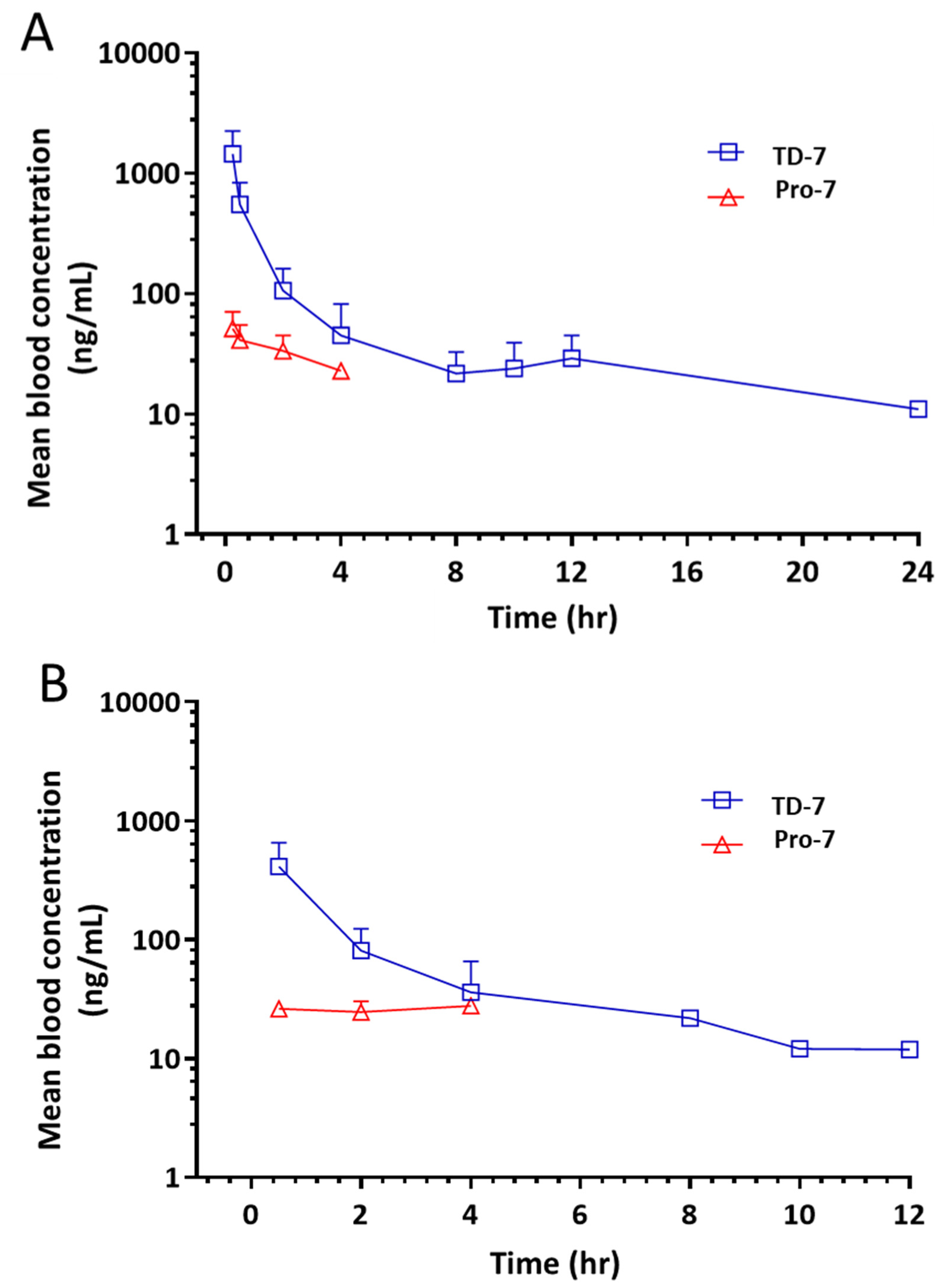
3.6. Pro-7 Is Capable of Releasing TD-7 in Blood
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| Hb | hemoglobin |
| AEH | allosteric effectors of hemoglobin |
| RBC | red blood cell |
| SCD | sickle cell disease; deoxyHb, deoxygenated hemoglobin |
| R-state | relaxed state |
| T-state | tense state |
References
- Ingram, V.M. Gene Mutations in Human Hæmoglobin: The Chemical Difference Between Normal and Sickle Cell Hæmoglobin. Nature 1957, 180, 326–328. [Google Scholar] [CrossRef]
- Pauling, L.; Itano, H.A.; Singer, S.J.; Wells, I.C. Sickle Cell Anemia, a Molecular Disease. Science 1949, 110, 543–548. [Google Scholar] [CrossRef] [PubMed]
- Belcher, J.D.; Bryant, C.J.; Nguyen, J.; Bowlin, P.R.; Kielbik, M.C.; Bischof, J.C.; Hebbel, R.P.; Vercellotti, G.M. Transgenic Sickle Mice Have Vascular Inflammation. Blood 2003, 101, 3953–3959. [Google Scholar] [CrossRef]
- Ferrone, F.A. Polymerization and Sickle Cell Disease: A Molecular View. Microcirculation 2004, 11, 115–128. [Google Scholar] [CrossRef] [PubMed]
- Aliyu, Z.Y.; Gordeuk, V.; Sachdev, V.; Babadoko, A.; Mamman, A.I.; Akpanpe, P.; Attah, E.; Suleiman, Y.; Aliyu, N.; Yusuf, J.; et al. Prevalence and Risk Factors for Pulmonary Artery Systolic Hypertension among Sickle Cell Disease Patients in Nigeria. Am. J. Hematol. 2008, 83, 485–490. [Google Scholar] [CrossRef]
- De Franceschi, L. Pathophisiology of Sickle Cell Disease and New Drugs for the Treatment. Mediterr. J. Hematol. Infect. Dis. 2009, 1, e2009024. [Google Scholar] [CrossRef]
- Akinsheye, I.; Klings, E.S. Sickle Cell Anemia and Vascular Dysfunction: The Nitric Oxide Connection. J. Cell. Physiol. 2010, 224, 620–625. [Google Scholar] [CrossRef] [PubMed]
- Kato, G.J. New Insights into Sickle Cell Disease: Mechanisms and Investigational Therapies. Curr. Opin. Hematol. 2016, 23, 224–232. [Google Scholar] [CrossRef] [PubMed]
- Piel, F.B.; Steinberg, M.H.; Rees, D.C. Sickle Cell Disease. N. Engl. J. Med. 2017, 377, 305. [Google Scholar] [CrossRef]
- Donkor, A.K.; Pagare, P.P.; Mughram, M.H.A.; Safo, M.K. X-ray Crystallography and Sickle Cell Disease Drug Discovery-a Tribute to Donald Abraham. Front. Mol. Biosci. 2023, 10, 1136970. [Google Scholar] [CrossRef]
- Huang, B.; Ghatge, M.S.; Donkor, A.K.; Musayev, F.N.; Deshpande, T.M.; Al-Awadh, M.; Alhashimi, R.T.; Zhu, H.; Omar, A.M.; Telen, M.J.; et al. Design, Synthesis, and Investigation of Novel Nitric Oxide (NO)-Releasing Aromatic Aldehydes as Drug Candidates for the Treatment of Sickle Cell Disease. Molecules 2022, 27, 6835. [Google Scholar] [CrossRef]
- Pagare, P.P.; Rastegar, A.; Abdulmalik, O.; Omar, A.M.; Zhang, Y.; Fleischman, A.; Safo, M.K. Modulating Hemoglobin Allostery for Treatment of Sickle Cell Disease: Current Progress and Intellectual Property. Expert Opin. Ther. Pat. 2022, 32, 115–130. [Google Scholar] [CrossRef]
- Piccin, A.; Murphy, C.; Eakins, E.; Rondinelli, M.B.; Daves, M.; Vecchiato, C.; Wolf, D.; Mc Mahon, C.; Smith, O.P. Insight into the Complex Pathophysiology of Sickle Cell Anaemia and Possible Treatment. Eur. J. Haematol. 2019, 102, 319–330. [Google Scholar] [CrossRef] [PubMed]
- Piccin, A.; Magzoub, I.; Hervig, T. The ‘Scintilla’ Starting Vaso-occlusion in Sickle Cell Disease. Br. J. Haematol. 2023, 201, 379–380. [Google Scholar] [CrossRef] [PubMed]
- Telen, M.J. Beyond Hydroxyurea: New and Old Drugs in the Pipeline for Sickle Cell Disease. Blood 2016, 127, 810–819. [Google Scholar] [CrossRef]
- Mvalo, T.; Topazian, H.; Kamthunzi, P.; Chen, J.; Kambalame, I.; Mafunga, P.; Mumba, N.; Chiume-Chiphaliwali, M.; Paseli, K.; Key, N.; et al. Increasing Hydroxyurea Use in Children with Sickle Cell Disease at Kamuzu Central Hospital, Malawi. Blood Adv. 2018, 2, 30–32. [Google Scholar] [CrossRef]
- Quinn, C.T. L-Glutamine for Sickle Cell Anemia: More Questions than Answers. Blood 2018, 132, 689–693. [Google Scholar] [CrossRef] [PubMed]
- Cieri-Hutcherson, N.E.; Hutcherson, T.C.; Conway-Habes, E.E.; Burns, B.N.; White, N.A. Systematic Review of L-Glutamine for Prevention of Vaso-Occlusive Pain Crisis in Patients with Sickle Cell Disease. Pharmacotherapy 2019, 39, 1095–1104. [Google Scholar] [CrossRef]
- Ataga, K.I.; Kutlar, A.; Kanter, J.; Liles, D.; Cancado, R.; Friedrisch, J.; Guthrie, T.H.; Knight-Madden, J.; Alvarez, O.A.; Gordeuk, V.R.; et al. Crizanlizumab for the Prevention of Pain Crises in Sickle Cell Disease. N. Engl. J. Med. 2017, 376, 429–439. [Google Scholar] [CrossRef]
- Oksenberg, D.; Dufu, K.; Patel, M.P.; Chuang, C.; Li, Z.; Xu, Q.; Silva-Garcia, A.; Zhou, C.; Hutchaleelaha, A.; Patskovska, L.; et al. GBT440 Increases Haemoglobin Oxygen Affinity, Reduces Sickling and Prolongs RBC Half-Life in a Murine Model of Sickle Cell Disease. Br. J. Haematol. 2016, 175, 141–153. [Google Scholar] [CrossRef]
- Metcalf, B.; Chuang, C.; Dufu, K.; Patel, M.P.; Silva-Garcia, A.; Johnson, C.; Lu, Q.; Partridge, J.R.; Patskovska, L.; Patskovsky, Y.; et al. Discovery of GBT440, an Orally Bioavailable R-State Stabilizer of Sickle Cell Hemoglobin. ACS Med. Chem. Lett. 2017, 8, 321–326. [Google Scholar] [CrossRef]
- Dufu, K.; Oksenberg, D. GBT440 Reverses Sickling of Sickled Red Blood Cells under Hypoxic Conditions in Vitro. Hematol. Rep. 2018, 10, 7419. [Google Scholar] [CrossRef] [PubMed]
- Vichinsky, E.; Hoppe, C.C.; Ataga, K.I.; Ware, R.E.; Nduba, V.; El-Beshlawy, A.; Hassab, H.; Achebe, M.M.; Alkindi, S.; Brown, R.C.; et al. A Phase 3 Randomized Trial of Voxelotor in Sickle Cell Disease. N. Engl. J. Med. 2019, 381, 509–519. [Google Scholar] [CrossRef] [PubMed]
- Abdulmalik, O.; Ghatge, M.S.; Musayev, F.N.; Parikh, A.; Chen, Q.; Yang, J.; Nnamani, I.; Danso-Danquah, R.; Eseonu, D.N.; Asakura, T.; et al. Crystallographic Analysis of Human Hemoglobin Elucidates the Structural Basis of the Potent and Dual Antisickling Activity of Pyridyl Derivatives of Vanillin. Acta Crystallogr. D Biol. Crystallogr. 2011, 67, 920–928. [Google Scholar] [CrossRef]
- Rolan, P.E.; Mercer, A.J.; Wootton, R.; Posner, J. Pharmacokinetics and Pharmacodynamics of Tucaresol, an Antisickling Agent, in Healthy Volunteers. Br. J. Clin. Pharmacol. 1995, 39, 375–380. [Google Scholar] [CrossRef] [PubMed]
- Arya, R.; Rolan, P.E.; Wootton, R.; Posner, J.; Bellingham, A.J. Tucaresol Increases Oxygen Affinity and Reduces Haemolysis in Subjects with Sickle Cell Anaemia. Br. J. Haematol. 1996, 93, 817–821. [Google Scholar] [CrossRef] [PubMed]
- Deshpande, T.M.; Pagare, P.P.; Ghatge, M.S.; Chen, Q.; Musayev, F.N.; Venitz, J.; Zhang, Y.; Abdulmalik, O.; Safo, M.K. Rational Modification of Vanillin Derivatives to Stereospecifically Destabilize Sickle Hemoglobin Polymer Formation. Acta Crystallogr. D Struct. Biol. 2018, 74, 956–964. [Google Scholar] [CrossRef] [PubMed]
- Stern, W.; Mathews, D.; McKew, J.; Shen, X.; Kato, G.J. A Phase 1, First-in-Man, Dose-Response Study of Aes-103 (5-HMF), an Anti-Sickling, Allosteric Modifier of Hemoglobin Oxygen Affinity in Healthy Norman Volunteers. Blood 2012, 120, 3210. [Google Scholar] [CrossRef]
- Kato, G.J.; Lawrence, M.P.; Mendelsohn, L.G.; Saiyed, R.; Wang, X.; Conrey, A.K.; Starling, J.M.; Grimes, G.; Taylor, J.G.; McKew, J.; et al. Phase 1 Clinical Trial of the Candidate Anti-Sickling Agent Aes-103 in Adults with Sickle Cell Anemia. Blood 2013, 122, 1009. [Google Scholar] [CrossRef]
- Abraham, D.J.; Mehanna, A.S.; Wireko, F.C.; Whitney, J.; Thomas, R.P.; Orringer, E.P. Vanillin, a Potential Agent for the Treatment of Sickle Cell Anemia. Blood 1991, 77, 1334–1341. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, A.; Rzhetsky, A.; Hsu, L.C.; Chang, C. Human Aldehyde Dehydrogenase Gene Family. Eur. J. Biochem. 1998, 251, 549–557. [Google Scholar] [CrossRef] [PubMed]
- Godfrey, V.B.; Chen, L.J.; Griffin, R.J.; Lebetkin, E.H.; Burka, L.T. Distribution and Metabolism of (5-Hydroxymethyl)Furfural in Male F344 Rats and B6C3F1 Mice after Oral Administration. J. Toxicol. Environ. Health A 1999, 57, 199–210. [Google Scholar] [CrossRef] [PubMed]
- Vasiliou, V.; Pappa, A.; Petersen, D.R. Role of Aldehyde Dehydrogenases in Endogenous and Xenobiotic Metabolism. Chem. Biol. Interact. 2000, 129, 1–19. [Google Scholar]
- Zhang, C.; Li, X.; Lian, L.; Chen, Q.; Abdulmalik, O.; Vassilev, V.; Lai, C.-S.; Asakura, T. Anti-Sickling Effect of MX-1520, a Prodrug of Vanillin: An in Vivo Study Using Rodents: Vanillin Prodrug for the Treatment of Sickle Cell Disease. Br. J. Haematol. 2004, 125, 788–795. [Google Scholar] [CrossRef] [PubMed]
- Fife, T.H.; Natarajan, R.; Shen, C.C.; Bembi, R. Mechanism of Thiazolidine Hydrolysis. Ring Opening and Hydrolysis of 1,3-Thiazolidine Derivatives of p-(Dimethylamino)Cinnamaldehyde. J. Am. Chem. Soc. 1991, 113, 3071–3079. [Google Scholar] [CrossRef]
- Omar, A.M.; Abdulmalik, O.; Ghatge, M.S.; Muhammad, Y.A.; Paredes, S.D.; El-Araby, M.E.; Safo, M.K. An Investigation of Structure-Activity Relationships of Azolylacryloyl Derivatives Yielded Potent and Long-Acting Hemoglobin Modulators for Reversing Erythrocyte Sickling. Biomolecules 2020, 10, 1508. [Google Scholar] [CrossRef]
- Sahoo, S.; Behera, A.; Malik, B.; Patil, S. Effect of Plasticizers on Various Characteristics of Eudragit Microspheres Formulated by Solvent Evaporation Method. Int. J. Drug Dev. Res. 2011, 3, 285–290. [Google Scholar]
- Obeidat, W.M.; Price, J.C. Preparation and Evaluation of Eudragit S 100 Microspheres as pH-Sensitive Release Preparations for Piroxicam and Theophylline Using the Emulsion-Solvent Evaporation Method. J. Microencapsul. 2006, 23, 195–202. [Google Scholar] [CrossRef]
- Ahmed, T.A.; El-Say, K.M.; Abd-Allah, F.I.; Omar, A.M.; El-Araby, M.E.; Muhammad, Y.A.; Pagare, P.P.; Zhang, Y.; Mohmmad, K.A.; Abdulmalik, O.; et al. Improving the Solubility and Oral Bioavailability of a Novel Aromatic Aldehyde Antisickling Agent (PP10) for the Treatment of Sickle Cell Disease. Pharmaceutics 2021, 13, 1148. [Google Scholar] [CrossRef]
- Önen-Bayram, F.E.; Buran, K.; Durmaz, I.; Berk, B.; Cetin-Atalay, R. 3-Propionyl-Thiazolidine-4-Carboxylic Acid Ethyl Esters: A Family of Antiproliferative Thiazolidines. Med. Chem. Commun. 2015, 6, 90–93. [Google Scholar] [CrossRef]
- USP-NF. United States Pharmacopeia General Chapter: <729> Globule Size Distribution in Lipid Injectable Emulsions; USP-NF: Rockville, MD, USA, 2023. [Google Scholar]
- Nilsson, N.; Nezvalova-Henriksen, K.; Tho, I. Emulsion Stability of Different Intravenous Propofol Formulations in Simulated Co-Administration with Remifentanil Hydrochloride. Pharm. Technol. Hosp. Pharm. 2019, 4, 77–87. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| %Hb Modification (Hb Adduct) b | %Hb O2 Affinity (P50 Shift) c | %Sickling Inhibition d | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Time (h) | TD-7 | Pro-7 | DMSO | TD-7 | Pro-7 | DMSO | TD-7 | Pro-7 | DMSO |
| 0 | 0.00 ± 0.00 | 0.00 ± 0.00 | 0.00 | ND | ND | ND | ND | ND | ND |
| 1 | 25.20 ± 1.20 | 7.10 ± 4.40 | 0.00 | 69.26 | 6.48 ± 1.54 | 0.00 | 76.30 | 12.53 | 0.00 |
| 2 | 23.40 ± 3.50 | 14.30 ± 6.80 | 0.00 | ND | ND | ND | ND | ND | ND |
| 3 | ND | ND | ND | 67.42 | 13.57 ± 5.53 | 0.00 | 59.62 | 40.76 | 0.00 |
| 4 | 41.70 ± 4.30 | 17.50 ± 7.10 | 0.00 | ND | ND | ND | ND | ND | ND |
| 6 | ND | ND | ND | 49.06 | 21.64 ± 10.93 | 0.00 | 54.02 | 52.04 | 0.00 |
| 8 | 47.30 ± 4.70 | 29.70 ± 10.9 | 0.00 | ND | ND | ND | ND | ND | ND |
| 12 | 48.30 ± 0.50 | 41.20 ± 15.8 | 0.00 | 49.58 | 41.04 ± 10.93 | 0.00 | ND | ND | ND |
| 24 | 43.30 ± 1.90 | 54.80 ± 21.8 | 0.00 | 48.56 | 43.87 ± 3.22 | 0.00 | ND | ND | ND |
| Pharmacokinetic Parameter | Intravenous Administration | Oral Administration |
|---|---|---|
| t½ (h) | 4.98 ± 1.89 | 4.73 ± 1.22 |
| Cmax (ng/mL) | 1426 ± 926 | 469 ± 322 |
| Tmax (h) | - | 0.5 ± 0.0 |
| AUC0–t (ng∙h/mL) | 1830 ± 737 | 672 ± 255 |
| AUC0–∞ (ng∙h/mL) | 1956 ± 792 | 730 ± 225 |
| Cl (mL/min/kg) | 238 ± 85.9 | 200.89 ± 34.3 |
| Vd (L/kg) | 98.9 ± 45.3 | 83.7 ± 22.1 |
| Fraction of drug absorbed (% F) | - | 18.4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alhashimi, R.T.; Ahmed, T.A.; Alghanem, L.; Pagare, P.P.; Huang, B.; Ghatge, M.S.; Omar, A.M.; Abdulmalik, O.; Zhang, Y.; Safo, M.K. Design, Synthesis, and Antisickling Investigation of a Thiazolidine Prodrug of TD-7 That Prolongs the Duration of Action of Antisickling Aromatic Aldehyde. Pharmaceutics 2023, 15, 2547. https://doi.org/10.3390/pharmaceutics15112547
Alhashimi RT, Ahmed TA, Alghanem L, Pagare PP, Huang B, Ghatge MS, Omar AM, Abdulmalik O, Zhang Y, Safo MK. Design, Synthesis, and Antisickling Investigation of a Thiazolidine Prodrug of TD-7 That Prolongs the Duration of Action of Antisickling Aromatic Aldehyde. Pharmaceutics. 2023; 15(11):2547. https://doi.org/10.3390/pharmaceutics15112547
Chicago/Turabian StyleAlhashimi, Rana T., Tarek A. Ahmed, Lamya Alghanem, Piyusha P. Pagare, Boshi Huang, Mohini S. Ghatge, Abdelsattar M. Omar, Osheiza Abdulmalik, Yan Zhang, and Martin K. Safo. 2023. "Design, Synthesis, and Antisickling Investigation of a Thiazolidine Prodrug of TD-7 That Prolongs the Duration of Action of Antisickling Aromatic Aldehyde" Pharmaceutics 15, no. 11: 2547. https://doi.org/10.3390/pharmaceutics15112547
APA StyleAlhashimi, R. T., Ahmed, T. A., Alghanem, L., Pagare, P. P., Huang, B., Ghatge, M. S., Omar, A. M., Abdulmalik, O., Zhang, Y., & Safo, M. K. (2023). Design, Synthesis, and Antisickling Investigation of a Thiazolidine Prodrug of TD-7 That Prolongs the Duration of Action of Antisickling Aromatic Aldehyde. Pharmaceutics, 15(11), 2547. https://doi.org/10.3390/pharmaceutics15112547