
Liposomal Amphotericin B for Treatment of Leishmaniasis: From the Identification of Critical Physicochemical Attributes to the Design of Effective Topical and Oral Formulations

 ,
,  , , and
, , and
Abstract

1. Introduction
2. Physicochemical Characteristics, Mechanisms of Action and Toxicity of AmB
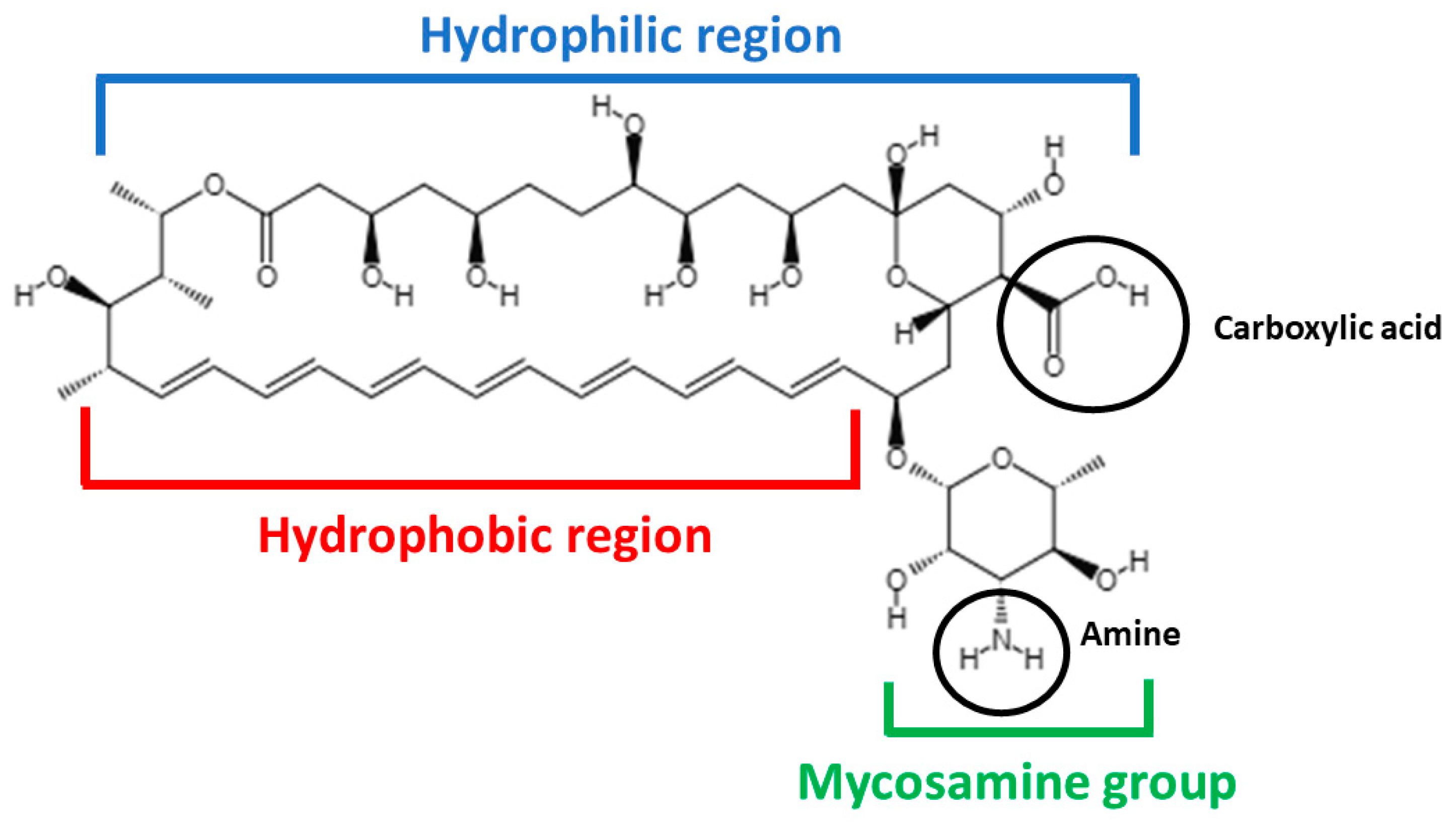
2.1. AmB Physicochemical Characteristics
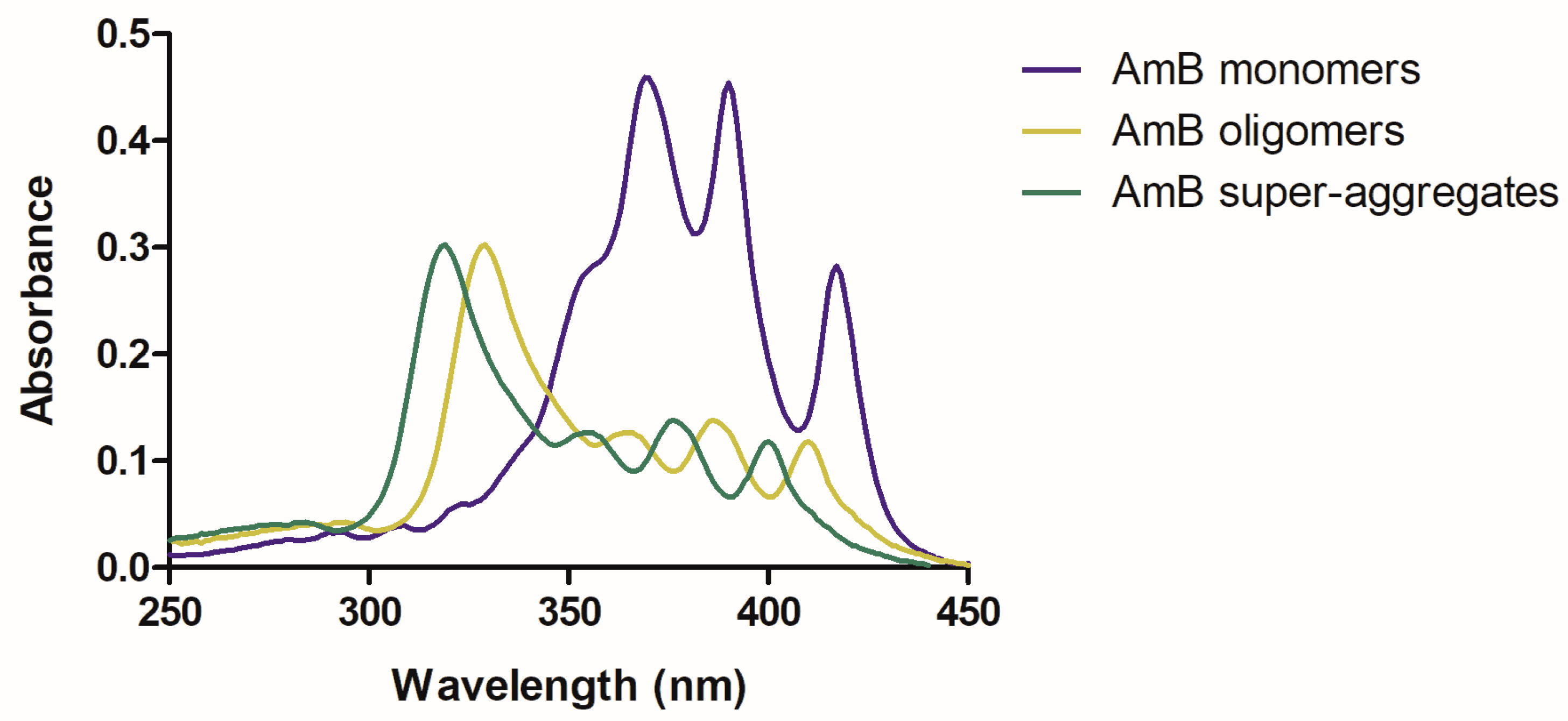
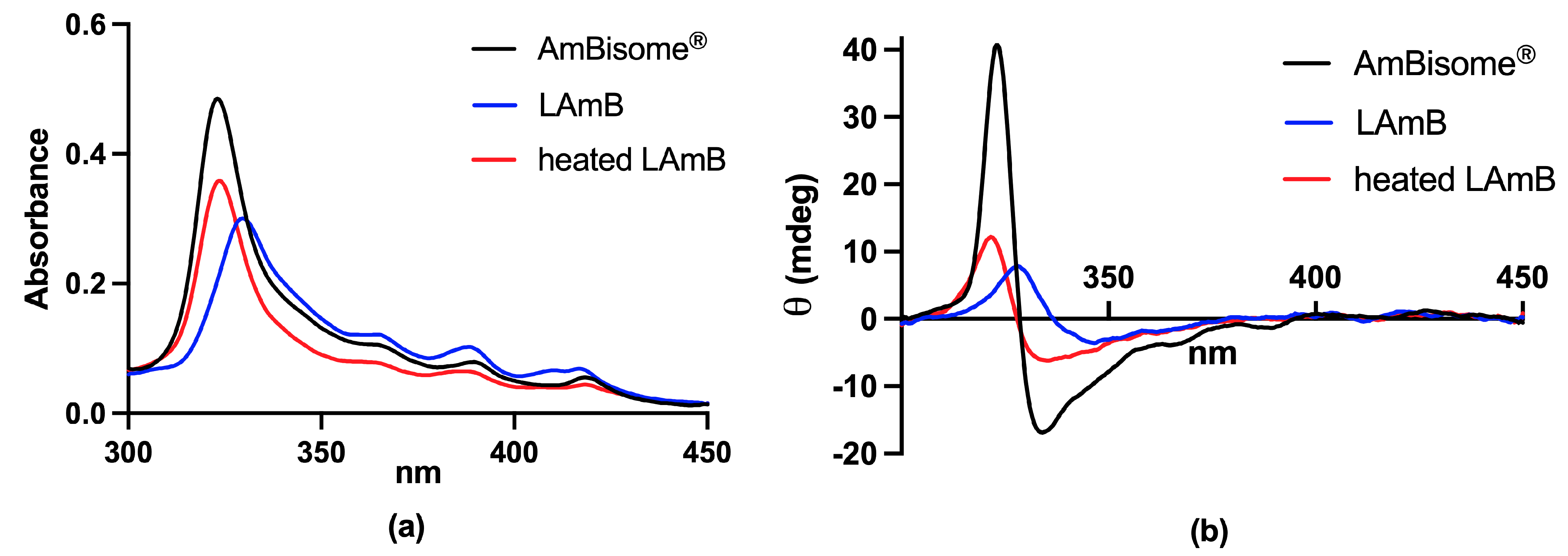
2.1.1. UV/Vis Electronic Absorption
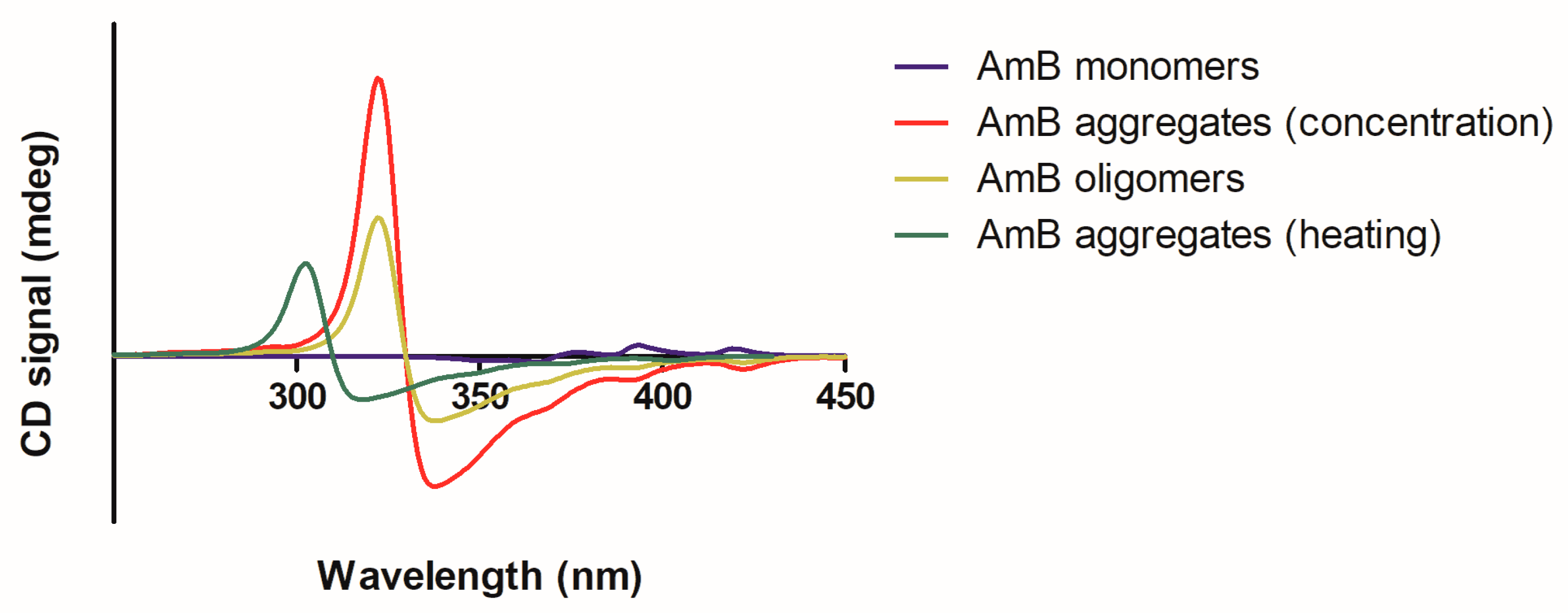
2.1.2. Circular Dichroism
2.1.3. Fluorescence Techniques
2.1.4. Dynamic Light Scattering for Particle Size Analysis
2.1.5. DSC and PXRD for Crystallinity Analysis
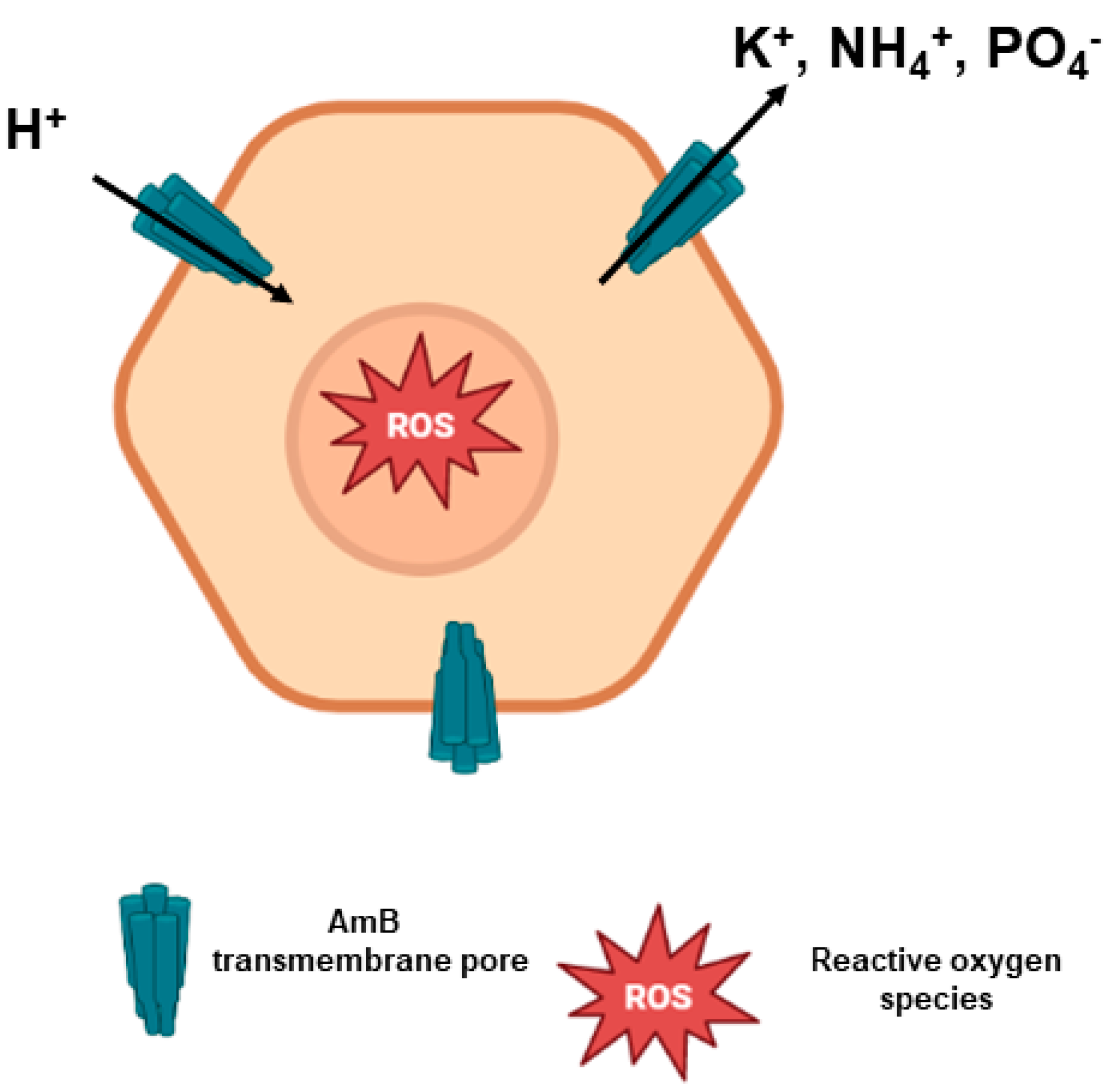
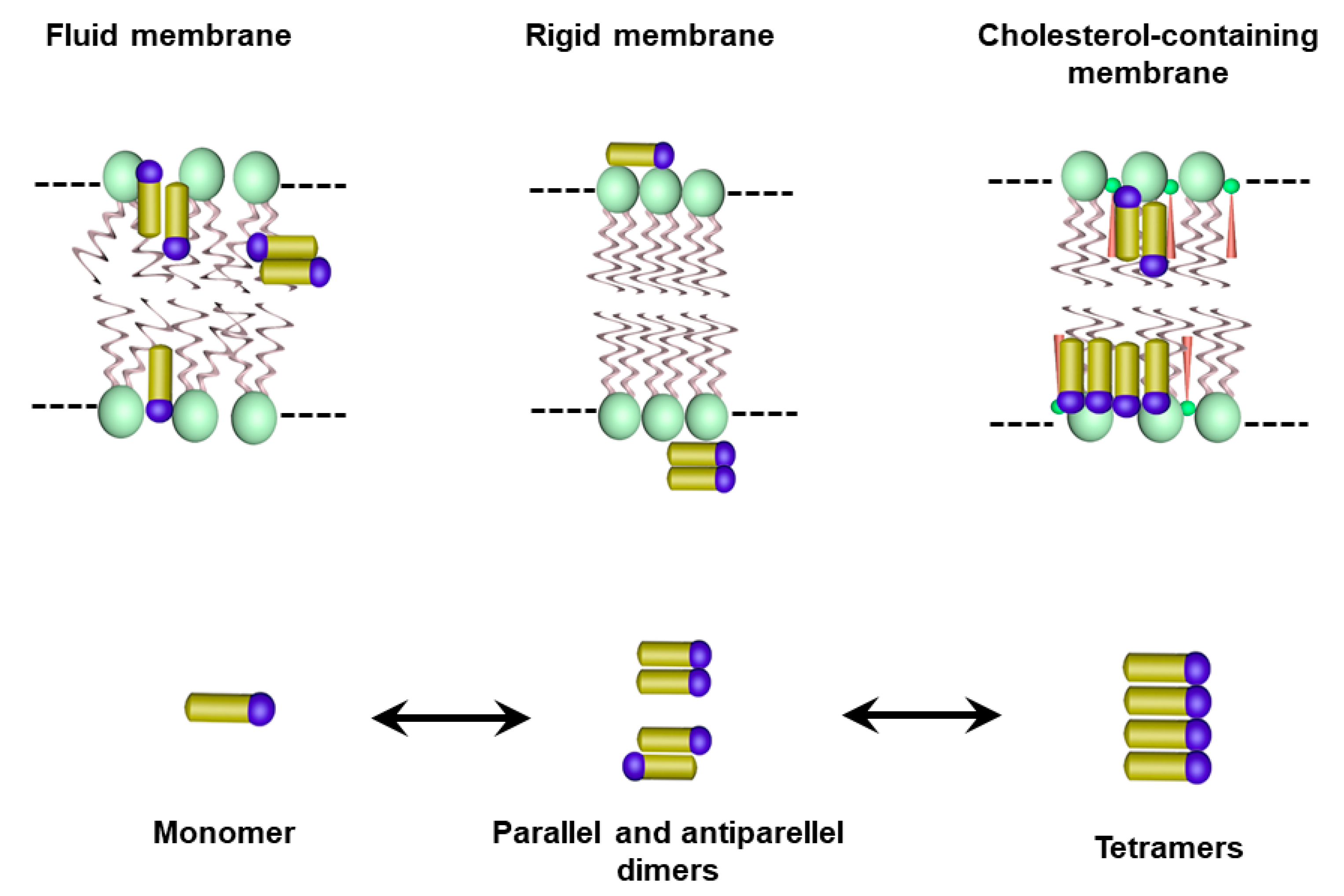
2.2. AmB Mechanism of Action
2.3. AmB Mechanism of Toxicity
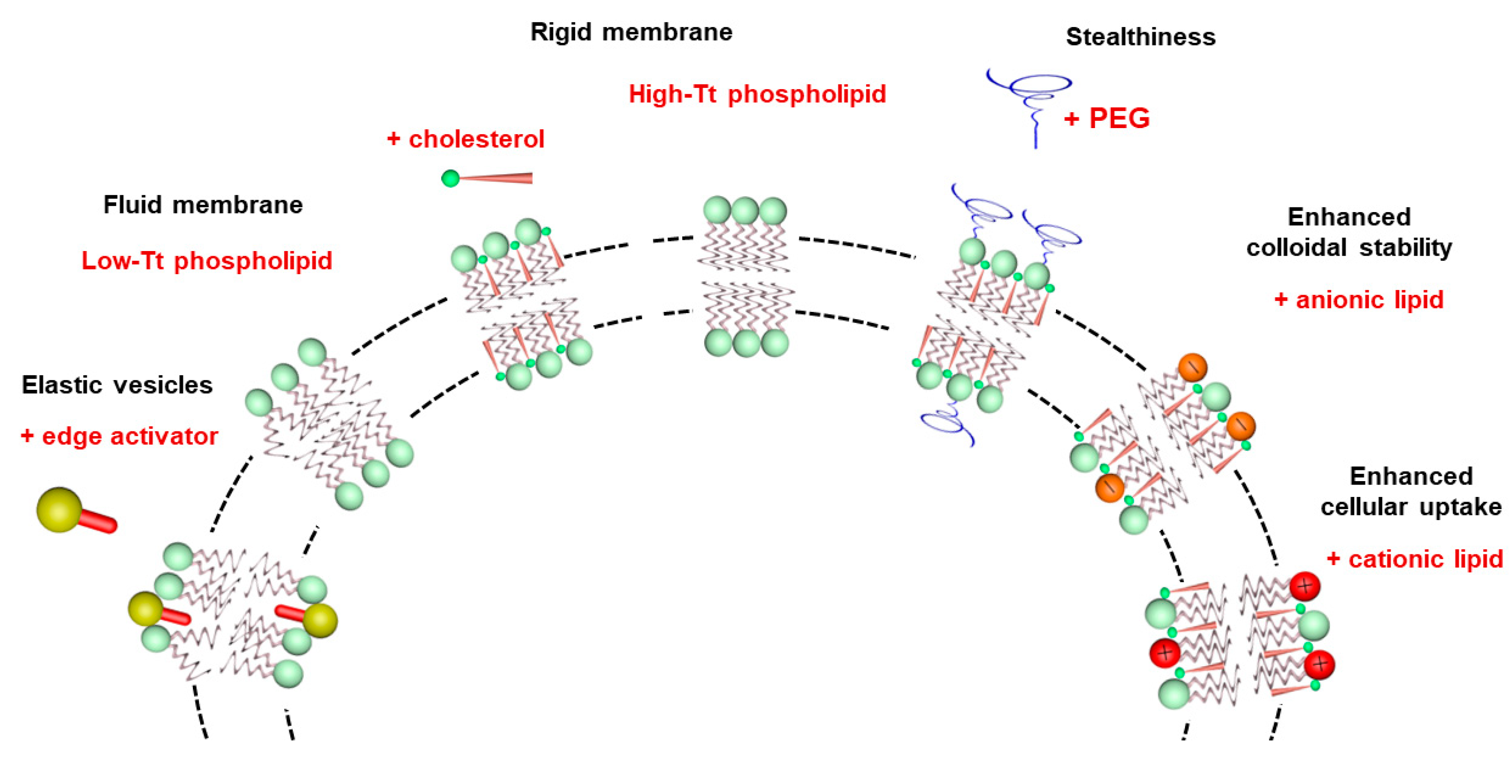
3. Basic Characteristics of Liposomes
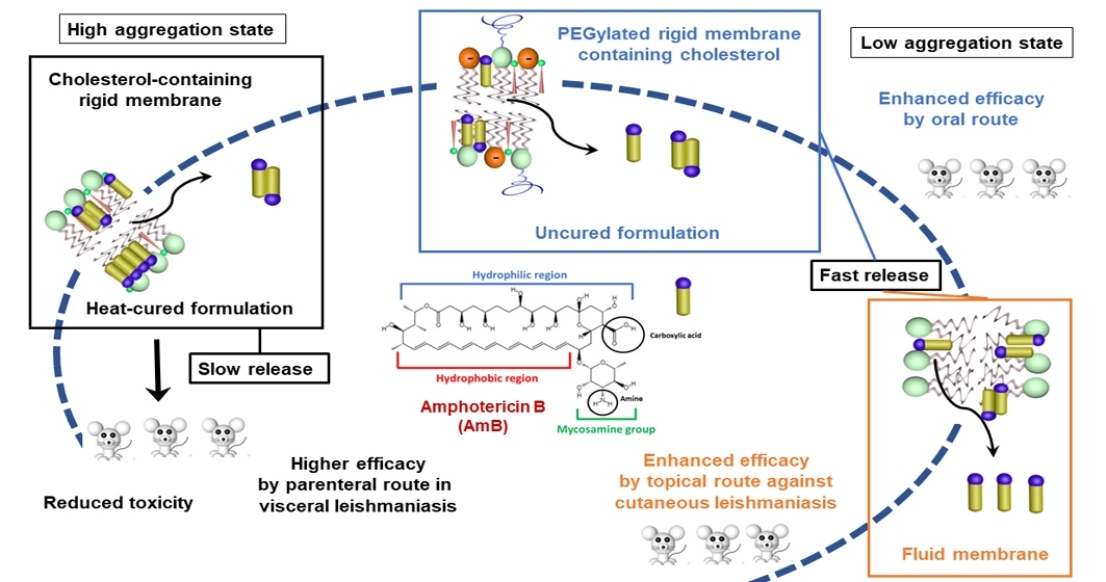
4. Reduced Toxicity of Liposomal AmB Formulations: Role of the Aggregation State of AmB and the Rate of Drug Release
5. Injectable Liposomal AmB Formulations: Factors Influencing the Antileishmanial Efficacy
5.1. Influence of the Lipid Composition
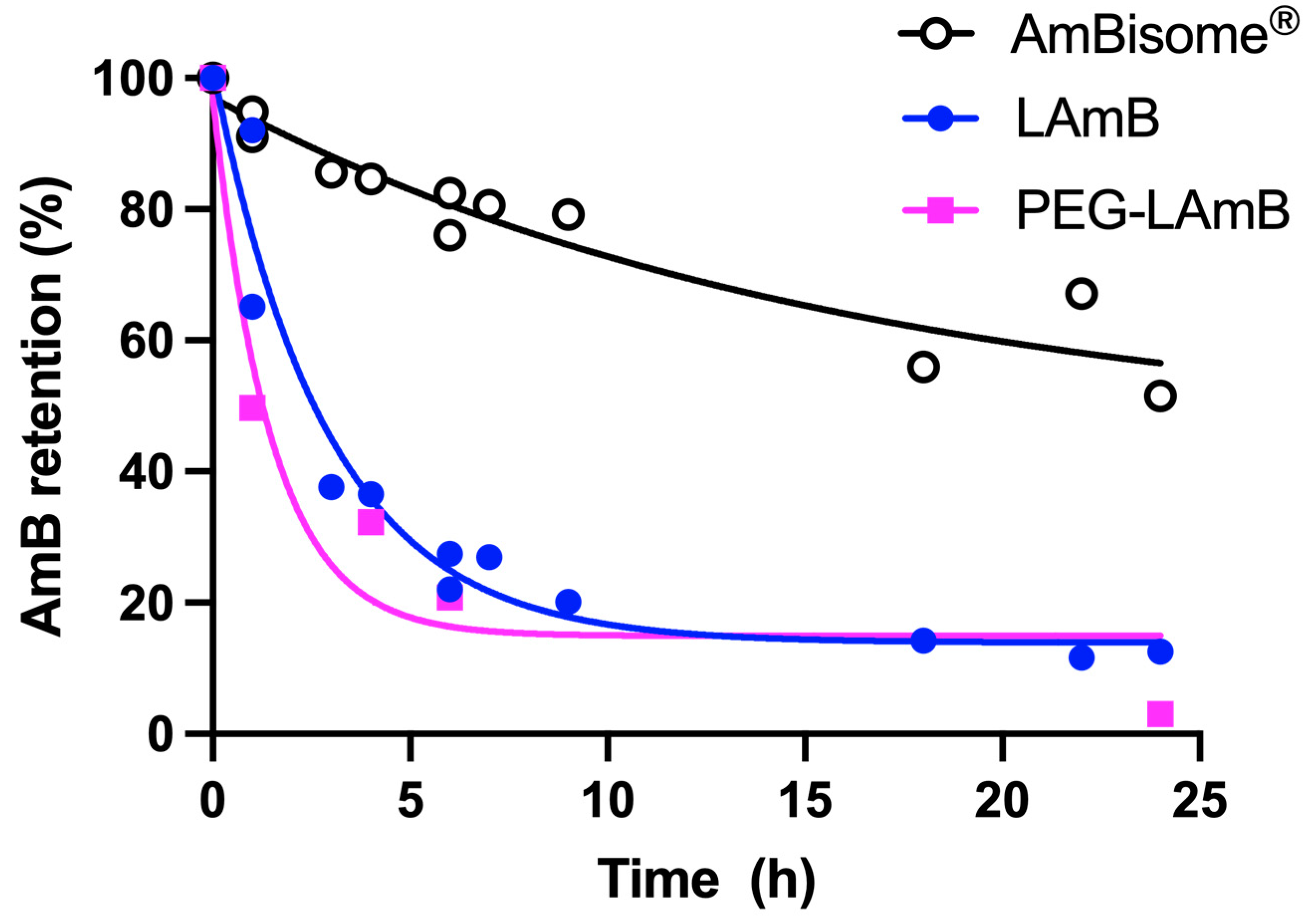
5.2. Influence of Liposome Size and PEGylation for CL
6. Topical Liposomal Formulations of AmB
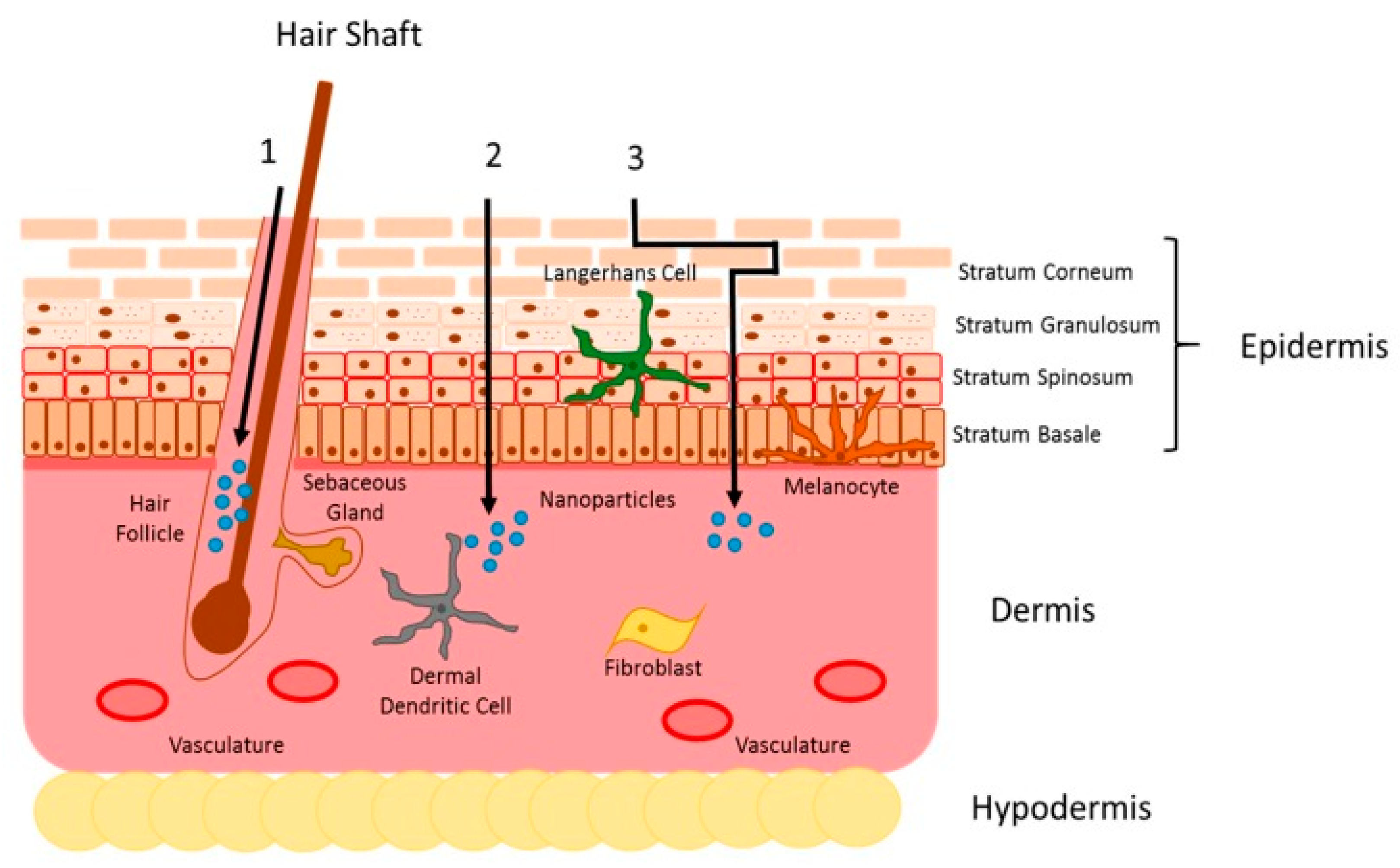
6.1. Topical Delivery

6.2. AmB Delivery: Liposomes for Topical Management of CL

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Composition | Permeation Studies Outcomes | Animal Model (Dose, Regimen) | In Vivo Outcomes | Reference |
|---|---|---|---|---|
| AmB; Soy phosphatidylcholine and Tween-80 | Higher AmB penetration in SC and in viable epidermis compared to AmBisome® after topical application in intact human skin. | NA * | NA * | [104] |
| AmB; Soy phosphatidylcholine and sodium cholate | Deeper penetration of AmB and to a larger extent compared to conventional liposomes, after topical application in intact human skin (Franz diffusion cell). | NA * | NA * | [88] |
| AmB; Soy phosphatidylcholine and Tween-80 | Increased drug retention in viable epidermis compared with free AmB, after topical application in intact pig skin (Franz diffusion cell). | NA * | NA * | [105] |
| AmB deoxycholate and meglumine antimoniate (Glucantime®); Span 40; Tween 40; cholesterol; Carbopol® 934 and triethanolamine | NA | BALB/c mice (twice daily for 30 days) | Significant reduction in lesion size after topical treatment with niosomes co-encapsulating AmB and Glucantime® compared to placebo gel (p < 0.001) and intramuscular Glucantime® in L. major-infected mice. Complete lesion healing not observed. | [95] |
| AmB and miltefosine; Phospholipon 90G; Tween-80; Carbopol® 934 and triethanolamine | 6-fold greater AmB permeation of AmB, compared to AmB simple gel applied topically in intact mouse skin (Franz diffusion cell). | BALB/c mice (AmB 1.5 mg/kg/day twice daily for 4 weeks) | Complete lesion resolution with no signs of scaring in L. mexicana-infected mice after topical treatment with co-loaded AmB-miltefosine deformable liposomes. Significant reduction in parasite load at lesion site compared to placebo gel control, AmB gel or single AmB in deformable liposomes. | [96] |
| AmB; sodium deoxycholate; Soy phosphatidylcholine; ethanol and mannitol | Enhanced permeation across intact mouse skin, compared to previously described liposomal formulations. The in vivo skin pharmacokinetic showed permeability and accumulation within the dermis at therapeutic concentrations for CL treatment. | BALB/c mice (0.5 mg/mL, 20 mg of formulation/day, once a day for 10 consecutive days) | Significant reduction in lesion size compared to the control group (untreated) and almost complete reduction in parasite load at lesion site, after topical treatment in L. amazonensis-infected mice. | [97] |
| AmB; Soy phosphatidylcholine; Cholesterol; Dimethyl sulfoxide; Propylene glycol; Oleic acid; Vitamin E; Methylparaben and Propylparaben | Greater amount of permeated AmB through skin from AmB-liposome (0.4%), compared formulation with lower AmB concentration in permeation study after topical application on intact BALB/c mouse skin (Franz diffusion cell). | BALB/c mice (50 mg liposomal AmB 0.4%, twice a day, for four weeks) | Higher efficacy of liposomal AmB formulation (0.4%) after topical treatment in L. major-infected mice, based on the significant reduction (p < 0.001) in lesion size and almost complete elimination of parasite load in skin and spleen compared to control groups (PBS or empty liposomes). | [101,103] |
7. Oral Liposomal Formulations of AmB
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- PAHO. Pan American Health Organization: Leishmaniasis. Available online: https://www.paho.org/en/topics/leishmaniasis (accessed on 11 October 2022).
- World Health Organization (WHO). Ending the Neglect to Attain the Sustainable Development Goals: A Road Map for Neglected Tropical Diseases 2021−2030; WHO: Geneva, Switzerland, 2020. [Google Scholar]
- Roatt, B.M.; de Oliveira Cardoso, J.M.; De Brito, R.C.F.; Coura-Vital, W.; de Oliveira Aguiar-Soares, R.D.; Reis, A.B. Recent advances and new strategies on leishmaniasis treatment. Appl. Microbiol. Biotechnol. 2020, 104, 8965–8977. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization (WHO). WHO Guideline for the Treatment of Visceral Leishmaniasis in HIV Co-Infected Patients in East Africa and South-East Asia; World Health Organization: Geneva, Switzerland, 2022. [Google Scholar]
- Burza, S.; Croft, S.L.; Boelaert, M. Leishmaniasis. Lancet 2018, 392, 951–970. [Google Scholar] [CrossRef] [PubMed]
- Berman, J. Liposomal Amphotericin B Treatment and the Leishmaniases. Am. J. Trop. Med. Hyg. 2019, 101, 727–728. [Google Scholar] [CrossRef]
- PAHO. Pan American Health Organization. Guideline for the Treatment of Leishmaniasis in the Americas, 2nd ed.; PAHO: Washington, DC, USA, 2022. [Google Scholar] [CrossRef]
- Frézard, F.; Demicheli, C.; Ribeiro, R.R. Pentavalent antimonials: New perspectives for old drugs. Molecules 2009, 14, 2317–2336. [Google Scholar] [CrossRef]
- Lanza, J.S.; Pomel, S.; Loiseau, P.M.; Frézard, F. Recent advances in amphotericin B delivery strategies for the treatment of leishmaniases. Expert Opin. Drug Deliv. 2019, 16, 1063–1079. [Google Scholar] [CrossRef] [PubMed]
- Novais, F.O.; Amorim, C.F.; Scott, P. Host-Directed Therapies for Cutaneous Leishmaniasis. Front. Immunol. 2021, 12, 660183. [Google Scholar] [CrossRef]
- Sundar, S.; Chakravarty, J.; Rai, V.K.; Agrawal, N.; Singh, S.P.; Chauhan, V.; Murray, H.W. Amphotericin B Treatment for Indian Visceral Leishmaniasis: Response to 15 Daily versus Alternate-Day Infusions. Clin. Infect. Dis. 2007, 45, 556–561. [Google Scholar] [CrossRef]
- Hamill, R.J. Amphotericin B Formulations: A Comparative Review of Efficacy and Toxicity. Drugs 2013, 73, 919–934. [Google Scholar] [CrossRef]
- Shah, A.; Gupta, S.S. Anti-leishmanial Nanotherapeutics: A Current Perspective. Curr. Drug Metab. 2019, 20, 473–482. [Google Scholar] [CrossRef]
- Jafari, M.; Abolmaali, S.S.; Tamaddon, A.M.; Zomorodian, K.; (Sarkari), B.S. Nanotechnology approaches for delivery and targeting of Amphotericin B in fungal and parasitic diseases. Nanomedicine 2021, 16, 857–877. [Google Scholar] [CrossRef]
- Yardley, V.; Croft, S.L. A comparison of the activities of three amphotericin B lipid formulations against experimental visceral and cutaneous leishmaniasis. Int. J. Antimicrob. Agents 2000, 13, 243–248. [Google Scholar] [CrossRef] [PubMed]
- Falci, D.R.; da Rosa, F.B.; Pasqualotto, A.C. Comparison of nephrotoxicity associated to different lipid formulations of amphotericin B: A real-life study. Mycoses 2015, 58, 104–112. [Google Scholar] [CrossRef] [PubMed]
- Adler-Moore, J.P.; Gangneux, J.-P.; Pappas, P.G. Comparison between liposomal formulations of amphotericin B. Med. Mycol. 2016, 54, 223–231. [Google Scholar] [CrossRef] [PubMed]
- Faustino, P. Lipid Systems for the Delivery of Amphotericin B in Antifungal Therapy. Pharmaceutics 2020, 12, 29. [Google Scholar] [CrossRef]
- Kumari, S.; Kumar, V.; Tiwari, R.K.; Ravidas, V.; Pandey, K.; Kumar, A. Amphotericin B: A drug of choice for Visceral Leishmaniasis. Acta Trop. 2022, 235, 106661. [Google Scholar] [CrossRef]
- Machado, P.R.L.; Rosa, M.E.A.; Guimarães, L.H.; Prates, F.V.O.; Queiroz, A.; Schriefer, A.; Carvalho, E.M. Treatment of disseminated leishmaniasis with liposomal amphotericin B. Clin. Infect. Dis. 2015, 61, 945–949. [Google Scholar] [CrossRef]
- Guery, R.; Henry, B.; Martin-Blondel, G.; Rouzaud, C.; Cordoliani, F.; Harms, G.; Gangneux, J.-P.; Foulet, F.; Bourrat, E.; Baccard, M.; et al. Liposomal amphotericin B in travelers with cutaneous and muco-cutaneous leishmaniasis: Not a panacea. PLoS Negl. Trop. Dis. 2017, 11, e0006094. [Google Scholar] [CrossRef]
- Senchyna, A.; Simon, S.; Cissé, H.; Ginouves, M.; Prevot, G.; Alcoba, G.; Demar, M.; Couppie, P.; Blaizot, R. American cutaneous leishmaniasis in French Guiana: A retrospective comparison between liposomal amphotericin B and meglumine antimoniate. Br. J. Dermatol. 2020, 183, 389–391. [Google Scholar] [CrossRef]
- Cavassin, F.B.; Baú-Carneiro, J.L.; Vilas-Boas, R.R.; Queiroz-Telles, F. Sixty years of Amphotericin B: An overview of the main antifungal agent used to treat invasive fungal infections. Infect. Dis. Ther. 2021, 10, 115–147. [Google Scholar] [CrossRef]
- Wang, Y.-H.; Zhang, J.-P.; Chang, Y.; Hu, C.-Q. A newly identified derivative of amphotericin B: Isolation, structure determination and primary evaluation of the activity and toxicity. J. Antibiot. 2010, 63, 553–557. [Google Scholar] [CrossRef]
- Kupetz, E.; Bunjes, H. Lipid nanoparticles: Drug localization is substance-specific and achievable load depends on the size and physical state of the particles. J. Control. Release 2014, 189, 54–64. [Google Scholar] [CrossRef] [PubMed]
- Waugh, C.D. Amphotericin B. In xPharm: The Comprehensive Pharmacology Reference; Enna, S.J., Bylund, D.B., Eds.; Elsevier: Amsterdam, The Netherlands, 2008; pp. 1–5. [Google Scholar] [CrossRef]
- Pham, T.T.H.; Barratt, G.; Michel, J.P.; Loiseau, P.M.; Saint-Pierre-Chazalet, M. Interactions of antileishmanial drugs with monolayers of lipids used in the development of amphotericin B–miltefosine-loaded nanocochleates. Colloids Surf. B Biointerfaces 2013, 106, 224–233. [Google Scholar] [CrossRef] [PubMed]
- Fernández-García, R.; Muñoz-García, J.C.; Wallace, M.; Fabian, L.; González-Burgos, E.; Gómez-Serranillos, M.P.; Raposo, R.; Bolás-Fernández, F.; Ballesteros, M.P.; Healy, A.M.; et al. Self-assembling, supramolecular chemistry and pharmacology of amphotericin B: Poly-aggregates, oligomers and monomers. J. Control. Release 2022, 341, 716–732. [Google Scholar] [CrossRef] [PubMed]
- Caldeira, L.R.; Fernandes, F.R.; Costa, D.F.; Frézard, F.; Afonso, L.C.C.; Ferreira, L.A.M. Nanoemulsions loaded with amphotericin B: A new approach for the treatment of leishmaniasis. Eur. J. Pharm. Sci. 2015, 70, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Gaboriau, F.; Chéron, M.; Leroy, L.; Bolard, J. Physico-chemical properties of the heat-induced ‘superaggregates’ of amphotericin B. Biophys. Chem. 1997, 66, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Belkherroubi-Sari, L.; Adida, H.; Seghir, A.; Boucherit, Z.; Boucherit, K. New strategy for enhancing the therapeutic index of Fungizone®. J. Mycol. Med. 2013, 23, 3–7. [Google Scholar] [CrossRef]
- Yao, H.; Wynendaele, E.; Xu, X.; Kosgei, A.; De Spiegeleer, B. Circular dichroism in functional quality evaluation of medicines. J. Pharm. Biomed. Anal. 2018, 147, 50–64. [Google Scholar] [CrossRef]
- Legrand, P.; Romero, E.A.; Cohen, B.E.; Bolard, J. Effects of aggregation and solvent on the toxicity of amphotericin B to human erythrocytes. Antimicrob. Agents Chemother. 1992, 36, 2518–2522. [Google Scholar] [CrossRef]
- Mazerski, J.; Bolard, J.; Borowski, E. Self-association of some polyene macrolide antibiotics in aqueous media. Biochim. Biophys. Acta 1982, 719, 11–17. [Google Scholar] [CrossRef]
- Rinnert, H.; Thirion, C.; Dupont, G.; Lematre, J. Structural studies on aqueous and hydroalcoholic solutions of a polyene antibiotic: Amphotericin B. Biopolymers 1977, 16, 2419–2427. [Google Scholar] [CrossRef]
- Gruszecki, W.I.; Luchowski, R.; Wasko, P.; Gryczynski, Z.; Gryczynski, I. Molecular organization of polyene antibiotic amphotericin B studied by means of fluorescence technique. Methods Mol. Biol. 2012, 875, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Wasko, P.; Luchowski, R.; Tutaj, K.; Grudzinski, W.; Adamkiewicz, P.; Gruszecki, W.I. Toward understanding of toxic side effects of a polyene antibiotic amphotericin B: Fluorescence spectroscopy reveals widespread formation of the specific supramolecular structures of the drug. Mol. Pharm. 2012, 9, 1511–1520. [Google Scholar] [CrossRef] [PubMed]
- Starzyk, J.; Gruszecki, M.; Tutaj, K.; Luchowski, R.; Szlazak, R.; Wasko, P.; Grudzinski, W.; Czub, J.; Gruszecki, W.I. Self-association of amphotericin B: Spontaneous formation of molecular structures responsible for the toxic side effects of the antibiotic. J. Phys. Chem. B 2014, 118, 13821–13832. [Google Scholar] [CrossRef] [PubMed]
- van Etten, E.W.M.; van Vianen, W.; Roovers, P.; Frederik, P. Mild heating of amphotericin B-desoxycholate: Effects on ultrastructure, in vitro activity and toxicity, and therapeutic efficacy in severe candidiasis in leukopenic mice. Antimicrob. Agents Chemother. 2000, 44, 1598–1603. [Google Scholar] [CrossRef][Green Version]
- Cuddihy, G.; Wasan, E.; Di, Y.; Wasan, K. The Development of oral amphotericin B to treat systemic fungal and parasitic infections: Has the myth been finally realized? Pharmaceutics 2019, 11, 99. [Google Scholar] [CrossRef]
- Zielińska, J.; Wieczór, M.; Bączek, T.; Gruszecki, M.; Czub, J. Thermodynamics and kinetics of amphotericin B self-association in aqueous solution characterized in molecular detail. Sci. Rep. 2016, 6, 19109. [Google Scholar] [CrossRef]
- Kamiński, D.M. Recent progress in the study of the interactions of amphotericin B with cholesterol and ergosterol in lipid environments. Eur. Biophys. J. 2014, 43, 453–467. [Google Scholar] [CrossRef]
- Carolus, H.; Pierson, S.; Lagrou, K.; Van Dijck, P. Amphotericin B and other polyenes—Discovery, clinical use, mode of action and drug resistance. J. Fungi 2020, 6, 321. [Google Scholar] [CrossRef]
- Vriens, K.; Kumar, P.T.; Struyfs, C.; Cools, T.L.; Spincemaille, P.; Kokalj, T.; Sampaio-Marques, B.; Ludovico, P.; Lammertyn, J.; Cammue, B.P.A.; et al. Increasing the fungicidal action of amphotericin B by inhibiting the nitric oxide-dependent tolerance pathway. Oxid. Med. Cell Longev. 2017, 2017, 4064628. [Google Scholar] [CrossRef]
- Mesa-Arango, A.C.; Trevijano-Contador, N.; Román, E.; Sánchez-Fresneda, R.; Casas, C.; Herrero, E.; Argüelles, J.C.; Pla, J.; Cuenca-Estrella, M.; Zaragoza, O. The production of reactive oxygen species is a universal action mechanism of amphotericin B against pathogenic yeasts and contributes to the fungicidal effect of this drug. Antimicrob. Agents Chemother. 2014, 58, 6627–6638. [Google Scholar] [CrossRef]
- Gaboriau, F.; Chéron, M.; Petit, C.; Bolard, J. Heat-induced superaggregation of amphotericin B reduces its in vitro toxicity: A new way to improve its therapeutic index. Antimicrob. Agents Chemother. 1997, 41, 2345–2351. [Google Scholar] [CrossRef] [PubMed]
- Blanco, E.; Shen, H.; Ferrari, M. Principles of nanoparticle design for overcoming biological barriers to drug delivery. Nat. Biotechnol. 2015, 33, 941–951. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.Y.; Gao, J.; Shin, D.H.; Alvarez, C.; Zhong, W.; Kwon, G.S. Pharmacokinetics and renal toxicity of monomeric amphotericin B in rats after a multiple dose regimen. Pharm. Nanotechnol. 2016, 4, 16–23. [Google Scholar] [CrossRef][Green Version]
- Silva-Filho, M.A.; Siqueira, S.D.; Freire, L.B.; Araújo, I.B.; Holanda e Silva, K.G.; Medeiros Ada, C.; Araújo-Filho, I.; Oliveira, A.G.; Egito, E.S. How can micelle systems be rebuilt by a heating process? Int. J. Nanomed. 2012, 7, 141–150. [Google Scholar] [CrossRef]
- Petit, C.; Yardley, V.; Gaboriau, F.; Bolard, J.; Croft, S.L. Activity of a heat-induced reformulation of amphotericin B deoxycholate (Fungizone) against Leishmania donovani. Antimicrob. Agents Chemother. 1999, 43, 390–392. [Google Scholar] [CrossRef]
- Das, S.; Devarajan, P.V. Enhancing safety and efficacy by altering the toxic aggregated state of amphotericin B in lipidic nanoformulations. Mol. Pharm. 2020, 17, 2186–2195. [Google Scholar] [CrossRef] [PubMed]
- Bangham, A.D.; Horne, R.W. Negative staining of phospholipids and their structural modification by surface-active agents as observed in the electron microscope. J. Mol. Biol. 1964, 8, 660-IN10. [Google Scholar] [CrossRef]
- Senior, J.; Gregoriadis, G. Stability of small unilamellar liposomes in serum and clearance from the circulation: The effect of the phospholipid and cholesterol components. Life Sci. 1982, 30, 2123–2136. [Google Scholar] [CrossRef]
- Richards, M.H.; Gardner, C.R. Effects of bile salts on the structural integrity of liposomes. Biochim. Biophys. Acta 1978, 543, 508–522. [Google Scholar] [CrossRef]
- Woodle, M.C.; Lasic, D.D. Sterically stabilized liposomes. Biochim. Biophys. Acta 1992, 1113, 171–199. [Google Scholar] [CrossRef]
- Li, H. Polyethylene glycol-coated liposomes for oral delivery of recombinant human epidermal growth factor. Int. J. Pharm. 2003, 258, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Neupane, Y.R.; Mangla, B.; Shafi, S.; Kohli, K. PEGylated nanoliposomes potentiated oral combination therapy for effective cancer treatment. Curr. Drug Deliv. 2020, 17, 728–735. [Google Scholar] [CrossRef] [PubMed]
- He, H.; Lu, Y.; Qi, J.; Zhu, Q.; Chen, Z.; Wu, W. Adapting liposomes for oral drug delivery. Acta Pharm. Sin. B 2019, 9, 36–48. [Google Scholar] [CrossRef] [PubMed]
- Mezei, M.; Gulasekharam, V. Liposomes—A selective drug delivery system for the topical route of administration. Lotion dosage form. Life Sci. 1980, 26, 1473–1477. [Google Scholar] [CrossRef] [PubMed]
- Verma, D.D.; Verma, S.; Blume, G.; Fahr, A. Liposomes increase skin penetration of entrapped and non-entrapped hydrophilic substances into human skin: A skin penetration and confocal laser scanning microscopy study. Eur. J. Pharm. Biopharm. 2003, 55, 271–277. [Google Scholar] [CrossRef]
- Cevc, G.; Blume, G. Lipid vesicles penetrate into intact skin owing to the transdermal osmotic gradients and hydration force. Biochim. Biophys. Acta Biomembr. (BBA)-Biomembr. 1992, 1104, 226–232. [Google Scholar] [CrossRef]
- Cevc, G. Lipid vesicles and other colloids as drug carriers on the skin. Adv. Drug Deliv. Rev. 2004, 56, 675–711. [Google Scholar] [CrossRef]
- New, R.R.C.; Chance, M.L.; Heath, S. Antileishmanial activity of amphotericin and other antifungal agents entrapped in liposomes. J. Antimicrob. Chemother. 1981, 8, 371–381. [Google Scholar] [CrossRef]
- Szoka, F.C.; Milholland, D.; Barza, M. Effect of lipid composition and liposome size on toxicity and in vitro fungicidal activity of liposome-intercalated amphotericin B. Antimicrob. Agents Chemother. 1987, 31, 421–429. [Google Scholar] [CrossRef]
- Adler-moore, J.P.; Proffitt, R.T. Development, characterization, efficacy and mode of action of Ambisome, a unilamellar liposomal formulation of amphotericin B. J. Liposome Res. 1993, 3, 429–450. [Google Scholar] [CrossRef]
- Bolard, J.; Vertut-Croquin, A.; Cybulska, B.E.; Gary-Bobo, C.M. Transfer of the polyene antibiotic amphotericin B between single-walled vesicles of dipalmitoylphosphatidylcholine and egg-yolk phosphatidylcholine. Biochim. Biophys. Act-Biomembr. 1981, 647, 241–248. [Google Scholar] [CrossRef]
- Witzke, N.M.; Bittman, R. Dissociation kinetics and equilibrium binding properties of polyene antibiotic complexes with phosphatidylcholine/sterol vesicles. Biochemistry 1984, 23, 1668–1674. [Google Scholar] [CrossRef] [PubMed]
- Gagoś, M.; Koper, R.; Gruszecki, W.I. Spectrophotometric analysis of organisation of dipalmitoylphosphatidylcholine bilayers containing the polyene antibiotic amphotericin B. Biochim. Biophys. Acta-Biomembr. 2001, 1511, 90–98. [Google Scholar] [CrossRef]
- Gruszecki, W.I.; Gagoś, M.; Hereć, M. Dimers of polyene antibiotic amphotericin B detected by means of fluorescence spectroscopy: Molecular organization in solution and in lipid membranes. J. Photochem. Photobiol. B 2003, 69, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Grudzinski, W.; Sagan, J.; Welc, R.; Luchowski, R.; Gruszecki, W.I. Molecular organization, localization and orientation of antifungal antibiotic amphotericin B in a single lipid bilayer. Sci. Rep. 2016, 6, 32780. [Google Scholar] [CrossRef]
- Adler-Moore, J.; Proffitt, R.T. AmBisome: Liposomal formulation, structure, mechanism of action and pre-clinical experience. J. Antimicrob. Chemother. 2002, 49 (Suppl. S1), 21–30. [Google Scholar] [CrossRef]
- Rivnay, B.; Wakim, J.; Avery, K.; Petrochenko, P.; Myung, J.H.; Kozak, D.; Yoon, S.; Landrau, N.; Nivorozhkin, A. Critical process parameters in manufacturing of liposomal formulations of amphotericin B. Int. J. Pharm. 2019, 565, 447–457. [Google Scholar] [CrossRef]
- Proffitt, R.T.; Adler-Moore, J.; Chiang, S.-M. Amphotericin B Liposome Preparation. US Patent WO 59651561999A, 19 October 1990. [Google Scholar]
- Adler-Moore, J.; Gamble, R.C.; Proffitt, R.T. Treatment of Systemic Fungal Infections with Phospholipid Particles Encapsulating Polyene Antibiotics. US Patent WO US5874104A, 23 September 1991. [Google Scholar]
- Tang, J.; Srinivasan, S.; Yuan, W.; Ming, R.; Liu, Y.; Dai, Z.; Noble, C.O.; Hayes, M.E.; Zheng, N.; Jiang, W.; et al. Development of a flow-through USP 4 apparatus drug release assay for the evaluation of amphotericin B liposome. Eur. J. Pharm. Biopharm. 2019, 134, 107–116. [Google Scholar] [CrossRef]
- Liu, Y.; Mei, Z.; Mei, L.; Tang, J.; Yuan, W.; Srinivasan, S.; Ackermann, R.; Schwendeman, A.S. Analytical method development and comparability study for AmBisome® and generic amphotericin B liposomal products. Eur. J. Pharm. Biopharm. 2020, 157, 241–249. [Google Scholar] [CrossRef]
- Ramos, G.S.; Vallejos, V.M.R.; Borges, G.S.M.; Almeida, R.M.; Alves, I.M.; Aguiar, M.M.G.; Fernandes, C.; Guimarães, P.P.G.; Fujiwara, R.T.; Loiseau, P.M.; et al. Formulation of amphotericin B in PEGylated liposomes for improved treatment of cutaneous leishmaniasis by parenteral and oral routes. Pharmaceutics 2022, 14, 989. [Google Scholar] [CrossRef]
- Iman, M.; Huang, Z.; Szoka, F.C.; Jaafari, M.R. Characterization of the colloidal properties, in vitro antifungal activity, antileishmanial activity and toxicity in mice of a distigmasterylhemisuccinoyl-glycero-phosphocholine liposome-intercalated amphotericin B. Int. J. Pharm. 2011, 408, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Iman, M.; Huang, Z.; Alavizadeh, S.H.; Szoka, F.C.; Jaafari, M.R. Biodistribution and in vivo antileishmanial activity of 1,2-distigmasterylhemisuccinoyl-sn-glycero-3-phosphocholine liposome-intercalated amphotericin B. Antimicrob. Agents Chemother. 2017, 61, e02525-16. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, A.; De, M.; Ali, N. Complete cure of experimental visceral leishmaniasis with amphotericin B in stearylamine-bearing cationic liposomes involves down-regulation of IL-10 and favorable t cell responses. J. Immunol. 2008, 181, 1386–1398. [Google Scholar] [CrossRef] [PubMed]
- Wijnant, G.-J.; Van Bocxlaer, K.; Yardley, V.; Harris, A.; Alavijeh, M.; Silva-Pedrosa, R.; Antunes, S.; Mauricio, I.; Murdan, S.; Croft, S.L. Comparative efficacy, toxicity and biodistribution of the liposomal amphotericin B formulations Fungisome® and AmBisome® in murine cutaneous leishmaniasis. Int. J. Parasitol. Drugs Drug Resist. 2018, 8, 223–228. [Google Scholar] [CrossRef] [PubMed]
- Güngör, S.; Kahraman, E. Nanocarriers Mediated Cutaneous Drug Delivery. Eur. J. Pharm. Sci. 2020, 158, 105638. [Google Scholar] [CrossRef]
- N’Da, D. Prodrug strategies for enhancing the percutaneous absorption of drugs. Molecules 2014, 19, 20780–20807. [Google Scholar] [CrossRef]
- Tiwari, N.; Osorio Blanco, E.; Sonzogni, A.; Esporrín-Ubieto, D.; Wang, H.; Calderon, M. Nanocarriers for skin applications: Where do we stand? Angew. Chem. Int. Ed. Engl. 2021, 61, e202107960. [Google Scholar] [CrossRef]
- Cui, M.; Wiraja, C.; Chew, S.W.T.; Xu, C. Nanodelivery systems for topical management of skin disorders. Mol. Pharm. 2021, 18, 491–505. [Google Scholar] [CrossRef]
- Elmowafy, M. Skin penetration/permeation success determinants of nanocarriers: Pursuit of a perfect formulation. Colloids Surf. B Biointerfaces 2021, 203, 111748. [Google Scholar] [CrossRef]
- Richard, C.; Cassel, S.; Blanzat, M. Vesicular systems for dermal and transdermal drug delivery. RSC Adv. 2021, 11, 442–451. [Google Scholar] [CrossRef]
- Peralta, M.F.; Guzmán, M.L.; Pérez, A.P.; Apezteguia, G.A.; Fórmica, M.L.; Romero, E.L.; Olivera, M.E.; Carrer, D.C. Liposomes can both enhance or reduce drugs penetration through the skin. Sci. Rep. 2018, 8, 13253. [Google Scholar] [CrossRef]
- Wijnant, G.-J.; Van Bocxlaer, K.; Fortes Francisco, A.; Yardley, V.; Harris, A.; Alavijeh, M.; Murdan, S.; Croft, S.L. Local skin inflammation in cutaneous leishmaniasis as a source of variable pharmacokinetics and therapeutic efficacy of liposomal amphotericin B. Antimicrob. Agents Chemother. 2018, 62, e00631-18. [Google Scholar] [CrossRef] [PubMed]
- Nieva, C.A.B.; Cid, A.G.; Romero, A.I.; García-Bustos, M.F.; Villegas, M.; Bermúdez, J.M. An appraisal of the scientific current situation and new perspectives in the treatment of cutaneous leishmaniasis. Acta Trop. 2021, 221, 105988. [Google Scholar] [CrossRef] [PubMed]
- Carneiro, G.; Aguiar, M.G.; Fernandes, A.P.; Ferreira, L.A.M. Drug delivery systems for the topical treatment of cutaneous leishmaniasis. Expert Opin. Drug Deliv. 2012, 9, 1083–1097. [Google Scholar] [CrossRef] [PubMed]
- Palmer, B.; De Louise, L. Nanoparticle-enabled transdermal drug delivery systems for enhanced dose control and tissue targeting. Molecules 2016, 21, 1719. [Google Scholar] [CrossRef]
- Ghasemiyeh, P.; Mohammadi-Samani, S. Potential of Nanoparticles as permeation enhancers and targeted delivery options for skin: Advantages and disadvantages. Drug Des. Dev. Ther. 2020, 14, 3271–3289. [Google Scholar] [CrossRef]
- López, L.; Vélez, I.; Asela, C.; Cruz, C.; Alves, F.; Robledo, S.; Arana, B. A phase II study to evaluate the safety and efficacy of topical 3% amphotericin B cream (Anfoleish) for the treatment of uncomplicated cutaneous leishmaniasis in Colombia. PLoS Negl. Trop. Dis. 2018, 12, e0006653. [Google Scholar] [CrossRef]
- Mostafavi, M.; Sharifi, I.; Farajzadeh, S.; Khazaeli, P.; Sharifi, H.; Pourseyedi, E.; Kakooei, S.; Bamorovat, M.; Keyhani, A.; Parizi, M.H.; et al. Niosomal formulation of amphotericin B alone and in combination with glucantime: In vitro and in vivo leishmanicidal effects. Biomed. Pharmacother. 2019, 116, 108942. [Google Scholar] [CrossRef]
- Dar, M.J.; Khalid, S.; McElroy, C.A.; Satoskar, A.R.; Khan, G.M. Topical treatment of cutaneous leishmaniasis with novel amphotericin B-miltefosine co-incorporated second generation ultra-deformable liposomes. Int. J. Pharm. 2020, 573, 118900. [Google Scholar] [CrossRef]
- Fernández-García, R.; Statts, L.; de Jesus, J.A.; Dea-Ayuela, M.A.; Bautista, L.; Simão, R.; Bolás-Fernández, F.; Ballesteros, M.P.; Laurenti, M.D.; Passero, L.F.D.; et al. Ultradeformable lipid vesicles localize amphotericin B in the dermis for the treatment of infectious skin diseases. ACS Infect. Dis. 2020, 6, 2647–2660. [Google Scholar] [CrossRef]
- Riaz, A.; Hendricks, S.; Elbrink, K.; Guy, C.; Maes, L.; Ahmed, N.; Kiekens, F.; Khan, G.M. Preparation and characterization of nanostructured lipid carriers for improved topical drug delivery: Evaluation in cutaneous leishmaniasis and vaginal candidiasis animal models. AAPS PharmSciTech 2020, 21, 185. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, H.K.; Serrano, D.R.; Dea-Ayuela, M.A.; Bilbao-Ramos, P.E.; Bolás-Fernández, F.; Torrado, J.J.; Molero, G. New amphotericin B-gamma cyclodextrin formulation for topical use with synergistic activity against diverse fungal species and Leishmania spp. Int. J. Pharm. 2014, 473, 148–157. [Google Scholar] [CrossRef] [PubMed]
- Malli, S.; Pomel, S.; Ayadi, Y.; Deloménie, C.; Da Costa, A.; Loiseau, P.M.; Bouchemal, K. Topically applied chitosan-coated poly(isobutylcyanoacrylate) nanoparticles are active against cutaneous leishmaniasis by accelerating lesion healing and reducing the parasitic load. ACS Appl. Biol. Mater. 2019, 2, 2573–2586. [Google Scholar] [CrossRef]
- Jaafari, M.R.; Hatamipour, M.; Alavizadeh, S.H.; Abbasi, A.; Saberi, Z.; Rafati, S.; Taslimi, Y.; Mohammadi, A.M.; Khamesipour, A. Development of a topical liposomal formulation of amphotericin B for the treatment of cutaneous leishmaniasis. Int. J. Parasitol. Drugs Drug Resist. 2019, 11, 156–165. [Google Scholar] [CrossRef]
- Eskandari, S.E.; Firooz, A.; Nassiri-Kashani, M.; Jaafari, M.R.; Javadi, A.; Miramin-Mohammadi, A.; Valian-Keshavarz, H.; Khamesipour, A. Safety Evaluation of nano-liposomal formulation of amphotericin B (SinaAmpholeish) in animal model as a candidate for treatment of cutaneous leishmaniasis. J. Arthropod. Borne Dis. 2018, 12, 269–275. [Google Scholar] [CrossRef]
- Jaafari, M.R.; Khamesipour, A. Topical Liposomal Compositions for Delivering Hydrophobic Drugs and Methods Preparing Same. US Patent US20150147382A1, 19 September 2014. [Google Scholar]
- Perez, A.P.; Altube, M.J.; Schilrreff, P.; Apezteguia, G.; Celes, F.S.; Zacchino, S.; de Oliveira, C.I.; Romero, E.L.; Morilla, M.J. Topical amphotericin B in ultradeformable liposomes: Formulation, skin penetration study, antifungal and antileishmanial activity in vitro. Colloids Surf. B Biointerfaces 2016, 139, 190–198. [Google Scholar] [CrossRef]
- Carvalheiro, M.; Vieira, J.; Faria-Silva, C.; Marto, J.; Simões, S. Amphotericin B-loaded deformable lipid vesicles for topical treatment of cutaneous leishmaniasis skin lesions. Drug Deliv. Transl. Res. 2021, 11, 717–728. [Google Scholar] [CrossRef]
- Kiryakova, S.; Dencheva-Zarkova, M.; Genova, J. Effect of Amphotericin B antibiotic on the properties of model lipid membrane. J. Phys. Conf. Ser. 2014, 558, 012027. [Google Scholar] [CrossRef]
- Khamesipour, A.; Mohammadi, A.; Jaafari, M.; Eskandari, S.; Tasbihi, M.; Javadi, A.; Afshari, F.; Mortazavi, H.; Firooz, A. Pilot study of safety and efficacy of topical liposomal amphotericin B for cutaneous leishmaniasis caused by Leishmania major in Islamic Republic of Iran. East Mediterr. Health J. 2022, 28, 658–663. [Google Scholar] [CrossRef]
- Eskandari, S.E.; firooz, A.; Nassiri-Kashani, M.; Jaafari, M.R.; Javadi, A.; Miramin Mohammadi, A.; Khamesipour, A. safety evaluation of topical application of nano-liposomal form of amphotericin B (SinaAmpholeish) on healthy volunteers; phase I clinical trial. Iran. J. Parasitol. 2019, 14, 197–203. [Google Scholar] [CrossRef]
- Horev, A.; Sagi, O.; Zur, E.; Ben-Shimol, S. Topical liposomal amphotericin B gel treatment for cutaneous leishmaniasis caused by Leishmania major: A double-blind, randomized, placebo-controlled, pilot study. Int. J. Dermatol. 2022. [Google Scholar] [CrossRef]
- Liu, M.; Chen, M.; Yang, Z. Design of amphotericin B oral formulation for antifungal therapy. Drug Deliv. 2017, 24, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Wasan, E.; Mandava, T.; Crespo-Moran, P.; Nagy, A.; Wasan, K.M. Review of novel oral amphotericin B formulations for the treatment of parasitic infections. Pharmaceutics 2022, 14, 2316. [Google Scholar] [CrossRef] [PubMed]
- Wasan, K.M. Development of an oral amphotericin B formulation as an alternative approach to parenteral amphotericin B administration in the treatment of blood-borne fungal infections. Curr. Pharm. Des. 2020, 26, 1521–1523. [Google Scholar] [CrossRef] [PubMed]
- Italia, J.L.; Yahya, M.M.; Singh, D.; Ravi Kumar, M.N.V. Biodegradable nanoparticles improve oral bioavailability of amphotericin B and show reduced nephrotoxicity compared to intravenous Fungizone®. Pharm. Res. 2009, 26, 1324–1331. [Google Scholar] [CrossRef]
- Radwan, M.A.; AlQuadeib, B.T.; Šiller, L.; Wright, M.C.; Horrocks, B. Oral administration of amphotericin B nanoparticles: Antifungal activity, bioavailability and toxicity in rats. Drug Deliv. 2017, 24, 40–50. [Google Scholar] [CrossRef]
- Jain, S.; Valvi, P.U.; Swarnakar, N.K.; Thanki, K. Gelatin coated hybrid lipid nanoparticles for oral delivery of amphotericin B. Mol. Pharm. 2012, 9, 2542–2553. [Google Scholar] [CrossRef]
- Patel, P.A.; Patravale, V.B. AmbiOnp: Solid lipid nanoparticles of amphotericin B for oral administration. J. Biomed. Nanotechnol. 2011, 7, 632–639. [Google Scholar] [CrossRef]
- Tan, J.S.L.; Roberts, C.; Billa, N. Pharmacokinetics and tissue distribution of an orally administered mucoadhesive chitosan-coated amphotericin B-loaded nanostructured lipid carrier (NLC) in rats. J. Biomater. Sci. Polym. Ed. 2019, 31, 141–154. [Google Scholar] [CrossRef]
- Yang, Z.; Chen, M.; Yang, M.; Chen, J.; Fang, W.; Xu, P. Evaluating the potential of cubosomal nanoparticles for oral delivery of amphotericin B in treating fungal infection. Int. J. Nanomed. 2014, 9, 327–336. [Google Scholar] [CrossRef]
- Wasan, K.M.; Wasan, E.K.; Gershkovich, P.; Zhu, X.; Tidwell, R.R.; Werbovetz, K.A.; Clement, J.G.; Thornton, S.J. Highly effective oral amphotericin B formulation against murine visceral leishmaniasis. J. Infect. Dis. 2009, 200, 357–360. [Google Scholar] [CrossRef] [PubMed]
- Wasan, E.K.; Gershkovich, P.; Zhao, J.; Zhu, X.; Werbovetz, K.; Tidwell, R.R.; Clement, J.G.; Thornton, S.J.; Wasan, K.M. A novel tropically stable oral amphotericin B formulation (iCo-010) exhibits efficacy against visceral leishmaniasis in a murine model. PLoS Negl. Trop. Dis. 2010, 4, e913. [Google Scholar] [CrossRef] [PubMed]
- Perlin, D.S. Amphotericin B cochleates: A vehicle for oral delivery. Curr. Opin. Investig. Drugs 2004, 5, 198–201. [Google Scholar] [PubMed]
- Lipa-Castro, A.; Nicolas, V.; Angelova, A.; Mekhloufi, G.; Prost, B.; Chéron, M.; Faivre, V.; Barratt, G. Cochleate formulations of amphotericin B designed for oral administration using a naturally occurring phospholipid. Int. J. Pharm. 2021, 603, 120688. [Google Scholar] [CrossRef] [PubMed]
- Lipa Castro, A.; Pomel, S.; Cailleau, C.; Fournier, N.; Dennemont, I.; Loiseau, P.M.; Barratt, G. Pharmacokinetics, biodistribution, and activity of amphotericin B-loaded nanocochleates on the Leishmania donovani murine visceral leishmaniasis model. Int. J. Pharm. 2022, 624, 121985. [Google Scholar] [CrossRef]
- Skipper, C.P.; Atukunda, M.; Stadelman, A.; Engen, N.W.; Bangdiwala, A.S.; Hullsiek, K.H.; Abassi, M.; Rhein, J.; Nicol, M.R.; Laker, E.; et al. Phase I EnACT Trial of the safety and tolerability of a novel oral formulation of amphotericin B. Antimicrob. Agents Chemother. 2020, 64, e00838-20. [Google Scholar] [CrossRef]
- Hnik, P.; Wasan, E.K.; Wasan, K.M. Safety, tolerability, and pharmacokinetics of a novel oral amphotericin B formulation (ICo-019) following single-dose administration to healthy human subjects: An alternative approach to parenteral amphotericin B administration. Antimicrob. Agents Chemother. 2020, 64, e01450-20. [Google Scholar] [CrossRef]









| Parameter | Observed Therapeutic Effect | Proposed Mechanism | Reference |
|---|---|---|---|
| Model of visceral leishmaniasis | |||
| Membrane fluidity | Rigid lipo. > fluid lipo. | Slower drug release from rigid liposomes | [63] |
| Inclusion of cholesterol | Lipo. with cholesterol > Lipo. without cholesterol | Higher affinity of the drug for cholesterol-containing membrane | [63] |
| Inclusion of ergosterol | Lipo. with ergosterol > Lipo. with cholesterol | Higher affinity of the drug for ergosterol-containing membrane | [63] |
| Model of cutaneous leishmaniasis | |||
| Liposome size | Small-sized lipo. > large-sized lipo. | Extended blood circulation of smaller liposomes and higher accumulation in the lesion | [81] |
| PEGylation of liposomes | PEGylated lipo. > conventional lipo. | Extended blood circulation time of PEGylated liposomes and higher accumulation in the lesion | [77] |
| Inclusion of stearylamine (SA) | SA-containing lipo. > SA-free lipo. | Combined leishmanicidal and immunodulatory actions | [80] |
| Gap of Knowledge or Challenge |
|---|
| 1. Impact of DSPG on the supramolecular organization of AmB in liposomal formulation. |
| 2. Systematic study of the aggregation state of AmB in topical liposomal formulations. |
| 3. Mechanisms responsible for the improved oral efficacy of PEGylated liposomes. |
| 4. Improving the shelf-life stability of liposomal AmB formulations, regarding the effect of temperature. |
| 5. Developing more effective and safe strategies combining the same liposomal system AmB and an immunomodulator. |
| 6. Oral efficacy of PEGylated liposomal AmB in visceral leishmaniasis. |
| 7. Optimizing liposomal AmB for oral delivery and exploring alternative lipid compositions and coating strategies of liposomes. |
| 8. Translating experimental findings into human applications, scaling-up the production and overcoming regulatory issues and the “Neglected Tropical Disease” barrier. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Frézard, F.; Aguiar, M.M.G.; Ferreira, L.A.M.; Ramos, G.S.; Santos, T.T.; Borges, G.S.M.; Vallejos, V.M.R.; De Morais, H.L.O. Liposomal Amphotericin B for Treatment of Leishmaniasis: From the Identification of Critical Physicochemical Attributes to the Design of Effective Topical and Oral Formulations. Pharmaceutics 2023, 15, 99. https://doi.org/10.3390/pharmaceutics15010099
Frézard F, Aguiar MMG, Ferreira LAM, Ramos GS, Santos TT, Borges GSM, Vallejos VMR, De Morais HLO. Liposomal Amphotericin B for Treatment of Leishmaniasis: From the Identification of Critical Physicochemical Attributes to the Design of Effective Topical and Oral Formulations. Pharmaceutics. 2023; 15(1):99. https://doi.org/10.3390/pharmaceutics15010099
Chicago/Turabian StyleFrézard, Frédéric, Marta M. G. Aguiar, Lucas A. M. Ferreira, Guilherme S. Ramos, Thais T. Santos, Gabriel S. M. Borges, Virgínia M. R. Vallejos, and Helane L. O. De Morais. 2023. "Liposomal Amphotericin B for Treatment of Leishmaniasis: From the Identification of Critical Physicochemical Attributes to the Design of Effective Topical and Oral Formulations" Pharmaceutics 15, no. 1: 99. https://doi.org/10.3390/pharmaceutics15010099
APA StyleFrézard, F., Aguiar, M. M. G., Ferreira, L. A. M., Ramos, G. S., Santos, T. T., Borges, G. S. M., Vallejos, V. M. R., & De Morais, H. L. O. (2023). Liposomal Amphotericin B for Treatment of Leishmaniasis: From the Identification of Critical Physicochemical Attributes to the Design of Effective Topical and Oral Formulations. Pharmaceutics, 15(1), 99. https://doi.org/10.3390/pharmaceutics15010099