

Physiologic Functions and Therapeutic Applications of α7 Nicotinic Acetylcholine Receptor in Brain Disorders

Abstract
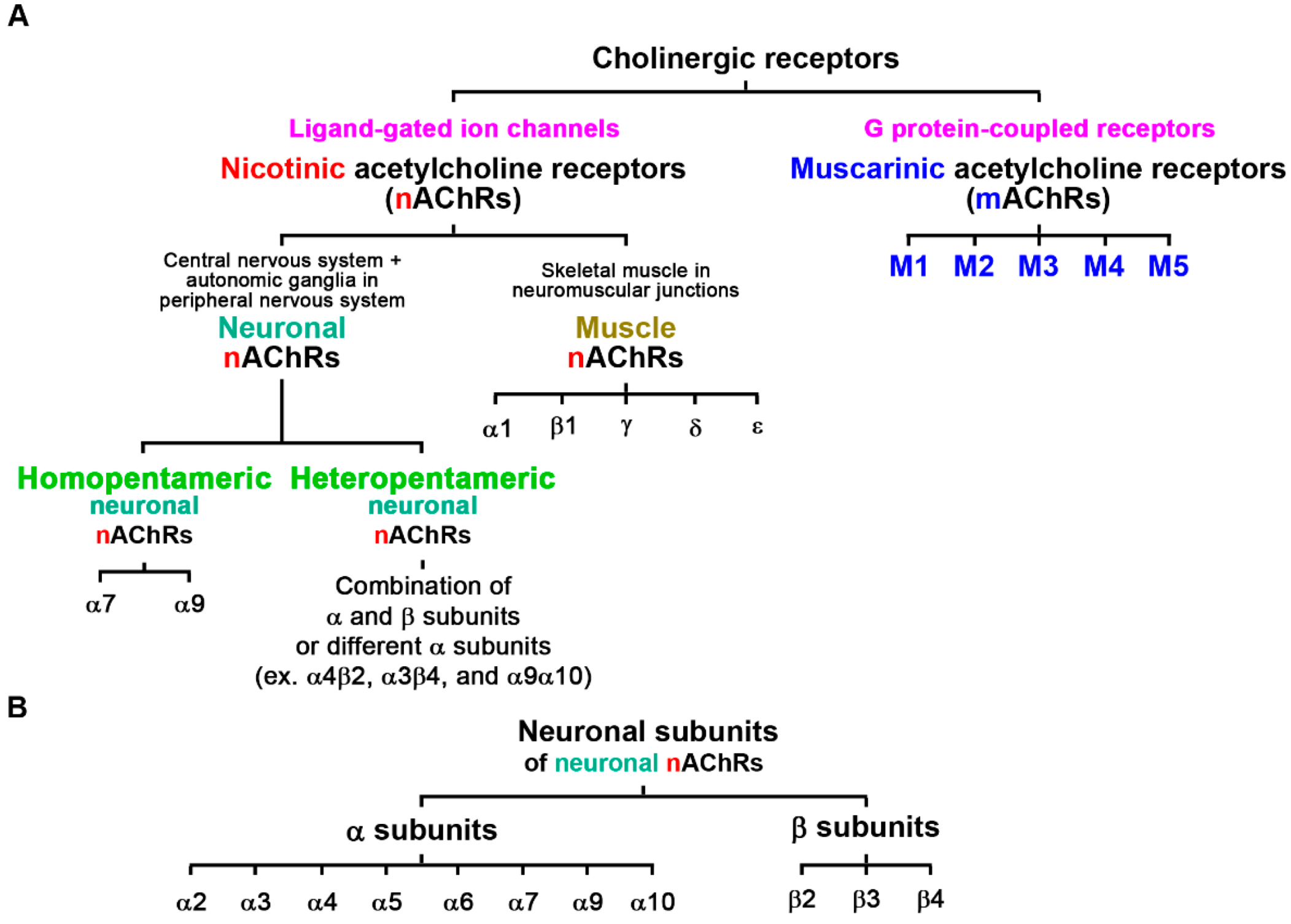
1. Introduction of Cholinergic Receptors and Nicotinic Acetylcholine Receptors (nAChRs)
2. α7 Nicotinic Acetylcholine Receptor (α7nAChR)
3. The CHRNA7 Gene
4. Transcriptional Factors, Promoter DNA Methylations, Tobacco Smoking, and Promoter Variants Regulate Human CHRNA7 mRNA Expression
5. Aberrant α7 Subunit Trafficking, Folding, and Assembly Reduces Cell-Surface Expression of Functional α7nACRs
6. Co-Assembly of the Dupα7 and α7 Subunits Impairs α7nAChR Functions
7. α7nAChR in AD and Therapeutic Applications
8. α7nAChR in PD and Therapeutic Applications
9. α7nAChR in Schizophrenia and Therapeutic Applications
10. α7nAChR in Depression and Therapeutic Applications
11. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Carlson, A.B.; Kraus, G.P. Physiology, Cholinergic Receptors; StatPearls: Treasure Island, FL, USA, 2022. [Google Scholar]
- Wess, J. Novel insights into muscarinic acetylcholine receptor function using gene targeting technology. Trends Pharmacol. Sci. 2003, 24, 414–420. [Google Scholar] [CrossRef] [PubMed]
- Le Novere, N.; Changeux, J.P. Molecular evolution of the nicotinic acetylcholine receptor: An example of multigene family in excitable cells. J. Mol. Evol. 1995, 40, 155–172. [Google Scholar] [CrossRef] [PubMed]
- Vijayaraghavan, S.; Pugh, P.C.; Zhang, Z.W.; Rathouz, M.M.; Berg, D.K. Nicotinic receptors that bind alpha-bungarotoxin on neurons raise intracellular free Ca2+. Neuron 1992, 8, 353–362. [Google Scholar] [CrossRef] [PubMed]
- Hogg, R.C.; Raggenbass, M.; Bertrand, D. Nicotinic acetylcholine receptors: From structure to brain function. In Reviews of Physiology, Biochemistry and Pharmacology; Springer: Berlin/Heidelberg, Germany, 2003; pp. 1–46. [Google Scholar] [CrossRef]
- Dani, J.A. Neuronal Nicotinic Acetylcholine Receptor Structure and Function and Response to Nicotine. Int. Rev. Neurobiol. 2015, 124, 3–19. [Google Scholar] [CrossRef] [PubMed]
- Toyohara, J.; Hashimoto, K. αlpha7 Nicotinic Receptor Agonists: Potential Therapeutic Drugs for Treatment of Cognitive Impairments in Schizophrenia and Alzheimer’s Disease. Open Med. Chem. J. 2010, 4, 37–56. [Google Scholar] [CrossRef][Green Version]
- Corradi, J.; Bouzat, C. Understanding the Bases of Function and Modulation of alpha7 Nicotinic Receptors: Implications for Drug Discovery. Mol. Pharmacol. 2016, 90, 288–299. [Google Scholar] [CrossRef]
- Quik, M.; Zhang, D.; McGregor, M.; Bordia, T. αlpha7 nicotinic receptors as therapeutic targets for Parkinson’s disease. Biochem. Pharmacol. 2015, 97, 399–407. [Google Scholar] [CrossRef]
- Wonnacott, S. αlpha-Bungarotoxin binds to low-affinity nicotine binding sites in rat brain. J. Neurochem. 1986, 47, 1706–1712. [Google Scholar] [CrossRef]
- Cooper, S.T.; Millar, N.S. Host cell-specific folding and assembly of the neuronal nicotinic acetylcholine receptor alpha7 subunit. J. Neurochem. 1997, 68, 2140–2151. [Google Scholar] [CrossRef]
- Broide, R.S.; Winzer-Serhan, U.H.; Chen, Y.; Leslie, F.M. Distribution of alpha7 Nicotinic Acetylcholine Receptor Subunit mRNA in the Developing Mouse. Front. Neuroanat. 2019, 13, 76. [Google Scholar] [CrossRef]
- Wang, H.; Yu, M.; Ochani, M.; Amella, C.A.; Tanovic, M.; Susarla, S.; Li, J.H.; Wang, H.; Yang, H.; Ulloa, L.; et al. Nicotinic acetylcholine receptor alpha7 subunit is an essential regulator of inflammation. Nature 2003, 421, 384–388. [Google Scholar] [CrossRef]
- Uteshev, V.V. α7 nicotinic ACh receptors as a ligand-gated source of Ca(2+) ions: The search for a Ca(2+) optimum. Adv. Exp. Med. Biol. 2012, 740, 603–638. [Google Scholar] [CrossRef]
- Gault, J.; Robinson, M.; Berger, R.; Drebing, C.; Logel, J.; Hopkins, J.; Moore, T.; Jacobs, S.; Meriwether, J.; Choi, M.J.; et al. Genomic organization and partial duplication of the human alpha7 neuronal nicotinic acetylcholine receptor gene (CHRNA7). Genomics 1998, 52, 173–185. [Google Scholar] [CrossRef]
- Araud, T.; Graw, S.; Berger, R.; Lee, M.; Neveu, E.; Bertrand, D.; Leonard, S. The chimeric gene CHRFAM7A, a partial duplication of the CHRNA7 gene, is a dominant negative regulator of alpha7*nAChR function. Biochem. Pharmacol. 2011, 82, 904–914. [Google Scholar] [CrossRef]
- Mexal, S.; Berger, R.; Logel, J.; Ross, R.G.; Freedman, R.; Leonard, S. Differential regulation of alpha7 nicotinic receptor gene (CHRNA7) expression in schizophrenic smokers. J. Mol. Neurosci. 2010, 40, 185–195. [Google Scholar] [CrossRef]
- Wang, Y.; Xiao, C.; Indersmitten, T.; Freedman, R.; Leonard, S.; Lester, H.A. The duplicated alpha7 subunits assemble and form functional nicotinic receptors with the full-length alpha7. J. Biol. Chem. 2014, 289, 26451–26463. [Google Scholar] [CrossRef]
- Fagerberg, L.; Hallstrom, B.M.; Oksvold, P.; Kampf, C.; Djureinovic, D.; Odeberg, J.; Habuka, M.; Tahmasebpoor, S.; Danielsson, A.; Edlund, K.; et al. Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol. Cell Proteom. 2014, 13, 397–406. [Google Scholar] [CrossRef]
- Bertrand, D.; Lee, C.H.; Flood, D.; Marger, F.; Donnelly-Roberts, D. Therapeutic Potential of alpha7 Nicotinic Acetylcholine Receptors. Pharmacol. Rev. 2015, 67, 1025–1073. [Google Scholar] [CrossRef]
- Finlay-Schultz, J.; Canastar, A.; Short, M.; El Gazzar, M.; Coughlan, C.; Leonard, S. Transcriptional repression of the alpha7 nicotinic acetylcholine receptor subunit gene (CHRNA7) by activating protein-2alpha (AP-2alpha). J. Biol. Chem. 2011, 286, 42123–42132. [Google Scholar] [CrossRef]
- Leonard, S.; Gault, J.; Hopkins, J.; Logel, J.; Vianzon, R.; Short, M.; Drebing, C.; Berger, R.; Venn, D.; Sirota, P.; et al. Association of promoter variants in the alpha7 nicotinic acetylcholine receptor subunit gene with an inhibitory deficit found in schizophrenia. Arch. Gen. Psychiatry 2002, 59, 1085–1096. [Google Scholar] [CrossRef]
- Valles, A.S.; Barrantes, F.J. Chaperoning alpha7 neuronal nicotinic acetylcholine receptors. Biochim. Biophys. Acta 2012, 1818, 718–729. [Google Scholar] [CrossRef]
- Williams, M.E.; Burton, B.; Urrutia, A.; Shcherbatko, A.; Chavez-Noriega, L.E.; Cohen, C.J.; Aiyar, J. Ric-3 promotes functional expression of the nicotinic acetylcholine receptor alpha7 subunit in mammalian cells. J. Biol. Chem. 2005, 280, 1257–1263. [Google Scholar] [CrossRef] [PubMed]
- Gu, S.; Matta, J.A.; Lord, B.; Harrington, A.W.; Sutton, S.W.; Davini, W.B.; Bredt, D.S. Brain alpha7 Nicotinic Acetylcholine Receptor Assembly Requires NACHO. Neuron 2016, 89, 948–955. [Google Scholar] [CrossRef] [PubMed]
- Miwa, J.M.; Ibanez-Tallon, I.; Crabtree, G.W.; Sanchez, R.; Sali, A.; Role, L.W.; Heintz, N. Lynx1, an endogenous toxin-like modulator of nicotinic acetylcholine receptors in the mammalian CNS. Neuron 1999, 23, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Ibanez-Tallon, I.; Miwa, J.M.; Wang, H.L.; Adams, N.C.; Crabtree, G.W.; Sine, S.M.; Heintz, N. Novel modulation of neuronal nicotinic acetylcholine receptors by association with the endogenous prototoxin lynx1. Neuron 2002, 33, 893–903. [Google Scholar] [CrossRef]
- Freedman, R.; Hall, M.; Adler, L.E.; Leonard, S. Evidence in postmortem brain tissue for decreased numbers of hippocampal nicotinic receptors in schizophrenia. Biol. Psychiatry 1995, 38, 22–33. [Google Scholar] [CrossRef]
- Peng, W.; Mao, L.; Dang, X. The emergence of the uniquely human alpha7 nicotinic acetylcholine receptor gene and its roles in inflammation. Gene 2022, 842, 146777. [Google Scholar] [CrossRef]
- Sinkus, M.L.; Graw, S.; Freedman, R.; Ross, R.G.; Lester, H.A.; Leonard, S. The human CHRNA7 and CHRFAM7A genes: A review of the genetics, regulation, and function. Neuropharmacology 2015, 96, 274–288. [Google Scholar] [CrossRef]
- Martín-Sánchez, C.; Alés, E.; Balseiro-Gómez, S.; Atienza, G.; Arnalich, F.; Bordas, A.; Cedillo, J.L.; Extremera, M.; Chavez-Reyes, A.; Montiel, C. The human-specific duplicated α7 gene inhibits the ancestral α7, negatively regulating nicotinic acetylcholine receptor-mediated transmitter release. J. Biol. Chem. 2021, 296, 100341. [Google Scholar] [CrossRef]
- Hung, S.-Y.; Fu, W.-M. Drug candidates in clinical trials for Alzheimer’s disease. J. Biomed. Sci. 2017, 24, 47. [Google Scholar] [CrossRef]
- Alzheimer’s Association. 2022 Alzheimer’s disease facts and figures. Alzheimers Dement. 2022, 18, 700–789. [Google Scholar] [CrossRef]
- Davies, P.; Maloney, A.J. Selective loss of central cholinergic neurons in Alzheimer’s disease. Lancet 1976, 2, 1403. [Google Scholar] [CrossRef]
- Davies, P.; Feisullin, S. Postmortem stability of alpha-bungarotoxin binding sites in mouse and human brain. Brain Res. 1981, 216, 449–454. [Google Scholar] [CrossRef]
- Hellstrom-Lindahl, E.; Mousavi, M.; Zhang, X.; Ravid, R.; Nordberg, A. Regional distribution of nicotinic receptor subunit mRNAs in human brain: Comparison between Alzheimer and normal brain. Brain Res. Mol. Brain Res. 1999, 66, 94–103. [Google Scholar] [CrossRef]
- Guan, Z.Z.; Zhang, X.; Ravid, R.; Nordberg, A. Decreased protein levels of nicotinic receptor subunits in the hippocampus and temporal cortex of patients with Alzheimer’s disease. J. Neurochem. 2000, 74, 237–243. [Google Scholar] [CrossRef]
- Qi, X.L.; Nordberg, A.; Xiu, J.; Guan, Z.Z. The consequences of reducing expression of the alpha7 nicotinic receptor by RNA interference and of stimulating its activity with an alpha7 agonist in SH-SY5Y cells indicate that this receptor plays a neuroprotective role in connection with the pathogenesis of Alzheimer’s disease. Neurochem. Int. 2007, 51, 377–383. [Google Scholar] [CrossRef]
- Kem, W.R. The brain alpha7 nicotinic receptor may be an important therapeutic target for the treatment of Alzheimer’s disease: Studies with DMXBA (GTS-21). Behav. Brain Res. 2000, 113, 169–181. [Google Scholar] [CrossRef]
- Nagele, R.G.; D’Andrea, M.R.; Anderson, W.J.; Wang, H.Y. Intracellular accumulation of beta-amyloid(1–42) in neurons is facilitated by the alpha 7 nicotinic acetylcholine receptor in Alzheimer’s disease. Neuroscience 2002, 110, 199–211. [Google Scholar] [CrossRef]
- The US Food and Drug Administration. Aducanumab (Marketed as Aduhelm) Information. Available online: https://www.fda.gov/drugs/postmarket-drug-safety-information-patients-and-providers/aducanumab-marketed-aduhelm-information (accessed on 5 December 2022).
- Wang, H.Y.; Lee, D.H.; D’Andrea, M.R.; Peterson, P.A.; Shank, R.P.; Reitz, A.B. beta-Amyloid(1–42) binds to alpha7 nicotinic acetylcholine receptor with high affinity. Implications for Alzheimer’s disease pathology. J. Biol. Chem. 2000, 275, 5626–5632. [Google Scholar] [CrossRef]
- Wang, H.Y.; Lee, D.H.; Davis, C.B.; Shank, R.P. Amyloid peptide Abeta(1–42) binds selectively and with picomolar affinity to alpha7 nicotinic acetylcholine receptors. J. Neurochem. 2000, 75, 1155–1161. [Google Scholar] [CrossRef]
- ALZFORUM. Simufilam. Available online: https://www.alzforum.org/therapeutics/simufilam (accessed on 5 December 2022).
- Yu, W.H.; Kumar, A.; Peterhoff, C.; Shapiro Kulnane, L.; Uchiyama, Y.; Lamb, B.T.; Cuervo, A.M.; Nixon, R.A. Autophagic vacuoles are enriched in amyloid precursor protein-secretase activities: Implications for beta-amyloid peptide over-production and localization in Alzheimer’s disease. Int. J. Biochem. Cell Biol. 2004, 36, 2531–2540. [Google Scholar] [CrossRef] [PubMed]
- Hung, S.-Y.; Huang, W.-P.; Liou, H.-C.; Fu, W.-M. Autophagy protects neuron from Aβ-induced cytotoxicity. Autophagy 2009, 5, 502–510. [Google Scholar] [CrossRef] [PubMed]
- Nixon, R.A. The role of autophagy in neurodegenerative disease. Nat. Med. 2013, 19, 983–997. [Google Scholar] [CrossRef] [PubMed]
- Hung, S.-Y.; Huang, W.-P.; Liou, H.-C.; Fu, W.-M. LC3 overexpression reduces Aβ neurotoxicity through increasing α7nAchR expression and autophagic activity in neurons and mice. Neuropharmacology 2015, 93, 243–251. [Google Scholar] [CrossRef] [PubMed]
- Santos, M.D.; Alkondon, M.; Pereira, E.F.; Aracava, Y.; Eisenberg, H.M.; Maelicke, A.; Albuquerque, E.X. The nicotinic allosteric potentiating ligand galantamine facilitates synaptic transmission in the mammalian central nervous system. Mol. Pharmacol. 2002, 61, 1222–1234. [Google Scholar] [CrossRef]
- Lilienfeld, S.; Parys, W. Galantamine: Additional benefits to patients with Alzheimer’s disease. Dement. Geriatr. Cogn. Disord. 2000, 11 (Suppl. S1), 19–27. [Google Scholar] [CrossRef]
- Dajas-Bailador, F.A.; Heimala, K.; Wonnacott, S. The allosteric potentiation of nicotinic acetylcholine receptors by galantamine is transduced into cellular responses in neurons: Ca2+ signals and neurotransmitter release. Mol. Pharmacol. 2003, 64, 1217–1226. [Google Scholar] [CrossRef]
- Lin, M.W.; Chen, Y.H.; Yang, H.B.; Lin, C.C.; Hung, S.Y. Galantamine Inhibits Aβ(1–42)-Induced Neurotoxicity by Enhancing α7nAChR Expression as a Cargo Carrier for LC3 Binding and Aβ(1–42) Engulfment During Autophagic Degradation. Neurotherapeutics 2020, 17, 676–689. [Google Scholar] [CrossRef]
- Mhyre, T.R.; Boyd, J.T.; Hamill, R.W.; Maguire-Zeiss, K.A. Parkinson’s disease. Subcell. Biochem. 2012, 65, 389–455. [Google Scholar] [CrossRef]
- GBD 2016 Neurology Collaborators. Global, regional, and national burden of neurological disorders, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019, 18, 459–480. [Google Scholar] [CrossRef]
- Lin, M.W.; Lin, C.C.; Chen, Y.H.; Yang, H.B.; Hung, S.Y. Celastrol Inhibits Dopaminergic Neuronal Death of Parkinson’s Disease through Activating Mitophagy. Antioxidants 2019, 9, 37. [Google Scholar] [CrossRef]
- Prasad, E.M.; Hung, S.Y. Current Therapies in Clinical Trials of Parkinson’s Disease: A 2021 Update. Pharmaceuticals 2021, 14, 717. [Google Scholar] [CrossRef]
- Prasad, E.M.; Hung, S.Y. Behavioral Tests in Neurotoxin-Induced Animal Models of Parkinson’s Disease. Antioxidants 2020, 9, 1007. [Google Scholar] [CrossRef]
- Aosaki, T.; Miura, M.; Suzuki, T.; Nishimura, K.; Masuda, M. Acetylcholine-dopamine balance hypothesis in the striatum: An update. Geriatr. Gerontol. Int. 2010, 10 (Suppl. S1), S148–S157. [Google Scholar] [CrossRef]
- Han, J.W.; Ahn, Y.D.; Kim, W.S.; Shin, C.M.; Jeong, S.J.; Song, Y.S.; Bae, Y.J.; Kim, J.M. Psychiatric Manifestation in Patients with Parkinson’s Disease. J. Korean Med. Sci. 2018, 33, e300. [Google Scholar] [CrossRef]
- Mercuri, N.B.; Bernardi, G. The ‘magic’ of L-dopa: Why is it the gold standard Parkinson’s disease therapy? Trends Pharmacol. Sci. 2005, 26, 341–344. [Google Scholar] [CrossRef]
- Obeso, J.A.; Grandas, F.; Vaamonde, J.; Luquin, M.R.; Artieda, J.; Lera, G.; Rodriguez, M.E.; Martinez-Lage, J.M. Motor complications associated with chronic levodopa therapy in Parkinson’s disease. Neurology 1989, 39, 11–19. [Google Scholar]
- Sweet, R.D.; McDowell, F.H. Five years’ treatment of Parkinson’s disease with levodopa. Therapeutic results and survival of 100 patients. Ann. Intern. Med. 1975, 83, 456–463. [Google Scholar] [CrossRef]
- Barbeau, A. Six years of high-level levodopa therapy in severely akinetic parkinsonian patients. Arch. Neurol. 1976, 33, 333–338. [Google Scholar] [CrossRef]
- Lewitt, P.A. Levodopa for the treatment of Parkinson’s disease. N. Engl. J. Med. 2008, 359, 2468–2476. [Google Scholar] [CrossRef]
- Fahn, S.; Oakes, D.; Shoulson, I.; Kieburtz, K.; Rudolph, A.; Lang, A.; Olanow, C.W.; Tanner, C.; Marek, K.; Parkinson Study, G. Levodopa and the progression of Parkinson’s disease. N. Engl. J. Med. 2004, 351, 2498–2508. [Google Scholar] [CrossRef]
- Bordia, T.; McGregor, M.; Papke, R.L.; Decker, M.W.; McIntosh, J.M.; Quik, M. The alpha7 nicotinic receptor agonist ABT-107 protects against nigrostriatal damage in rats with unilateral 6-hydroxydopamine lesions. Exp. Neurol. 2015, 263, 277–284. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, S.; Kawamata, J.; Matsushita, T.; Matsumura, A.; Hisahara, S.; Takata, K.; Kitamura, Y.; Kem, W.; Shimohama, S. 3-[(2,4-Dimethoxy)benzylidene]-anabaseine dihydrochloride protects against 6-hydroxydopamine-induced parkinsonian neurodegeneration through alpha7 nicotinic acetylcholine receptor stimulation in rats. J. Neurosci. Res. 2013, 91, 462–471. [Google Scholar] [CrossRef] [PubMed]
- Stuckenholz, V.; Bacher, M.; Balzer-Geldsetzer, M.; Alvarez-Fischer, D.; Oertel, W.H.; Dodel, R.C.; Noelker, C. The alpha7 nAChR agonist PNU-282987 reduces inflammation and MPTP-induced nigral dopaminergic cell loss in mice. J. Park. Dis. 2013, 3, 161–172. [Google Scholar] [CrossRef]
- Han, B.; Li, X.; Hao, J. The cholinergic anti-inflammatory pathway: An innovative treatment strategy for neurological diseases. Neurosci. Biobehav. Rev. 2017, 77, 358–368. [Google Scholar] [CrossRef]
- Thanvi, B.; Lo, N.; Robinson, T. Levodopa-induced dyskinesia in Parkinson’s disease: Clinical features, pathogenesis, prevention and treatment. Postgrad. Med. J. 2007, 83, 384–388. [Google Scholar] [CrossRef]
- Paik, J.; Keam, S.J. Amantadine Extended-Release (GOCOVRITM): A Review in Levodopa-Induced Dyskinesia in Parkinson’s Disease. CNS Drugs 2018, 32, 797–806. [Google Scholar] [CrossRef]
- Zhang, D.; McGregor, M.; Decker, M.W.; Quik, M. The alpha7 nicotinic receptor agonist ABT-107 decreases L-Dopa-induced dyskinesias in parkinsonian monkeys. J. Pharmacol. Exp. Ther. 2014, 351, 25–32. [Google Scholar] [CrossRef]
- Di Paolo, T.; Gregoire, L.; Feuerbach, D.; Elbast, W.; Weiss, M.; Gomez-Mancilla, B. AQW051, a novel and selective nicotinic acetylcholine receptor alpha7 partial agonist, reduces l-Dopa-induced dyskinesias and extends the duration of l-Dopa effects in parkinsonian monkeys. Park. Relat. Disord. 2014, 20, 1119–1123. [Google Scholar] [CrossRef]
- Zhang, D.; McGregor, M.; Bordia, T.; Perez, X.A.; McIntosh, J.M.; Decker, M.W.; Quik, M. αlpha nicotinic receptor agonists reduce levodopa-induced dyskinesias with severe nigrostriatal damage. Mov. Disord. 2015, 30, 1901–1911. [Google Scholar] [CrossRef]
- World Health Organization. Schizophrenia. 2022. Available online: https://www.who.int/news-room/fact-sheets/detail/schizophrenia (accessed on 5 December 2022).
- Javitt, D.C. Balancing therapeutic safety and efficacy to improve clinical and economic outcomes in schizophrenia: A clinical overview. Am. J. Manag. Care 2014, 20, S160–S165. [Google Scholar]
- Gejman, P.V.; Sanders, A.R.; Duan, J. The role of genetics in the etiology of schizophrenia. Psychiatr. Clin. N. Am. 2010, 33, 35–66. [Google Scholar] [CrossRef]
- Patel, K.R.; Cherian, J.; Gohil, K.; Atkinson, D. Schizophrenia: Overview and treatment options. P T 2014, 39, 638–645. [Google Scholar]
- Sanders, A.R.; Duan, J.; Levinson, D.F.; Shi, J.; He, D.; Hou, C.; Burrell, G.J.; Rice, J.P.; Nertney, D.A.; Olincy, A.; et al. No significant association of 14 candidate genes with schizophrenia in a large European ancestry sample: Implications for psychiatric genetics. Am. J. Psychiatry 2008, 165, 497–506. [Google Scholar] [CrossRef]
- Grinchii, D.; Dremencov, E. Mechanism of Action of Atypical Antipsychotic Drugs in Mood Disorders. Int. J. Mol. Sci. 2020, 21, 9532. [Google Scholar] [CrossRef]
- Recio-Barbero, M.; Segarra, R.; Zabala, A.; Gonzalez-Fraile, E.; Gonzalez-Pinto, A.; Ballesteros, J. Cognitive Enhancers in Schizophrenia: A Systematic Review and Meta-Analysis of Alpha-7 Nicotinic Acetylcholine Receptor Agonists for Cognitive Deficits and Negative Symptoms. Front. Psychiatry 2021, 12, 631589. [Google Scholar] [CrossRef]
- Peritogiannis, V.; Ninou, A.; Samakouri, M. Mortality in Schizophrenia-Spectrum Disorders: Recent Advances in Understanding and Management. Healthcare 2022, 10, 2366. [Google Scholar] [CrossRef]
- Wallace, T.L.; Bertrand, D. αlpha neuronal nicotinic receptors as a drug target in schizophrenia. Expert Opin. Ther. Targets 2013, 17, 139–155. [Google Scholar] [CrossRef]
- Freedman, R.; Coon, H.; Myles-Worsley, M.; Orr-Urtreger, A.; Olincy, A.; Davis, A.; Polymeropoulos, M.; Holik, J.; Hopkins, J.; Hoff, M.; et al. Linkage of a neurophysiological deficit in schizophrenia to a chromosome 15 locus. Proc. Natl. Acad. Sci. USA 1997, 94, 587–592. [Google Scholar] [CrossRef]
- Guan, Z.Z.; Zhang, X.; Blennow, K.; Nordberg, A. Decreased protein level of nicotinic receptor alpha7 subunit in the frontal cortex from schizophrenic brain. Neuroreport 1999, 10, 1779–1782. [Google Scholar] [CrossRef]
- ALZFORUM. Encenicline Misses Endpoints in Two Phase 3 Schizophrenia Trials. 2016. Available online: https://www.alzforum.org/news/research-news/encenicline-misses-endpoints-two-phase-3-schizophrenia-trials (accessed on 5 December 2022).
- Olincy, A.; Harris, J.G.; Johnson, L.L.; Pender, V.; Kongs, S.; Allensworth, D.; Ellis, J.; Zerbe, G.O.; Leonard, S.; Stevens, K.E.; et al. Proof-of-concept trial of an alpha7 nicotinic agonist in schizophrenia. Arch. Gen. Psychiatry 2006, 63, 630–638. [Google Scholar] [CrossRef] [PubMed]
- Freedman, R.; Olincy, A.; Buchanan, R.W.; Harris, J.G.; Gold, J.M.; Johnson, L.; Allensworth, D.; Guzman-Bonilla, A.; Clement, B.; Ball, M.P.; et al. Initial phase 2 trial of a nicotinic agonist in schizophrenia. Am. J. Psychiatry 2008, 165, 1040–1047. [Google Scholar] [CrossRef] [PubMed]
- Lieberman, J.A.; Dunbar, G.; Segreti, A.C.; Girgis, R.R.; Seoane, F.; Beaver, J.S.; Duan, N.; Hosford, D.A. A randomized exploratory trial of an alpha-7 nicotinic receptor agonist (TC-5619) for cognitive enhancement in schizophrenia. Neuropsychopharmacology 2013, 38, 968–975. [Google Scholar] [CrossRef] [PubMed]
- Walling, D.; Marder, S.R.; Kane, J.; Fleischhacker, W.W.; Keefe, R.S.; Hosford, D.A.; Dvergsten, C.; Segreti, A.C.; Beaver, J.S.; Toler, S.M.; et al. Phase 2 Trial of an Alpha-7 Nicotinic Receptor Agonist (TC-5619) in Negative and Cognitive Symptoms of Schizophrenia. Schizophr. Bull. 2016, 42, 335–343. [Google Scholar] [CrossRef] [PubMed]
- Kantrowitz, J.T.; Javitt, D.C.; Freedman, R.; Sehatpour, P.; Kegeles, L.S.; Carlson, M.; Sobeih, T.; Wall, M.M.; Choo, T.H.; Vail, B.; et al. Double blind, two dose, randomized, placebo-controlled, cross-over clinical trial of the positive allosteric modulator at the alpha7 nicotinic cholinergic receptor AVL-3288 in schizophrenia patients. Neuropsychopharmacology 2020, 45, 1339–1345. [Google Scholar] [CrossRef]
- World Health Organization. Depression. 2021. Available online: https://www.who.int/news-room/fact-sheets/detail/depression (accessed on 5 December 2022).
- Chand, S.P.; Arif, H. Depression; StatPearls: Treasure Island, FL, USA, 2022. [Google Scholar]
- Zhao, D.; Xu, X.; Pan, L.; Zhu, W.; Fu, X.; Guo, L.; Lu, Q.; Wang, J. Pharmacologic activation of cholinergic alpha7 nicotinic receptors mitigates depressive-like behavior in a mouse model of chronic stress. J. Neuroinflammation 2017, 14, 234. [Google Scholar] [CrossRef]
- Janowsky, D.S.; El-Yousef, M.K.; Davis, J.M.; Sekerke, H.J. A cholinergic-adrenergic hypothesis of mania and depression. Lancet 1972, 2, 632–635. [Google Scholar] [CrossRef]
- Fitzgerald, P.J.; Hale, P.J.; Ghimire, A.; Watson, B.O. Repurposing Cholinesterase Inhibitors as Antidepressants? Dose and Stress-Sensitivity May Be Critical to Opening Possibilities. Front. Behav. Neurosci. 2020, 14, 620119. [Google Scholar] [CrossRef]
- Mineur, Y.S.; Mose, T.N.; Blakeman, S.; Picciotto, M.R. Hippocampal alpha7 nicotinic ACh receptors contribute to modulation of depression-like behaviour in C57BL/6J mice. Br. J. Pharmacol. 2018, 175, 1903–1914. [Google Scholar] [CrossRef]
- Davidson, M.; Levi, L.; Park, J.; Nastas, I.; Ford, L.; Rassnick, S.; Canuso, C.; Davis, J.M.; Weiser, M. The effects of JNJ-39393406 a positive allosteric nicotine modulator on mood and cognition in patients with unipolar depression: A double-blind, add-on, placebo-controlled trial. Eur. Neuropsychopharmacol. 2021, 51, 33–42. [Google Scholar] [CrossRef]
- Thomsen, M.S.; Weyn, A.; Mikkelsen, J.D. Hippocampal alpha7 nicotinic acetylcholine receptor levels in patients with schizophrenia, bipolar disorder, or major depressive disorder. Bipolar Disord. 2011, 13, 701–707. [Google Scholar] [CrossRef]


{kind=link}
| Neuronal Subunit (Protein Accession of UniProtKB) | Peptide Length (Amino Acids) | Gene (Reference Sequence of NCBI) | Gene Locus (Data from GRCh38/hg38) | DNA Strand Orientation | Cytogenetic Band by HUGO Gene Nomenclature Committee (HGNC) | mRNA Length (NCBI Reference Sequence) |
|---|---|---|---|---|---|---|
| α2 (Q15822) | 529 | CHRNA2 (NC_000008.11, NC_060932.1) | chr8:27,459,756–27,479,883 | Minus strand | 8p21.2 | 4037-bases/NM_000742.4 3992-bases/NM_001282455.2 3987-bases/NM_001347705.2 4032-bases/NM_001347706.2 3916-bases/NM_001347707.2 3904-bases/NM_001347708.2 |
| α3 (P32297) | 505 | CHRNA3 (NC_000015.10) | chr15:78,593,052–78,621,295 | Minus strand | 15q25.1 | 3015-bases/NM_000743.5 1731-bases/NM_001166694.2 |
| α4 (P43681) | 627 | CHRNA4 (NC_000020.11) | chr20:63,343,223–63,378,401 | Minus strand | 20q13.33 | 5583-bases/NM_000744.7 5514-bases/NM_001256573.2 |
| α5 (P30532) | 468 | CHRNA5 (NC_000015.10) | chr15:78,565,520–78,595,269 | Plus strand | 15q25.1 | 3623-bases/NM_000745.4 2836-bases/NM_001307945.2 3493-bases/NM_001395171.1 2969-bases/NM_001395172.1 3091-bases/NM_001395173.1 3085-bases/NM_001395174.1 2833-bases/NM_001395175.1 |
| α6 (Q15825) | 494 | CHRNA6 (NC_000008.11) | chr8:42,752,620–42,796,392 | Minus strand | 8p11.21 | 2355-bases/NM_001199279.1 2400-bases/NM_004198.3 |
| α7 (P36544) | 502 | CHRNA7 (NC_000015.10) | chr15:31,923,438–32,173,018 | Plus strand | 15q13.3 | 6149-bases/NM_000746.6 6236-bases/NM_001190455.3 |
| α9 (Q9UGM1) | 479 | CHRNA9 (NC_000004.12) | chr4:40,335,333–40,355,217 | Plus strand | 4p14 | 2272-bases/NM_017581.4 |
| α10 (Q9GZZ6) | 450 | CHRNA10 (NC_000011.10) | chr11:3,665,587–3,671,384 | Minus strand | 11p15.4 | 2007-bases/NM_001303034.2 1940-bases/NM_001303035.2 1945-bases/NM_020402.4 |
| β2 (P17787) | 502 | CHRNB2 (NC_000001.11) | chr1:154,567,778–154,580,013 | Plus strand | 1q21.3 | 5857-bases/NM_000748.3 |
| β3 (Q05901) | 458 | CHRNB3 (NC_000008.11) | chr8:42,697,366–42,737,407 | Plus strand | 8p11.21 | 2347-bases/NM_000749.5 2480-bases/NM_001347717.2 |
| β4 (P30926) | 498 | CHRNB4 (NC_000015.10) | chr15:78,624,111–78,727,754 | Minus strand | 15q25.1 | 2596-bases/NM_000750.5 1617-bases/NM_001256567.3 |
| Dupα7 (Q494W8) | 412 | CHRFAM7A (NC_000015.10) | chr15:30,360,566–30,393,900 | Minus strand | 15q13.2 | 3411-bases/NM_139320.2 2794-bases/NM_148911.1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, C.-H.; Hung, S.-Y. Physiologic Functions and Therapeutic Applications of α7 Nicotinic Acetylcholine Receptor in Brain Disorders. Pharmaceutics 2023, 15, 31. https://doi.org/10.3390/pharmaceutics15010031
Lee C-H, Hung S-Y. Physiologic Functions and Therapeutic Applications of α7 Nicotinic Acetylcholine Receptor in Brain Disorders. Pharmaceutics. 2023; 15(1):31. https://doi.org/10.3390/pharmaceutics15010031
Chicago/Turabian StyleLee, Chien-Hsing, and Shih-Ya Hung. 2023. "Physiologic Functions and Therapeutic Applications of α7 Nicotinic Acetylcholine Receptor in Brain Disorders" Pharmaceutics 15, no. 1: 31. https://doi.org/10.3390/pharmaceutics15010031
APA StyleLee, C.-H., & Hung, S.-Y. (2023). Physiologic Functions and Therapeutic Applications of α7 Nicotinic Acetylcholine Receptor in Brain Disorders. Pharmaceutics, 15(1), 31. https://doi.org/10.3390/pharmaceutics15010031