
Conformational Sampling Deciphers the Chameleonic Properties of a VHL-Based Degrader

 , , ,
, , , 
Abstract
1. Introduction
2. Materials and Methods
3. Results and Discussion
3.1. Analysis of NMR Data
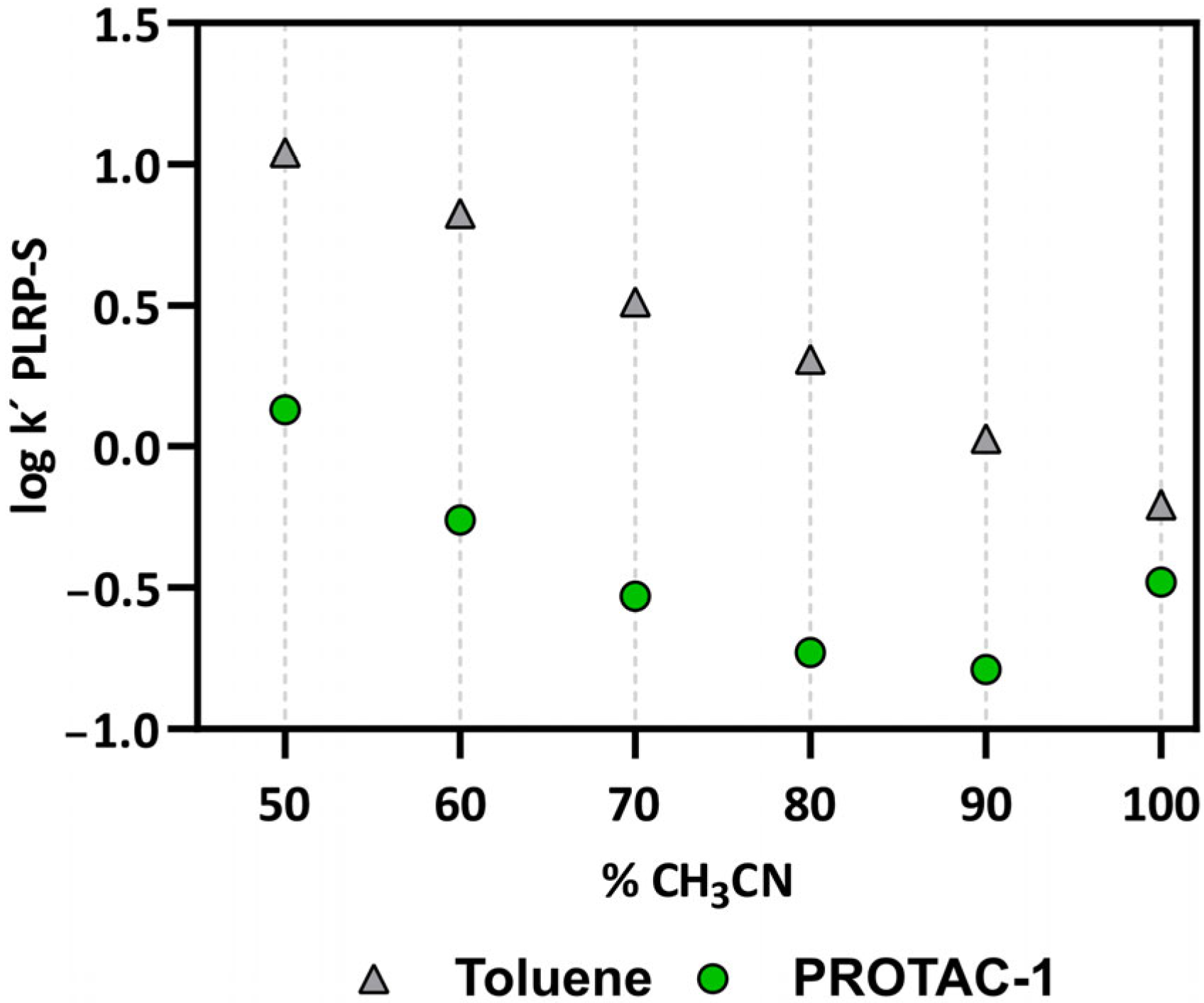
3.2. Experimental Physicochemical Data to Monitor the Chameleonicity of PROTAC-1
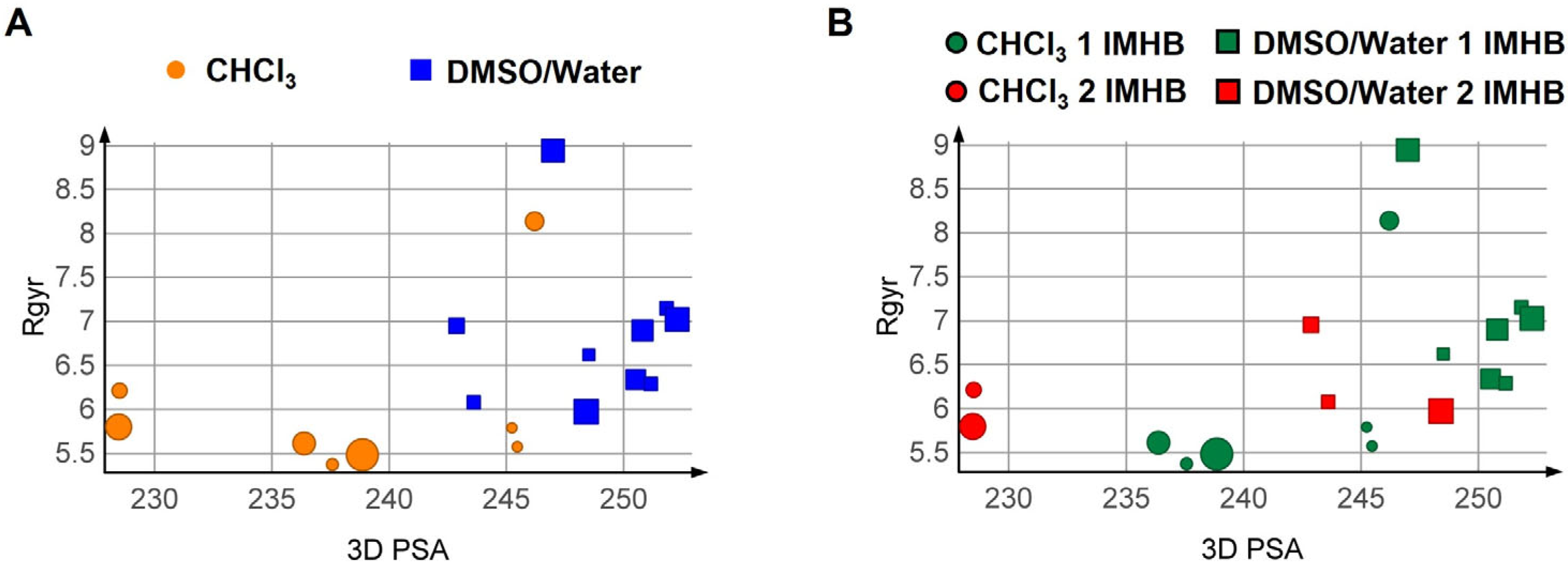
3.3. Computational Analysis
3.3.1. Conformational Analysis Strategies
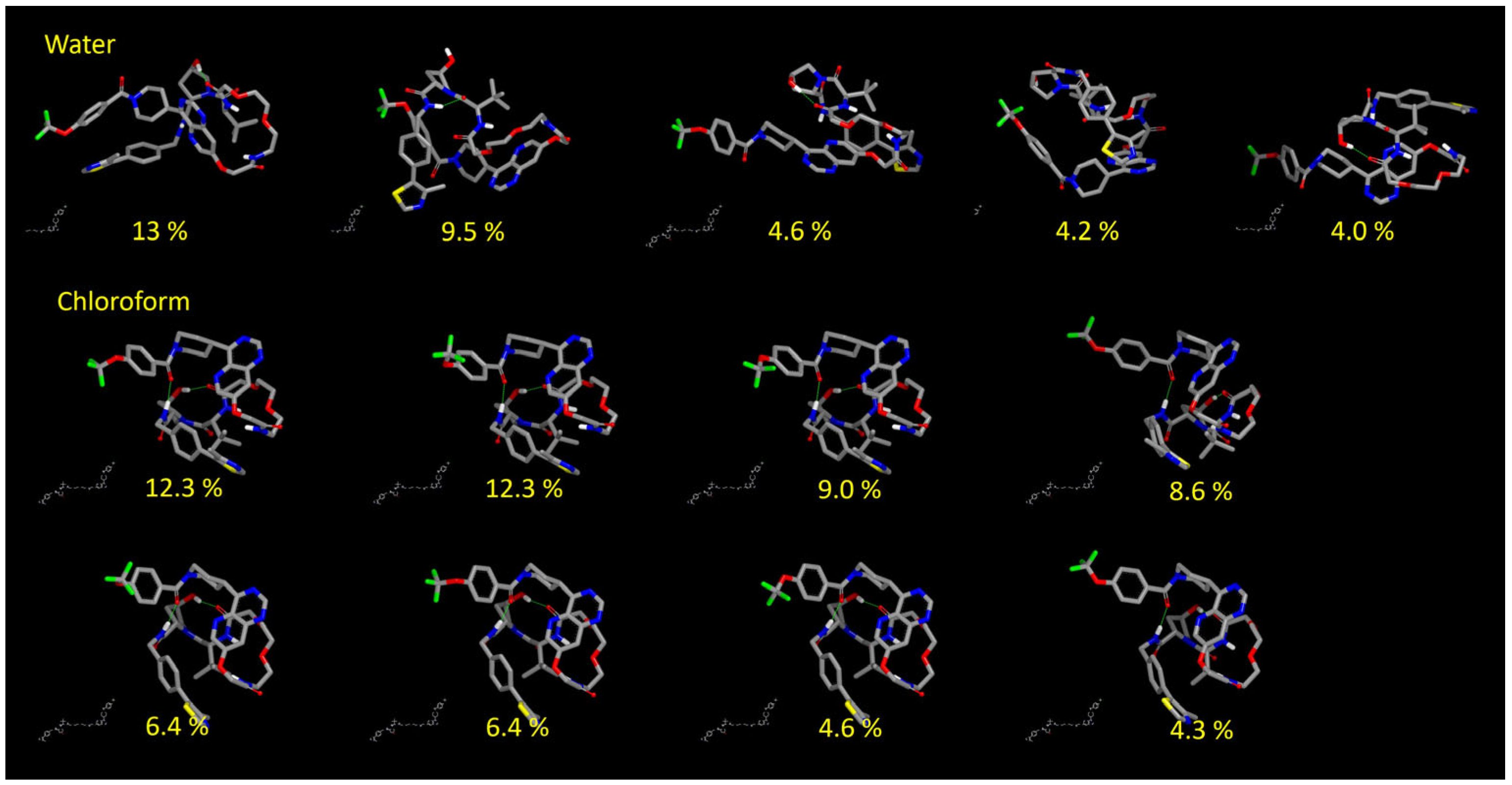
3.3.2. Results of the Conformational Sampling
3.3.3. Post Minimization with MOPAC
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Doak, B.C.; Over, B.; Giordanetto, F.; Kihlberg, J. Oral Druggable Space beyond the Rule of 5: Insights from Drugs and Clinical Candidates. Chem. Biol. 2014, 21, 1115–1142. [Google Scholar] [CrossRef]
- Doak, B.C.; Zheng, J.; Dobritzsch, D.; Kihlberg, J. How Beyond Rule of 5 Drugs and Clinical Candidates Bind to Their Targets. J. Med. Chem. 2016, 59, 2312–2327. [Google Scholar] [CrossRef] [PubMed]
- Doak, B.C.; Kihlberg, J. Drug Discovery beyond the Rule of 5—Opportunities and Challenges. Expert. Opin. Drug Discov. 2017, 12, 115–119. [Google Scholar] [CrossRef] [PubMed]
- Békés, M.; Langley, D.R.; Crews, C.M. PROTAC Targeted Protein Degraders: The Past Is Prologue. Nat. Rev. Drug Discov. 2022, 21, 181–200. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and Computational Approaches to Estimate Solubility and Permeability in Drug Discovery and Development Settings. Adv. Drug Deliv. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Shultz, M.D. Two Decades under the Influence of the Rule of Five and the Changing Properties of Approved Oral Drugs. J. Med. Chem. 2019, 62, 1701–1714. [Google Scholar] [CrossRef]
- DeGoey, D.A.; Chen, H.-J.; Cox, P.B.; Wendt, M.D. Beyond the Rule of 5: Lessons Learned from AbbVie’s Drugs and Compound Collection. J. Med. Chem. 2018, 61, 2636–2651. [Google Scholar] [CrossRef]
- Matsson, P.; Doak, B.C.; Over, B.; Kihlberg, J. Cell Permeability beyond the Rule of 5. Adv. Drug Deliv. Rev. 2016, 101, 42–61. [Google Scholar] [CrossRef]
- Guimarães, C.R.W.; Mathiowetz, A.M.; Shalaeva, M.; Goetz, G.; Liras, S. Use of 3D Properties to Characterize beyond Rule-of-5 Property Space for Passive Permeation. J. Chem. Inf. Model. 2012, 52, 882–890. [Google Scholar] [CrossRef]
- Ermondi, G.; Vallaro, M.; Goetz, G.; Shalaeva, M.; Caron, G. Updating the Portfolio of Physicochemical Descriptors Related to Permeability in the beyond the Rule of 5 Chemical Space. Eur. J. Pharm. Sci. 2020, 146, 105274. [Google Scholar] [CrossRef]
- Ermondi, G.; Vallaro, M.; Goetz, G.; Shalaeva, M.; Caron, G. Experimental Lipophilicity for beyond Rule of 5 Compounds. Future Drug Discov. 2019, 1, 1. [Google Scholar] [CrossRef]
- Digiesi, V.; de la Oliva Roque, V.; Vallaro, M.; Caron, G.; Ermondi, G. Permeability Prediction in the Beyond-Rule-of 5 Chemical Space: Focus on Cyclic Hexapeptides. Eur. J. Pharm. Biopharm. 2021, 165, 259–270. [Google Scholar] [CrossRef] [PubMed]
- Rossi Sebastiano, M.; Doak, B.C.; Backlund, M.; Poongavanam, V.; Over, B.; Ermondi, G.; Caron, G.; Matsson, P.; Kihlberg, J. Impact of Dynamically Exposed Polarity on Permeability and Solubility of Chameleonic Drugs beyond the Rule of 5. J. Med. Chem. 2018, 61, 4189–4202. [Google Scholar] [CrossRef] [PubMed]
- Ermondi, G.; Lavore, F.; Vallaro, M.; Tiana, G.; Vasile, F.; Caron, G. Managing Experimental 3D Structures in the Beyond-Rule-of-5 Chemical Space: The Case of Rifampicin. Chem.—A Eur. J. 2021, 27, 10394–10404. [Google Scholar] [CrossRef]
- Ono, S.; Naylor, M.R.; Townsend, C.E.; Okumura, C.; Okada, O.; Lee, H.W.; Lokey, R.S. Cyclosporin A: Conformational Complexity and Chameleonicity. J. Chem. Inf. Model. 2021, 61, 5601–5613. [Google Scholar] [CrossRef]
- Whitty, A.; Zhong, M.; Viarengo, L.; Beglov, D.; Hall, D.R.; Vajda, S. Quantifying the Chameleonic Properties of Macrocycles and Other High-Molecular-Weight Drugs. Drug Discov. Today 2016, 21, 712–717. [Google Scholar] [CrossRef]
- Carrupt, P.A.; Testa, B.; Bechalany, A.; el Tayar, N.; Descas, P.; Perrissoud, D. Morphine 6-Glucuronide and Morphine 3-Glucuronide as Molecular Chameleons with Unexpected Lipophilicity. J. Med. Chem. 1991, 34, 1272–1275. [Google Scholar] [CrossRef]
- Over, B.; Mccarren, P.; Artursson, P.; Foley, M.; Giordanetto, F.; Gro, G.; Hilgendorf, C.; Lee, M.D.; Muncipinto, G.; Perry, M.W.D.; et al. Impact of Stereospeci Fi c Intramolecular Hydrogen Bonding on Cell Permeability and Physicochemical Properties. J. Med. Chem. 2014, 57, 2746–2754. [Google Scholar] [CrossRef]
- Atilaw, Y.; Poongavanam, V.; Svensson Nilsson, C.; Nguyen, D.; Giese, A.; Meibom, D.; Erdelyi, M.; Kihlberg, J. Solution Conformations Shed Light on PROTAC Cell Permeability. ACS Med. Chem. Lett. 2021, 12, 107–114. [Google Scholar] [CrossRef]
- Poongavanam, V.; Danelius, E.; Peintner, S.; Alcaraz, L.; Caron, G.; Cummings, M.D.; Wlodek, S.; Erdelyi, M.; Hawkins, P.C.D.; Ermondi, G.; et al. Conformational Sampling of Macrocyclic Drugs in Different Environments: Can We Find the Relevant Conformations? ACS Omega 2018, 3, 11742–11757. [Google Scholar] [CrossRef]
- Caron, G.; Kihlberg, J.; Goetz, G.; Ratkova, E.; Poongavanam, V.; Ermondi, G. Steering New Drug Discovery Campaigns: Permeability, Solubility, and Physicochemical Properties in the BRo5 Chemical Space. ACS Med. Chem. Lett. 2021, 12, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Rezai, T.; Bock, J.E.; Zhou, M.V.; Kalyanaraman, C.; Lokey, R.S.; Jacobson, M.P. Conformational Flexibility, Internal Hydrogen Bonding, and Passive Membrane Permeability: Successful in Silico Prediction of the Relative Permeabilities of Cyclic Peptides. J. Am. Chem. Soc. 2006, 128, 14073–14080. [Google Scholar] [CrossRef] [PubMed]
- Caron, G.; Digiesi, V.; Solaro, S.; Ermondi, G. Flexibility in Early Drug Discovery: Focus on the beyond-Rule-of-5 Chemical Space. Drug Discov. Today 2020, 25, 621–627. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, B.; Mohr, P.; Stahl, M. Intramolecular Hydrogen Bonding in Medicinal Chemistry. J. Med. Chem. 2010, 53, 2601–2611. [Google Scholar] [CrossRef] [PubMed]
- Testa, B.; Carrupt, P.A.; Guillard, P.; Tsai, R.S. Intramolecular Interactions Encoded in Lipophilicity: Their Nature and Significance. Lipophilicity Drug Action Toxicol. 2008, 4, 49–71. [Google Scholar] [CrossRef]
- Caron, G.; Kihlberg, J.; Ermondi, G. Intramolecular Hydrogen Bonding: An Opportunity for Improved Design in Medicinal Chemistry. Med. Res. Rev. 2019, 39, 1707–1729. [Google Scholar] [CrossRef]
- Hyung, S.; Feng, X.; Che, Y.; Stroh, J.G.; Shapiro, M. Detection of Conformation Types of Cyclosporin Retaining Intramolecular Hydrogen Bonds by Mass Spectrometry. Anal. Bioanal. Chem. 2014, 406, 5785–5794. [Google Scholar] [CrossRef]
- Alex, A.; Millan, D.S.; Perez, M.; Wakenhut, F.; Whitlock, G.A. Intramolecular Hydrogen Bonding to Improve Membrane Permeability and Absorption in beyond Rule of Five Chemical Space. Medchemcomm 2011, 2, 669. [Google Scholar] [CrossRef]
- Danelius, E.; Poongavanam, V.; Peintner, S.; Wieske, L.H.E.; Erdélyi, M.; Kihlberg, J. Solution Conformations Explain the Chameleonic Behaviour of Macrocyclic Drugs. Chem. A Eur. J. 2020, 26, 5231–5244. [Google Scholar] [CrossRef]
- Rossi Sebastiano, M.; Garcia Jimenez, D.; Vallaro, M.; Caron, G.; Ermondi, G. Refinement of Computational Access to Molecular Physicochemical Properties: From Ro5 to BRo5. J. Med. Chem. 2022, 65, 12068–12083. [Google Scholar] [CrossRef]
- Ermondi, G.; Vallaro, M.; Caron, G. Degraders Early Developability Assessment: Face-to-Face with Molecular Properties. Drug Discov. Today 2020, 25, 1585–1591. [Google Scholar] [CrossRef] [PubMed]
- Caron, G.; Vallaro, M.; Ermondi, G. Log P as a Tool in Intramolecular Hydrogen Bond Considerations. Drug Discov. Today Technol. 2018, 27, 65–70. [Google Scholar] [CrossRef] [PubMed]
- Goetz, G.H.; Farrell, W.; Shalaeva, M.; Sciabola, S.; Anderson, D.P.; Yan, J.; Philippe, L.; Shapiro, M.J. A High Throughput Method for the Indirect Detection of Intramolecular Hydrogen Bonding. J. Med. Chem. 2014, 57, 2920–2929. [Google Scholar] [CrossRef]
- Shalaeva, M.; Caron, G.; Abramov, Y.A.; O’Connell, T.N.; Plummer, M.S.; Yalamanchi, G.; Farley, K.A.; Goetz, G.H.; Philippe, L.; Shapiro, M.J. Integrating Intramolecular Hydrogen Bonding (IMHB) Considerations in Drug Discovery Using ΔlogP as a Tool. J. Med. Chem. 2013, 56, 4870–4879. [Google Scholar] [CrossRef]
- Abraham, M.H.; Abraham, R.J.; Acree, W.E.; Aliev, A.E.; Leo, A.J.; Whaley, W.L. An NMR Method for the Quantitative Assessment of Intramolecular Hydrogen Bonding; Application to Physicochemical, Environmental, and Biochemical Properties. J. Org. Chem. 2014, 79, 11075–11083. [Google Scholar] [CrossRef]
- Pereira, D.M.; Rodrigues, C.M.P. Targeted Avenues for Cancer Treatment: The MEK5–ERK5 Signaling Pathway. Trends Mol. Med. 2020, 26, 394–407. [Google Scholar] [CrossRef]
- Stecca, B.; Rovida, E. Impact of ERK5 on the Hallmarks of Cancer. Int. J. Mol. Sci. 2019, 20, 1426. [Google Scholar] [CrossRef] [PubMed]
- Ertl, P.; Rohde, B.; Selzer, P. Fast Calculation of Molecular Polar Surface Area as a Sum of Fragment-Based Contributions and Its Application to the Prediction of Drug Transport Properties. J. Med. Chem. 2000, 43, 3714–3717. [Google Scholar] [CrossRef]
- Clark, D.E. Rapid Calculation of Polar Molecular Surface Area and Its Application to the Prediction of Transport Phenomena. 2. Prediction of Blood-Brain Barrier Penetration. J. Pharm. Sci. 1999, 88, 815–821. [Google Scholar] [CrossRef]
- Clark, D.E. What Has Polar Surface Area Ever Done for Drug Discovery? Future Med. Chem. 2011, 3, 469–484. [Google Scholar] [CrossRef]
- García Jiménez, D.; Rossi Sebastiano, M.; Vallaro, M.; Mileo, V.; Pizzirani, D.; Moretti, E.; Ermondi, G.; Caron, G. Designing Soluble PROTACs: Strategies and Preliminary Guidelines. J. Med. Chem. 2022, 65, 12639–12649. [Google Scholar] [CrossRef]
- Watts, K.S.; Dalal, P.; Murphy, R.B.; Sherman, W.; Friesner, R.A.; Shelley, J.C. ConfGen: A Conformational Search Method for Efficient Generation of Bioactive Conformers. J. Chem. Inf. Model. 2010, 50, 534–546. [Google Scholar] [CrossRef] [PubMed]
- Shivakumar, D.; Williams, J.; Wu, Y.; Damm, W.; Shelley, J.; Sherman, W. Prediction of Absolute Solvation Free Energies Using Molecular Dynamics Free Energy Perturbation and the OPLS Force Field. J. Chem. Theory Comput. 2010, 6, 1509–1519. [Google Scholar] [CrossRef]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An Open Chemical Toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef] [PubMed]
- Pedretti, A.; Mazzolari, A.; Gervasoni, S.; Fumagalli, L.; Vistoli, G. The VEGA Suite of Programs: An Versatile Platform for Cheminformatics and Drug Design Projects. Bioinformatics 2021, 37, 1174–1175. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A Visualization System for Exploratory Research and Analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Mills, J.E.J.; Dean, P.M. Three-Dimensional Hydrogen-Bond Geometry and Probability Information from a Crystal Survey. J. Comput. Aided Mol. Des. 1996, 10, 607–622. [Google Scholar] [CrossRef] [PubMed]
- Cicero, D.O.; Barbato, G.; Bazzo, R. NMR Analysis of Molecular Flexibility in Solution: A New Method for the Study of Complex Distributions of Rapidly Exchanging Conformations. Application to a 13-Residue Peptide with an 8-Residue Loop. J. Am. Chem. Soc. 1995, 117, 1027–1033. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Maxwell, D.S.; Tirado-Rives, J. Development and Testing of the OPLS All-Atom Force Field on Conformational Energetics and Properties of Organic Liquids. J. Am. Chem. Soc. 1996, 118, 11225–11236. [Google Scholar] [CrossRef]
- Stewart, J.J.P. Optimization of Parameters for Semiempirical Methods VI: More Modifications to the NDDO Approximations and Re-Optimization of Parameters. J. Mol. Model. 2013, 19, 1–32. [Google Scholar] [CrossRef]
- Witek, J.; Keller, B.G.; Blatter, M.; Meissner, A.; Wagner, T.; Riniker, S. Kinetic Models of Cyclosporin A in Polar and Apolar Environments Reveal Multiple Congruent Conformational States. J. Chem. Inf. Model. 2016, 56, 1547–1562. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CS Protocols | ||||
|---|---|---|---|---|
| Software | Protocol Label | Algorithm | Parameters | FF; Solvation |
| Schrodinger | Sc01 | Mixed torsional/low-mode sampling. LMOD | max number steps = 103 | OPLS3e; Generalized-Born/Surface-Area (GB/SA) model |
| Schrodinger | Sc02 | Mixed torsional/low-mode sampling. LMOD | max number steps = 104 | OPLS3e; Generalized-Born/Surface-Area (GB/SA) model |
| Schrodinger | Sc03 | Mixed torsional/low-mode sampling. LMOD | max number steps = 105 | OPLS3e; Generalized-Born/Surface-Area (GB/SA) model |
| Schrodinger | Sc04 | Torsional sampling (MCMM) | max number steps = 103 | OPLS3e; Generalized-Born/Surface-Area (GB/SA) model |
| Schrodinger | Sc05 | Torsional sampling (MCMM) | max number steps = 104 | OPLS3e; Generalized-Born/Surface-Area (GB/SA) model |
| Schrodinger | Sc06 | Torsional sampling (MCMM) | max number steps = 105 | OPLS3e; Generalized-Born/Surface-Area (GB/SA) model |
| Post-CS Minimization | ||||
| Software | Method Label | Starting Conformations | Parameters | Hamiltonian; Solvation |
| MOPAC | MO1 | Clusters from Sc01 | Default | PM7; Conductor-like Screening Model |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ermondi, G.; Jimenez, D.G.; Rossi Sebastiano, M.; Kihlberg, J.; Caron, G. Conformational Sampling Deciphers the Chameleonic Properties of a VHL-Based Degrader. Pharmaceutics 2023, 15, 272. https://doi.org/10.3390/pharmaceutics15010272
Ermondi G, Jimenez DG, Rossi Sebastiano M, Kihlberg J, Caron G. Conformational Sampling Deciphers the Chameleonic Properties of a VHL-Based Degrader. Pharmaceutics. 2023; 15(1):272. https://doi.org/10.3390/pharmaceutics15010272
Chicago/Turabian StyleErmondi, Giuseppe, Diego Garcia Jimenez, Matteo Rossi Sebastiano, Jan Kihlberg, and Giulia Caron. 2023. "Conformational Sampling Deciphers the Chameleonic Properties of a VHL-Based Degrader" Pharmaceutics 15, no. 1: 272. https://doi.org/10.3390/pharmaceutics15010272
APA StyleErmondi, G., Jimenez, D. G., Rossi Sebastiano, M., Kihlberg, J., & Caron, G. (2023). Conformational Sampling Deciphers the Chameleonic Properties of a VHL-Based Degrader. Pharmaceutics, 15(1), 272. https://doi.org/10.3390/pharmaceutics15010272