
Assessment of Diverse Solid−State Accelerated Autoxidation Methods for Droperidol


Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
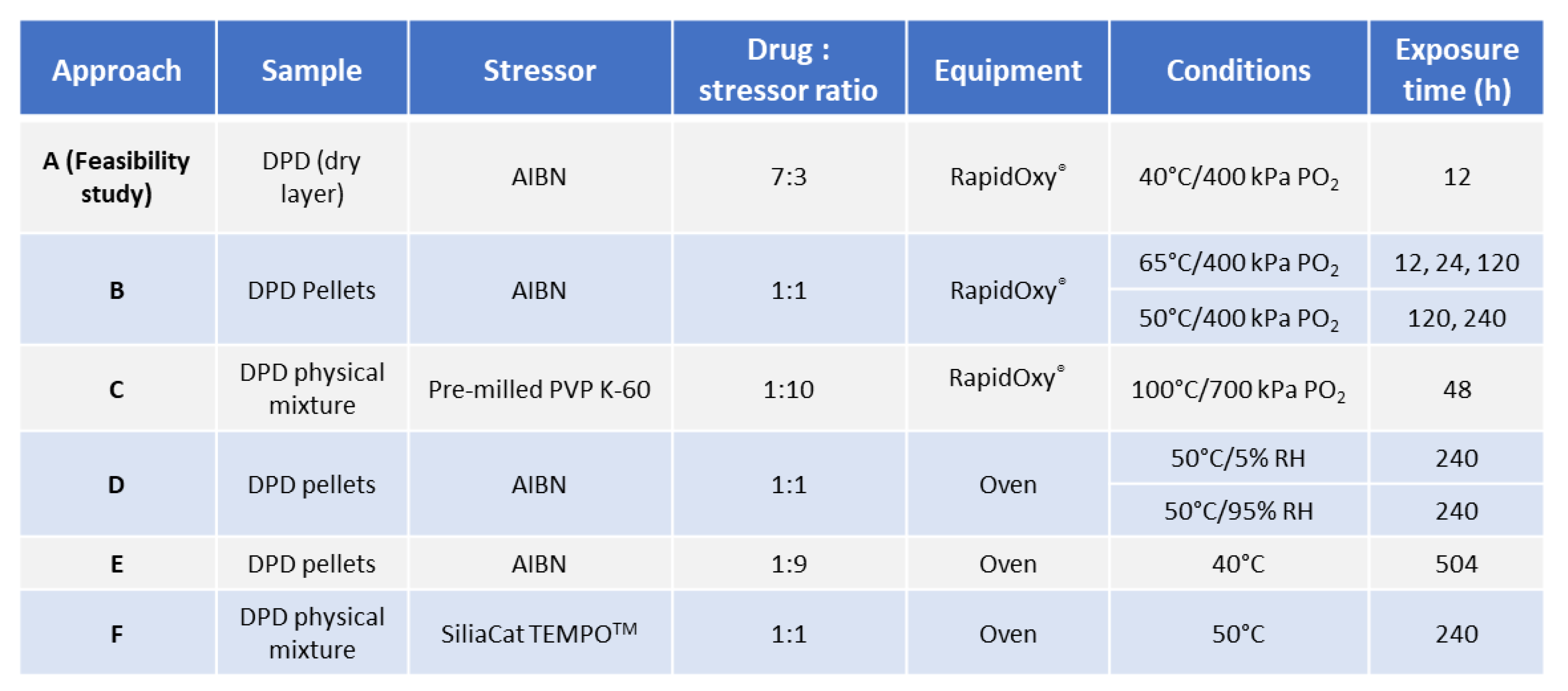
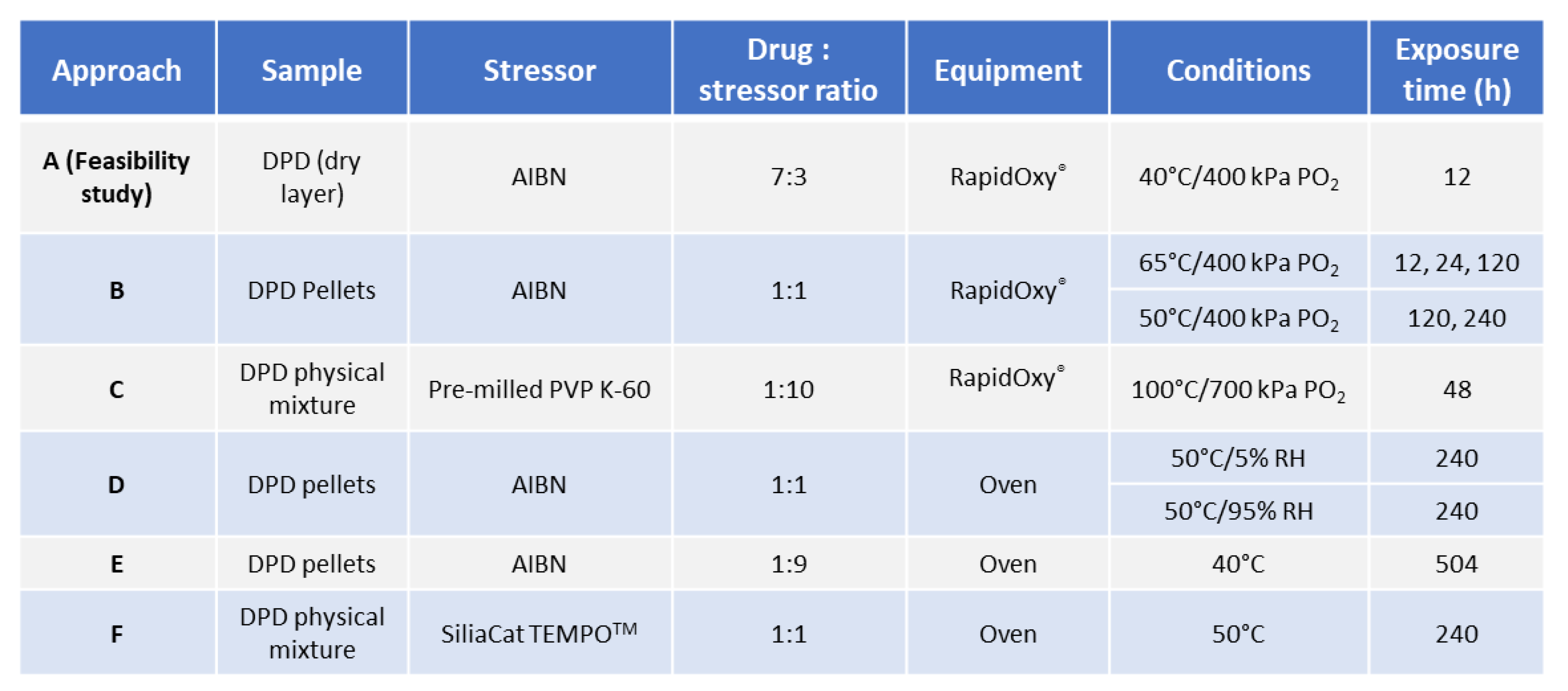
2.2.1. General Workflow for Testing Autoxidation
2.2.2. Investigation of Solid−state Degradation in DPD Using RapidOxy®
2.2.3. Investigation of Solid−state Degradation in DPD Using an Oven
2.2.4. Liquid Chromatography
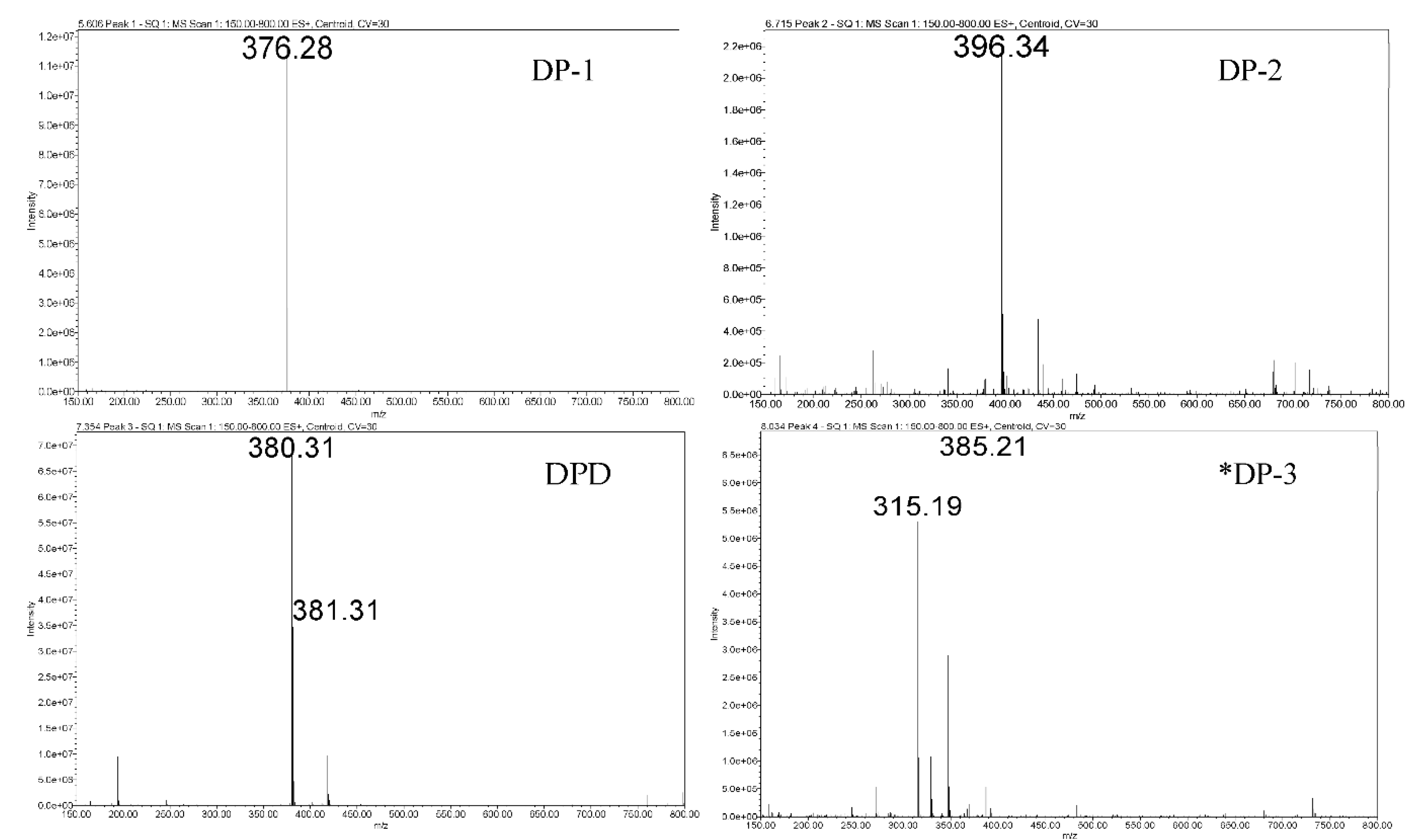
2.2.5. Liquid Chromatography−Single Quadrupole Mass Spectrometry (LC−SqD MS)
3. Results
3.1. Approach A−Feasibility Study
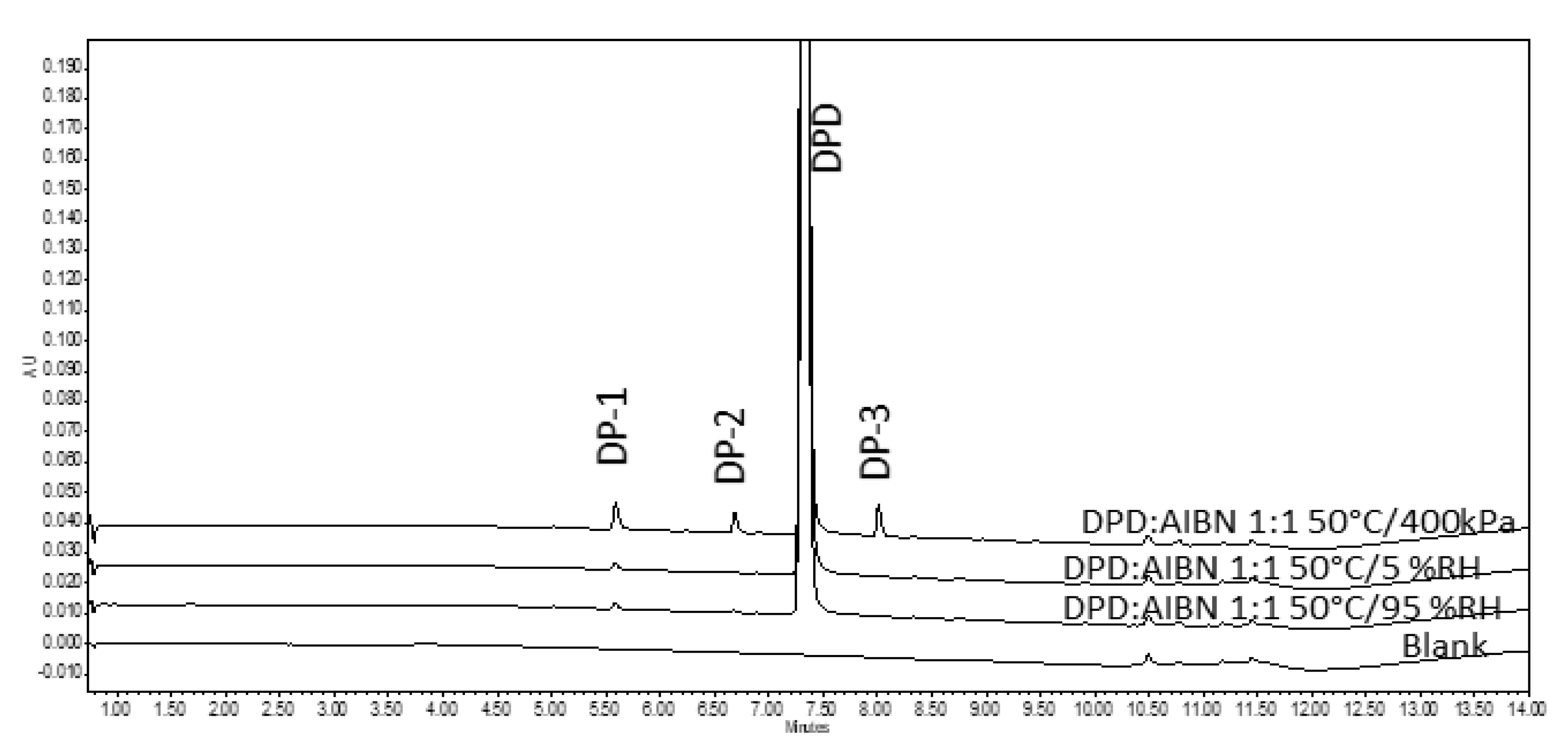
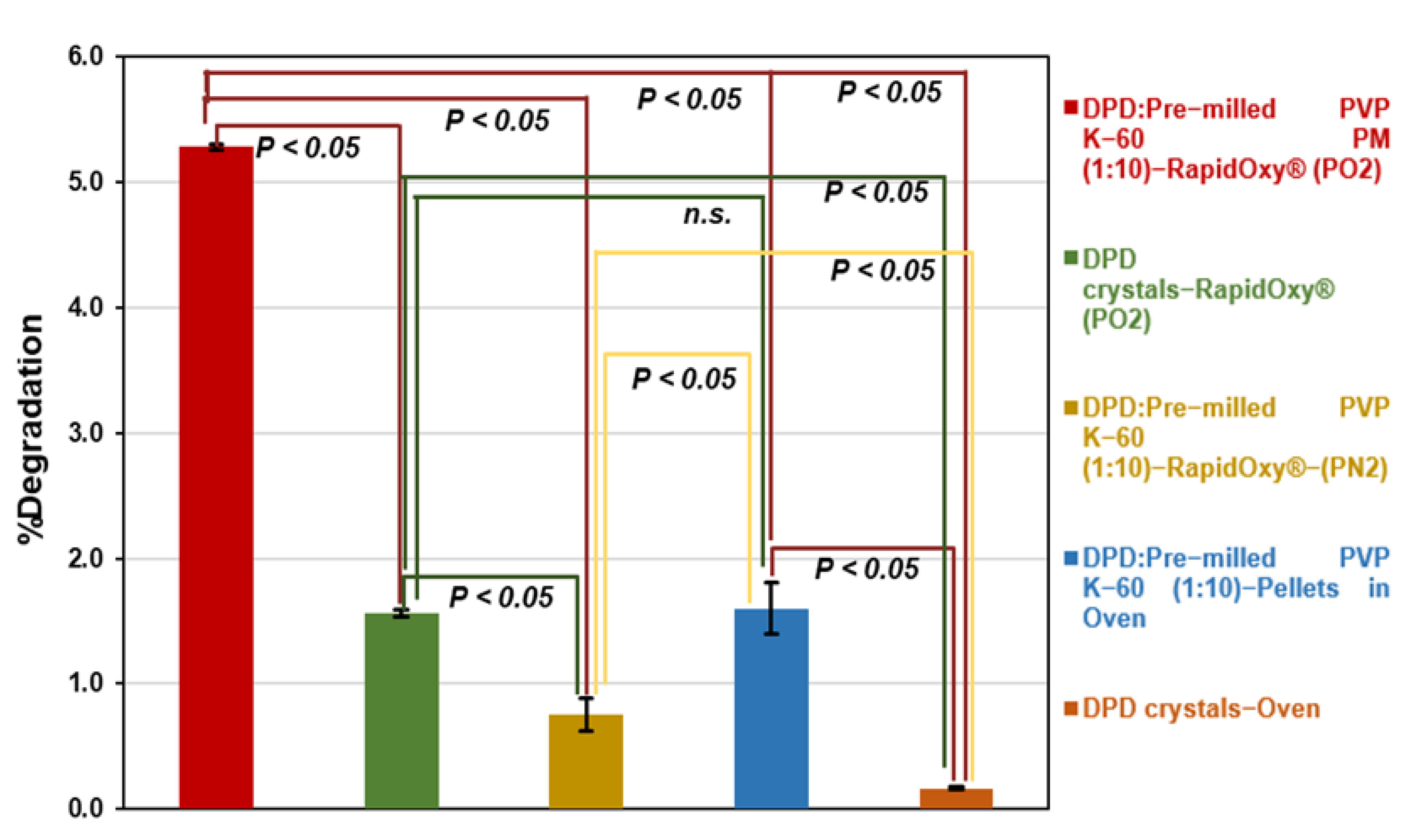
3.2. Approaches−B, D, E, and F
3.2.1. Assessment of Pellets Exposed in RapidOxy®
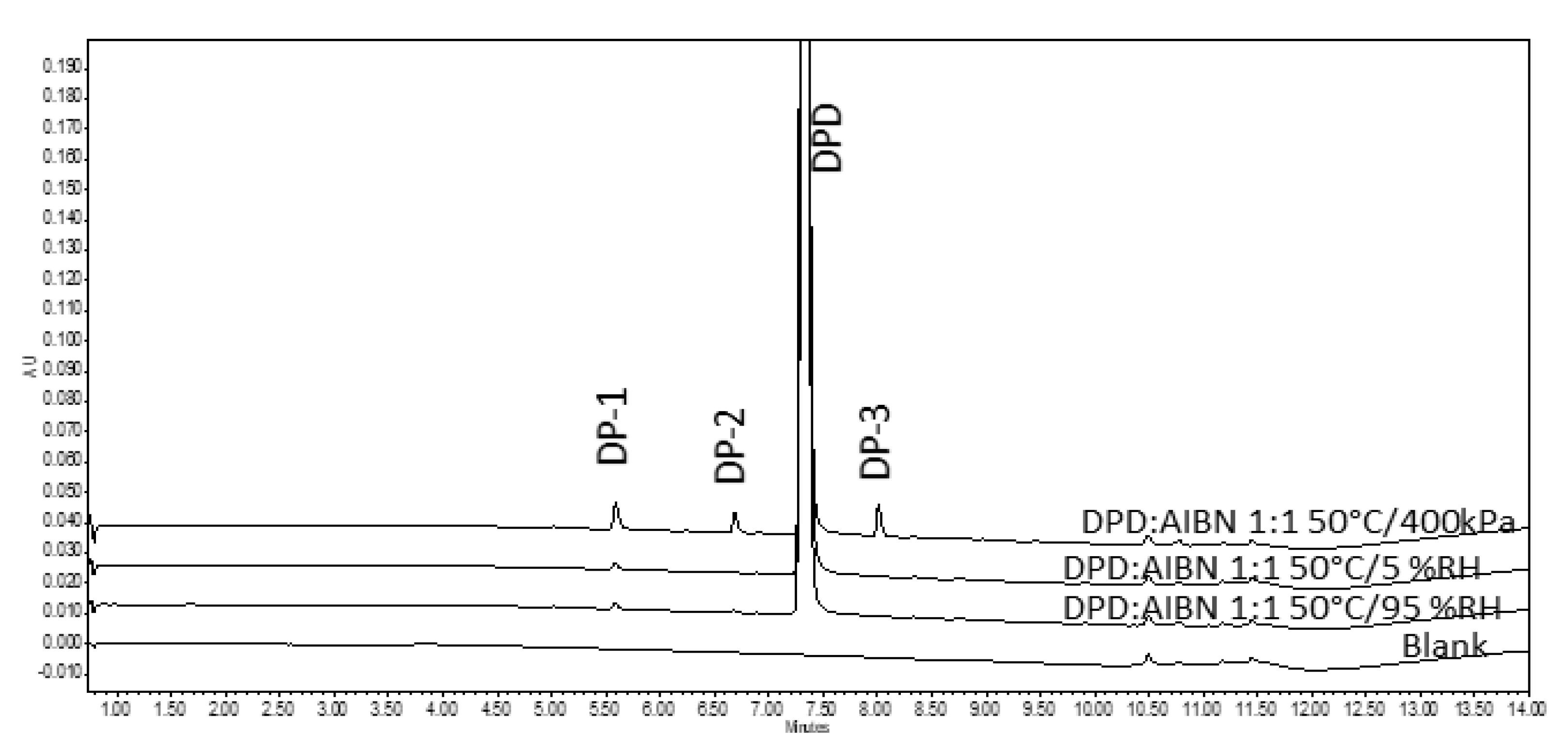
3.2.2. Assessment of Pellets Exposed to Oven
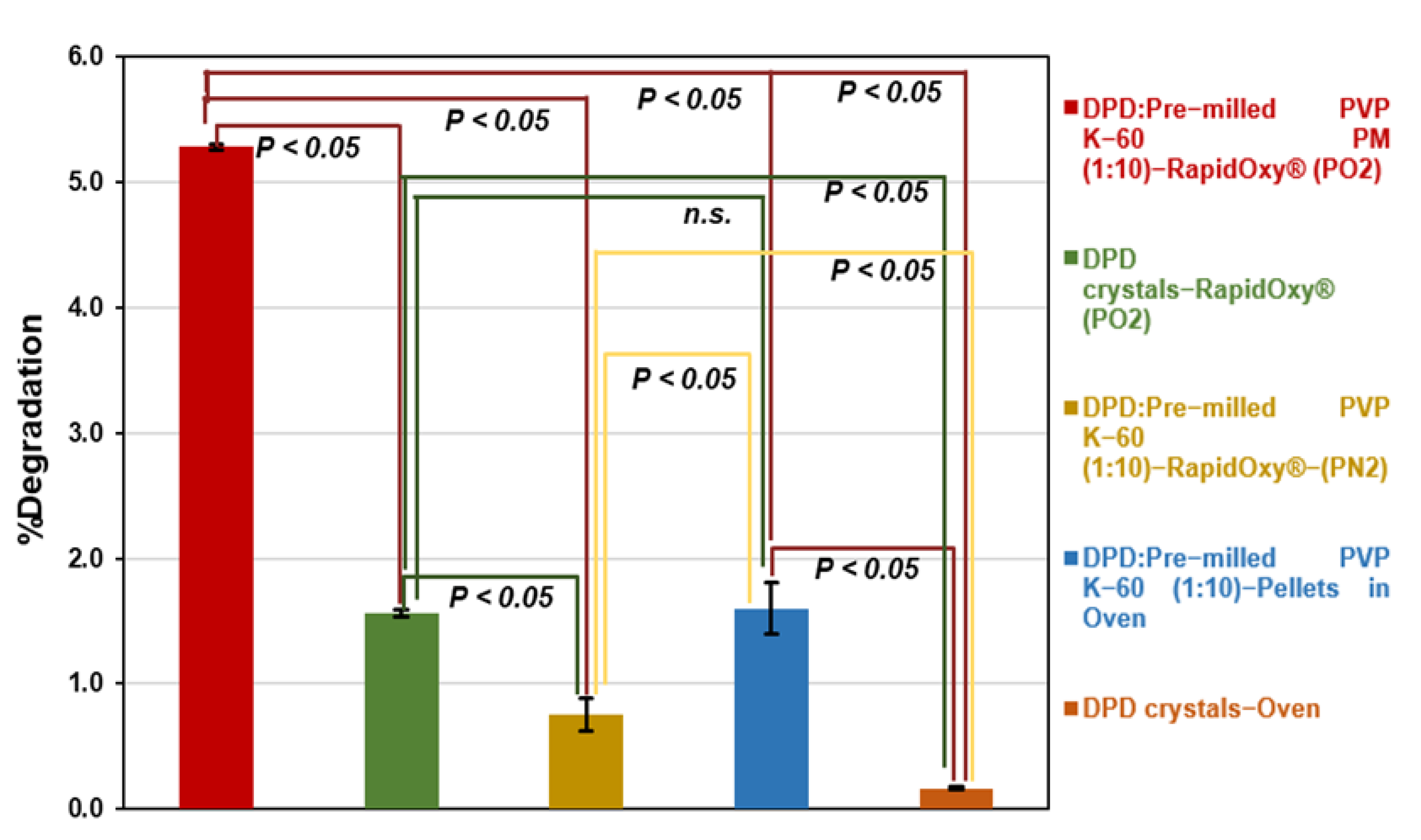
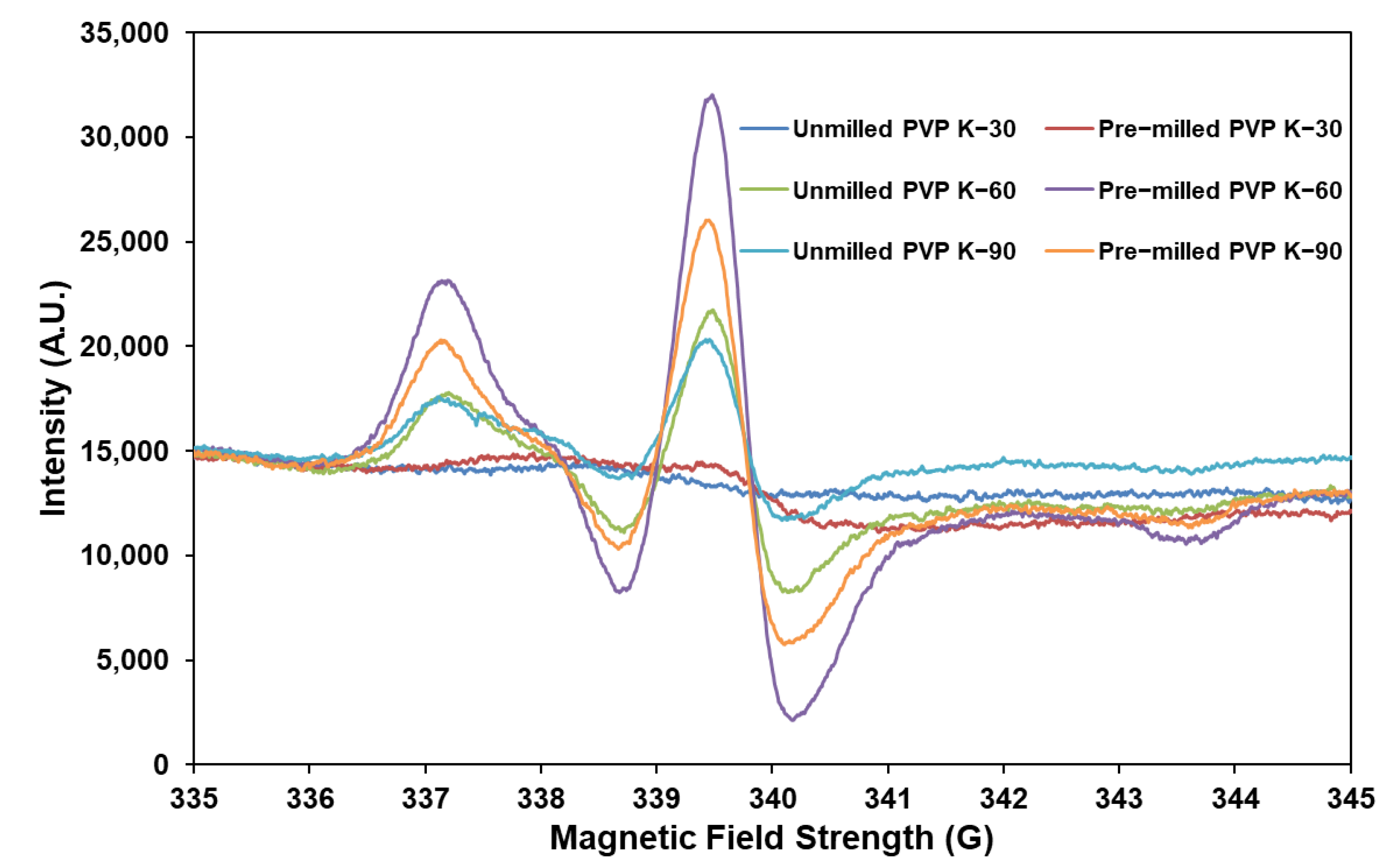
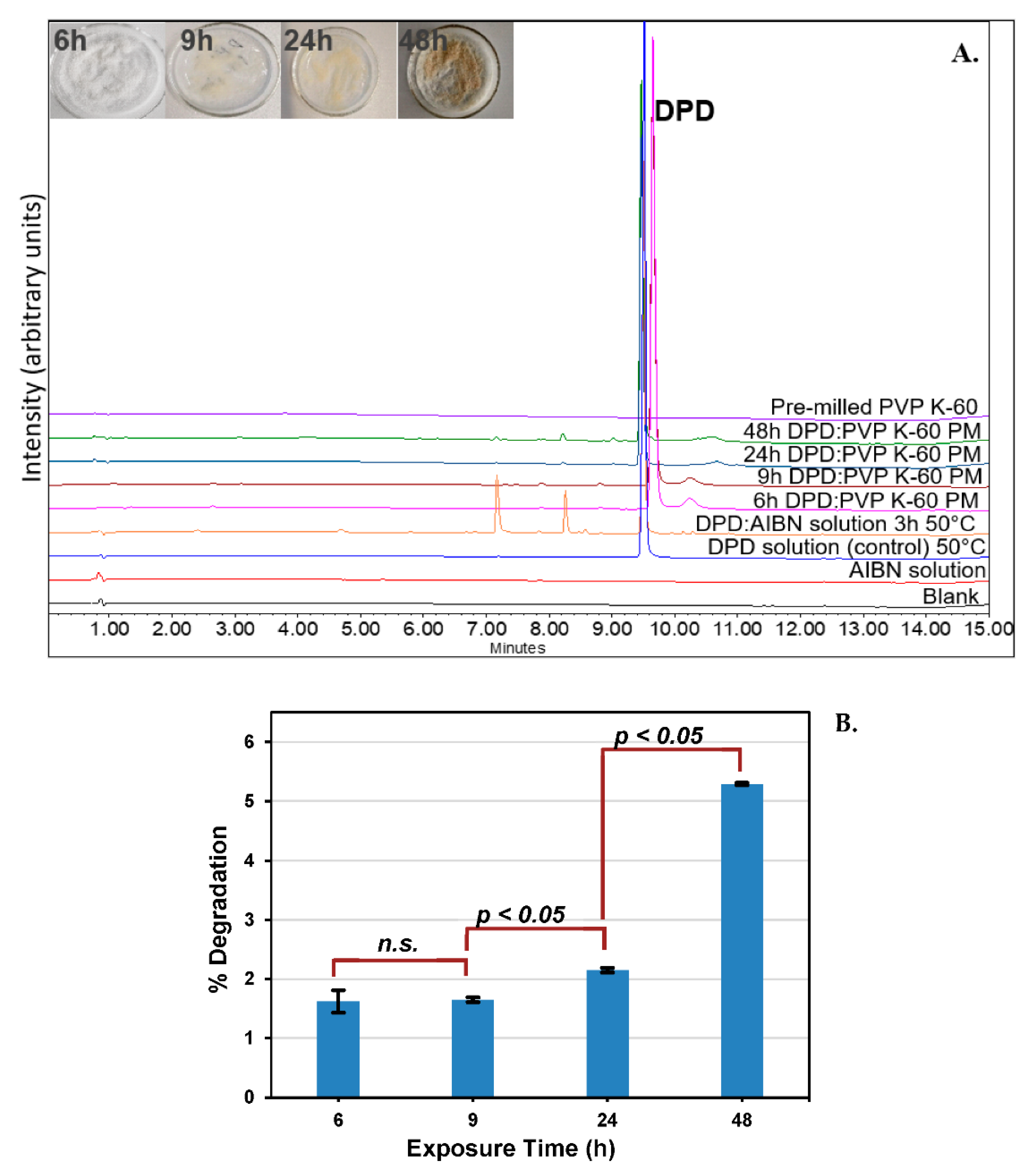
3.2.3. Approach C (Binary Mixture of Pre−milled PVP K−60 and DPD (10:1))
4. Discussion
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hovorka, S.W.; Schöneich, C. Oxidative Degradation of Pharmaceuticals: Theory, Mechanisms and Inhibition. J. Pharm. Sci. 2001, 90, 253–269. [Google Scholar] [CrossRef]
- Roman, R. Stability Kinetics. In Pharmaceutical Dosage Foms: Tablets, Volume 1:Unit Operations and Mechanical Properties; Augsburger, L.L., Hoag, S.W., Eds.; Informa Healthcare, USA, Inc.: New York, NY, USA, 2008; pp. 485–517. [Google Scholar]
- Lienard, P.; Gavartin, J.; Boccardi, G.; Meunier, M. Predicting drug substances autoxidation. Pharm. Res. 2015, 32, 300–310. [Google Scholar] [CrossRef] [PubMed]
- WHO Expert Committee on Specifications for Pharmaceutical Preparations. Report Stability testing of active pharmaceutical ingredients and finished pharmaceutical products. Annex 10 2018, 52, 309–352. [Google Scholar]
- Modhave, D.; Barrios, B.; Paudel, A. PVP-H2O2 Complex as a New Stressor for the Accelerated Oxidation Study of Pharmaceutical Solids. Pharmaceutics 2019, 11, 457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Correa, P.E.; Hardy, G.; Riley, D.P. Selective Autoxidation of Electron-Rich Substrates under Elevated Oxygen Pressures. J. Org. Chem. 1988, 53, 1695–1702. [Google Scholar] [CrossRef]
- Marteau, C.; Ruyffelaere, F.; Aubry, J.M.; Penverne, C.; Favier, D.; Nardello-Rataj, V. Oxidative degradation of fragrant aldehydes. Autoxidation by molecular oxygen. Tetrahedron 2013, 69, 2268–2275. [Google Scholar] [CrossRef]
- Reid, D.L.; Calvitt, C.J.; Zell, M.T.; Miller, K.G.; Kingsmill, C.A. Early prediction of pharmaceutical oxidation pathways by computational chemistry and forced degradation. Pharm. Res. 2004, 21, 1708–1717. [Google Scholar] [CrossRef]
- Boccardi, G. Autoxidation of drugs: Prediction of degradation impurities from results of reaction with radical chain initiators. Farmaco 1994, 49, 431–435. [Google Scholar]
- Baertschi, S.W.; Jansen, P.J.; Alsante, K.M.; Santafianos, D.; Harmon, P.; Boccardi, G. Chapter 2. Stress Testing: A predictive tool. Chapter 3. Stress Testing: The chemistry of drug degradation. Chapter 6. Oxidative susceptibility testing. In Pharmaceutical Stress Testing- Predicting Drug Degradation; Baertschi, S.W., Alsante, K.M., Reed, R.A., Eds.; Informa Healthcare, USA, Inc.: London, UK, 2011; pp. 23, 104–105, 169. ISBN 9781616310011. [Google Scholar]
- Betigeri, S.; Thakur, A.; Raghavan, K. Use of 2,2′-Azobis(2-amidinopropane) dihydrochloride as a reagent tool for evaluation of oxidative stability of drugs. Pharm. Res. 2005, 22, 310–317. [Google Scholar] [CrossRef]
- Boccardi, G.; Deleuze, C. Autoxidation of Tetrazepam in Tablets: Prediction of Degradation Impurities from the Oxidative Behavior in Solution. J. Pharm. Sci. 1992, 81, 183–185. [Google Scholar]
- Grinberg Dana, A.; Wu, H.; Ranasinghe, D.S.; Pickard, F.C.; Wood, G.P.F.; Zelesky, T.; Sluggett, G.W.; Mustakis, J.; Green, W.H. Kinetic Modeling of API Oxidation: (1) The AIBN/H2O/CH3OH Radical “Soup”. Mol. Pharm. 2021, 18, 3037–3049. [Google Scholar] [CrossRef] [PubMed]
- Ku, M.S. Chapter 4. Preformulation Consideration For Drugs in Oral CR Formulation. In Oral Controlled Release Formulation Design And Drug Delivery: Theory and Practice; Wen, H., Park, K., Eds.; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2010; pp. 52–55. [Google Scholar]
- Caron, S.; Dugger, R.W.; Ruggeri, S.G.; Ragan, J.A.; Brown Ripin, D.H. Large-scale oxidations in the pharmaceutical industry. Chem. Rev. 2006, 106, 2943–2989. [Google Scholar] [CrossRef] [PubMed]
- Carlsson, D.O.; Hua, K.; Forsgren, J.; Mihranyan, A. Aspirin degradation in surface-charged TEMPO-oxidized mesoporous crystalline nanocellulose. Int. J. Pharm. 2014, 461, 74–81. [Google Scholar] [CrossRef] [PubMed]
- Byrn, S.R.; Xu, W.; Newman, A.W. Chemical reactivity in solid-state pharmaceuticals: Formulation implications. Adv. Drug Deliv. Rev. 2001, 48, 115–136. [Google Scholar] [CrossRef]
- Modhave, D.; Laggner, P.; Brunsteiner, M.; Paudel, A. Solid-State Reactivity of Mechano-Activated Simvastatin: Atypical Relation to Powder Crystallinity. J. Pharm. Sci. 2019, 108, 3272–3280. [Google Scholar] [CrossRef]
- Li, M. Chapter 3. Oxidative degradation. In Organic Chemistry of Drug Degradation; Thurston, D., Ed.; Drug Discovery Series No. 29; Royal Society of Chemistry: Cambridge, UK, 2012; pp. 54–56. ISBN 978-1-84973-421-9. [Google Scholar]
- European Pharmacopoeia, 9th edition. Available online: https://pheur.edqm.eu/home (accessed on 2 December 2021).
- Raijada, D.K.; Prasad, B.; Paudel, A.; Shah, R.P.; Singh, S. Characterization of degradation products of amorphous and polymorphic forms of clopidogrel bisulphate under solid state stress conditions. J. Pharm. Biomed. Anal. 2010, 52, 332–344. [Google Scholar] [CrossRef]
- Lazár, M.; Ambrovič, P.; Mikovič, J. Thermal Decomposition of azo-bis-isobutyronitrile in the solid phase. J. Therm. Anal. 1973, 5, 415–425. [Google Scholar] [CrossRef]
- Li, X.R.; Wang, X.L.; Koseki, H. Study on thermal decomposition characteristics of AIBN. J. Hazard. Mater. 2008, 159, 13–18. [Google Scholar] [CrossRef]
- Byrn, S.R. Solid State Organic Chemistry and Drug Stability. Annu. Rep. Med. Chem. 1985, 20, 287–294. [Google Scholar] [CrossRef]
- Scott, G. Initiation processes in polymer degradation. Polym. Degrad. Stab. 1995, 48, 315–324. [Google Scholar] [CrossRef]
- Berkowitz, J.; Ellison, G.B.; Gutman, D. Three methods to measure RH bond energies. J. Phys. Chem. 1994, 98, 2744–2765. [Google Scholar] [CrossRef] [Green Version]
- Sharp, T.R. Calculated carbon-hydrogen bond dissociation enthalpies for predicting oxidative susceptibility of drug substance molecules. Int. J. Pharm. 2011, 418, 304–317. [Google Scholar] [CrossRef] [PubMed]
- Kieffer, J.; Brémond, É.; Lienard, P.; Boccardi, G. In silico assessment of drug substances chemical stability. J. Mol. Struct. THEOCHEM 2010, 954, 75–79. [Google Scholar] [CrossRef]
- Andersson, T.; Broo, A.; Evertsson, E. Prediction of Drug Candidates ’ Sensitivity Toward Autoxidation: Computational Estimation of C–H Dissociation Energies of Carbon-Centered Radicals. J. Pharm. Sci. 2014, 103, 1949–1955. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Approach | Method | AIBN:DPD Ratio | Conditions | %Degradation (100-%Area of DPs) |
|---|---|---|---|---|
| A | RapidOxy® | 1:1 | 40 °C/12 h/400 kPa | NIL |
| B * | RapidOxy® | 1:1 | 65 °C/12 h/400 kPa | 0.39 ± 0.14 |
| 65 °C/24 h/400 kPa | 0.42 ± 0.10 | |||
| 65 °C/120 h/400 kPa | 1.57 | |||
| 50 °C/120 h/400 kPa | 0.67 | |||
| 50 °C/240 h/400 kPa | 1.71 | |||
| D | Oven | 1:1 | 50 °C/5% RH/240 h | 0.34 ± 0.11 |
| 50 °C/95% RH/240 h | 0.43 ± 0.08 | |||
| E | Oven | 9:1 | 40 °C/5% RH/504 h | 0.51 ± 0.01 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Iyer, J.; Saraf, I.; Ray, A.; Brunsteiner, M.; Paudel, A. Assessment of Diverse Solid−State Accelerated Autoxidation Methods for Droperidol. Pharmaceutics 2022, 14, 1114. https://doi.org/10.3390/pharmaceutics14061114
Iyer J, Saraf I, Ray A, Brunsteiner M, Paudel A. Assessment of Diverse Solid−State Accelerated Autoxidation Methods for Droperidol. Pharmaceutics. 2022; 14(6):1114. https://doi.org/10.3390/pharmaceutics14061114
Chicago/Turabian StyleIyer, Jayant, Isha Saraf, Andrew Ray, Michael Brunsteiner, and Amrit Paudel. 2022. "Assessment of Diverse Solid−State Accelerated Autoxidation Methods for Droperidol" Pharmaceutics 14, no. 6: 1114. https://doi.org/10.3390/pharmaceutics14061114
APA StyleIyer, J., Saraf, I., Ray, A., Brunsteiner, M., & Paudel, A. (2022). Assessment of Diverse Solid−State Accelerated Autoxidation Methods for Droperidol. Pharmaceutics, 14(6), 1114. https://doi.org/10.3390/pharmaceutics14061114