
A Comparison of Evans Blue and 4-(p-Iodophenyl)butyryl Albumin Binding Moieties on an Integrin αvβ6 Binding Peptide



Abstract
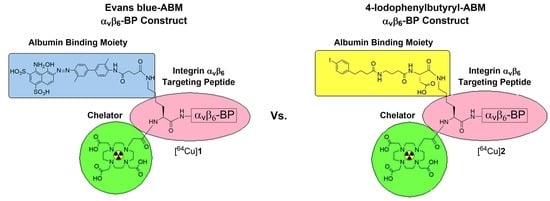
:
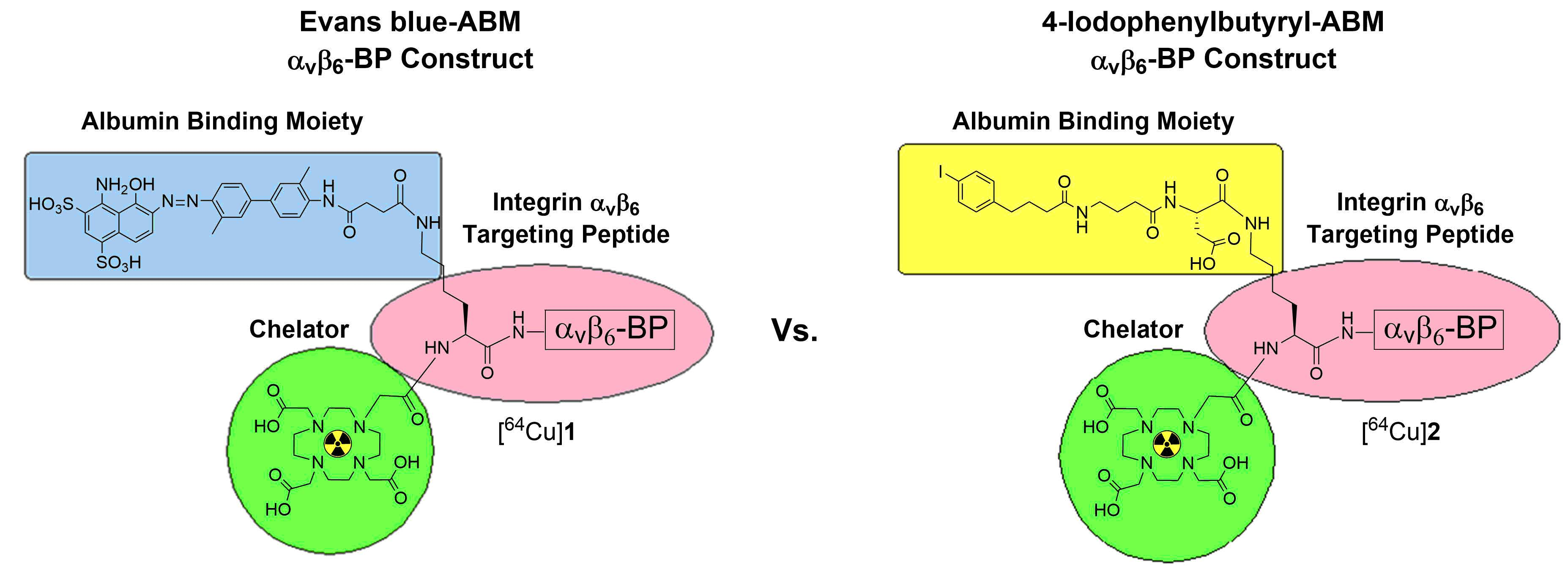{kind=link}
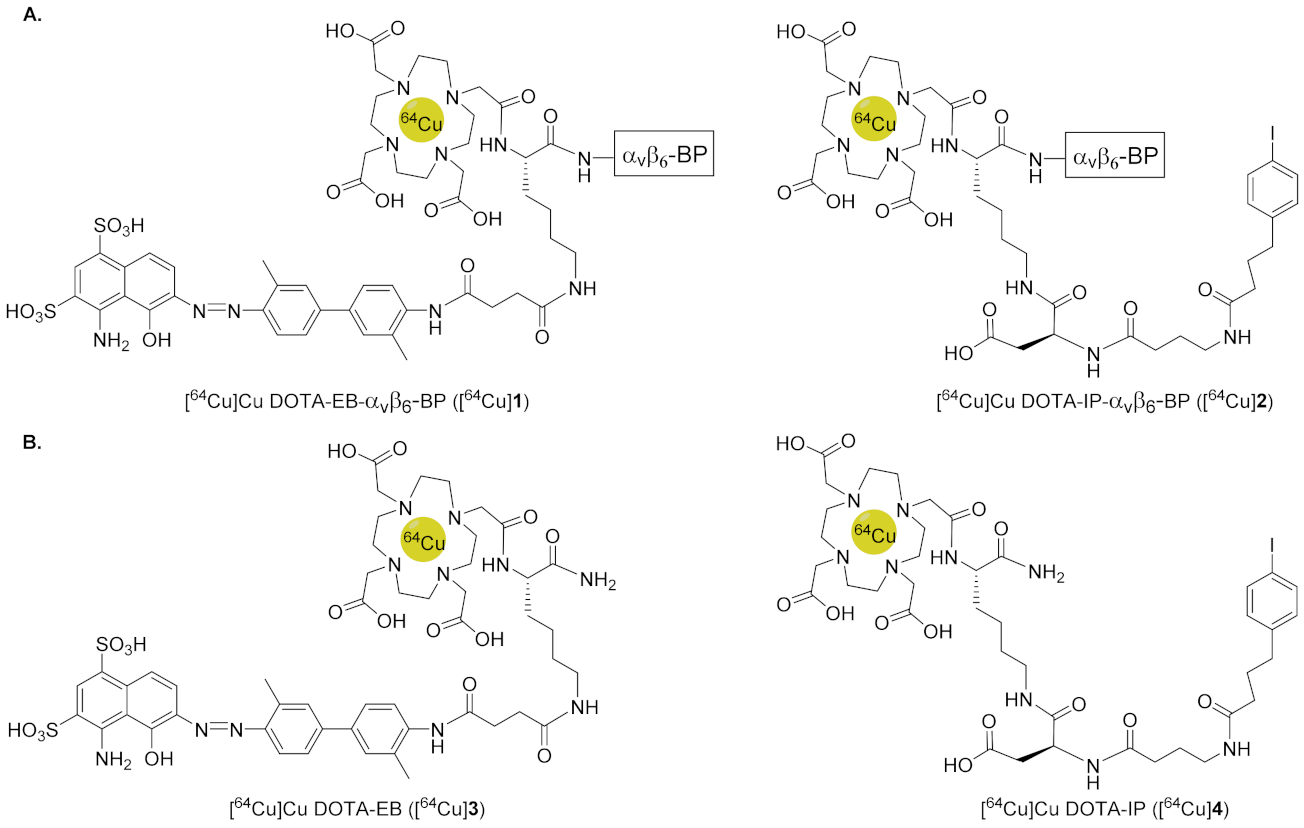{kind=link}
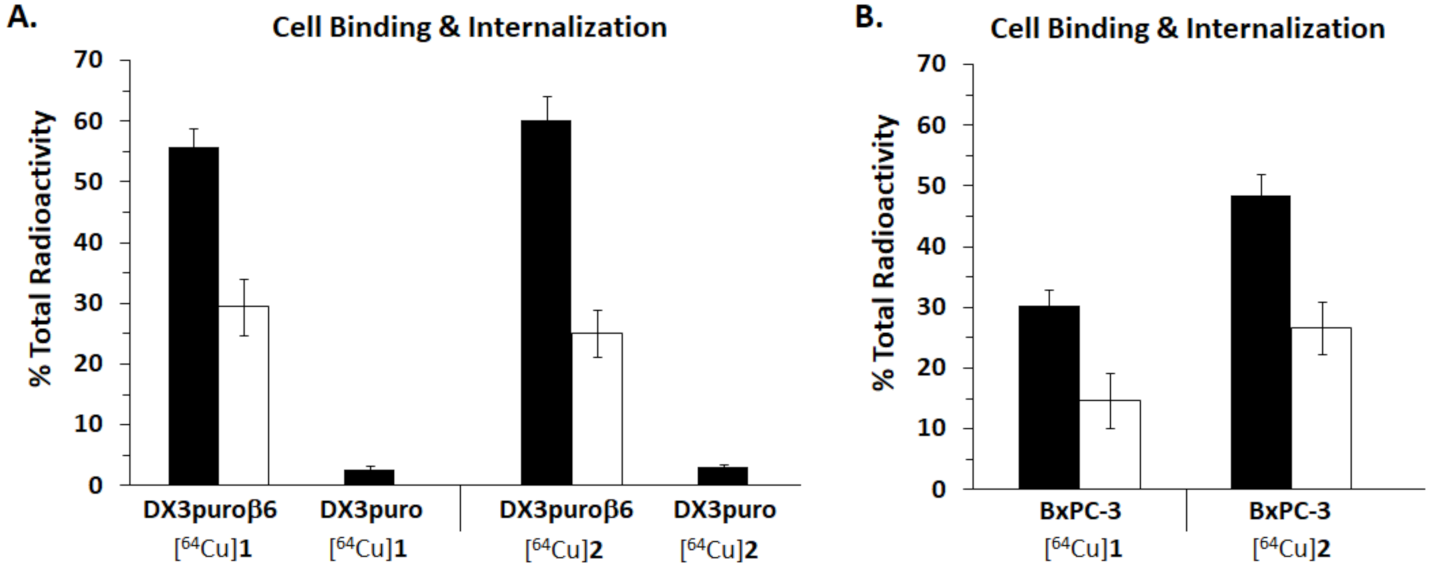{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Materials and General Information
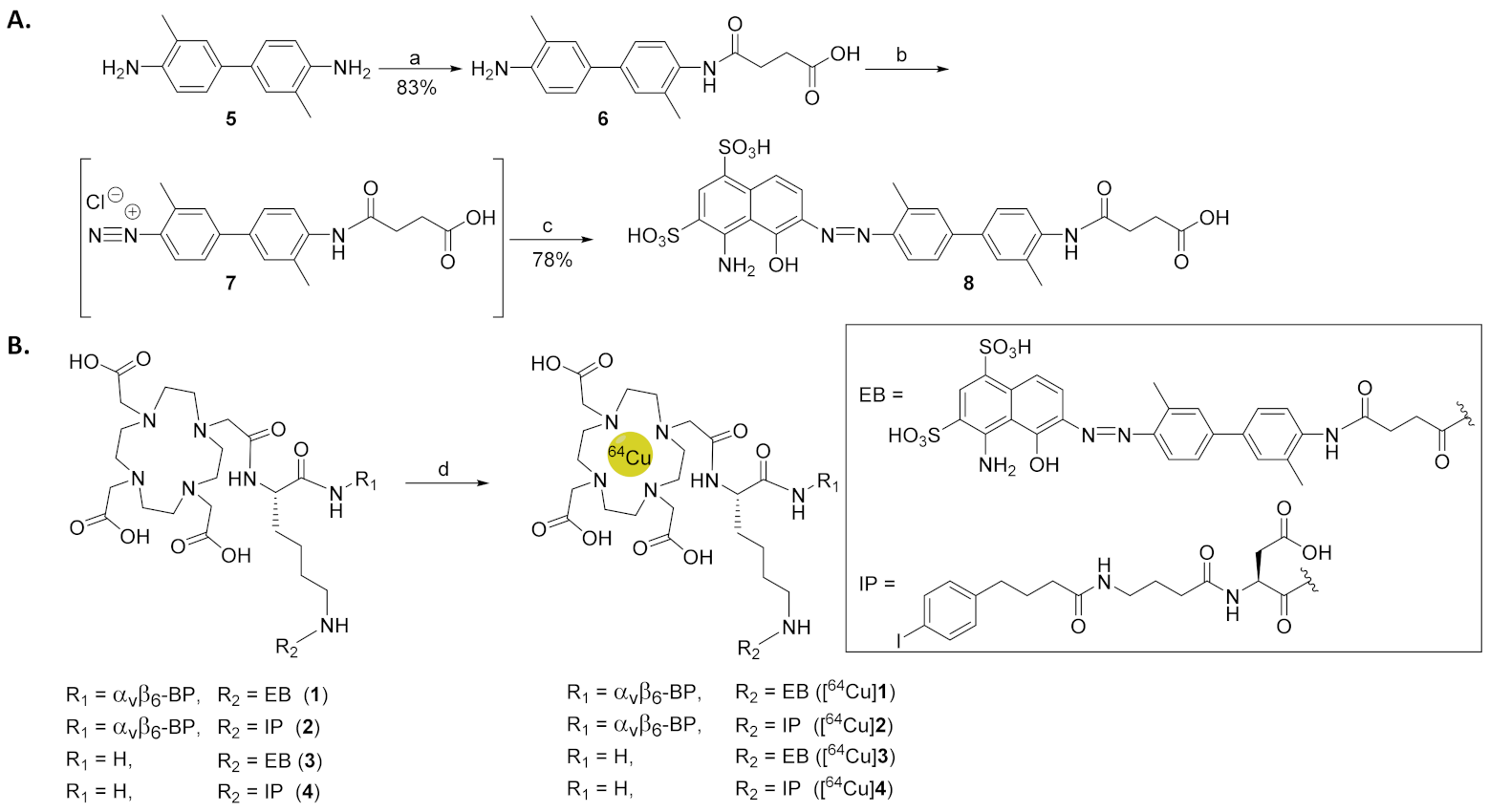
2.2. Synthesis of EB-ABM 8
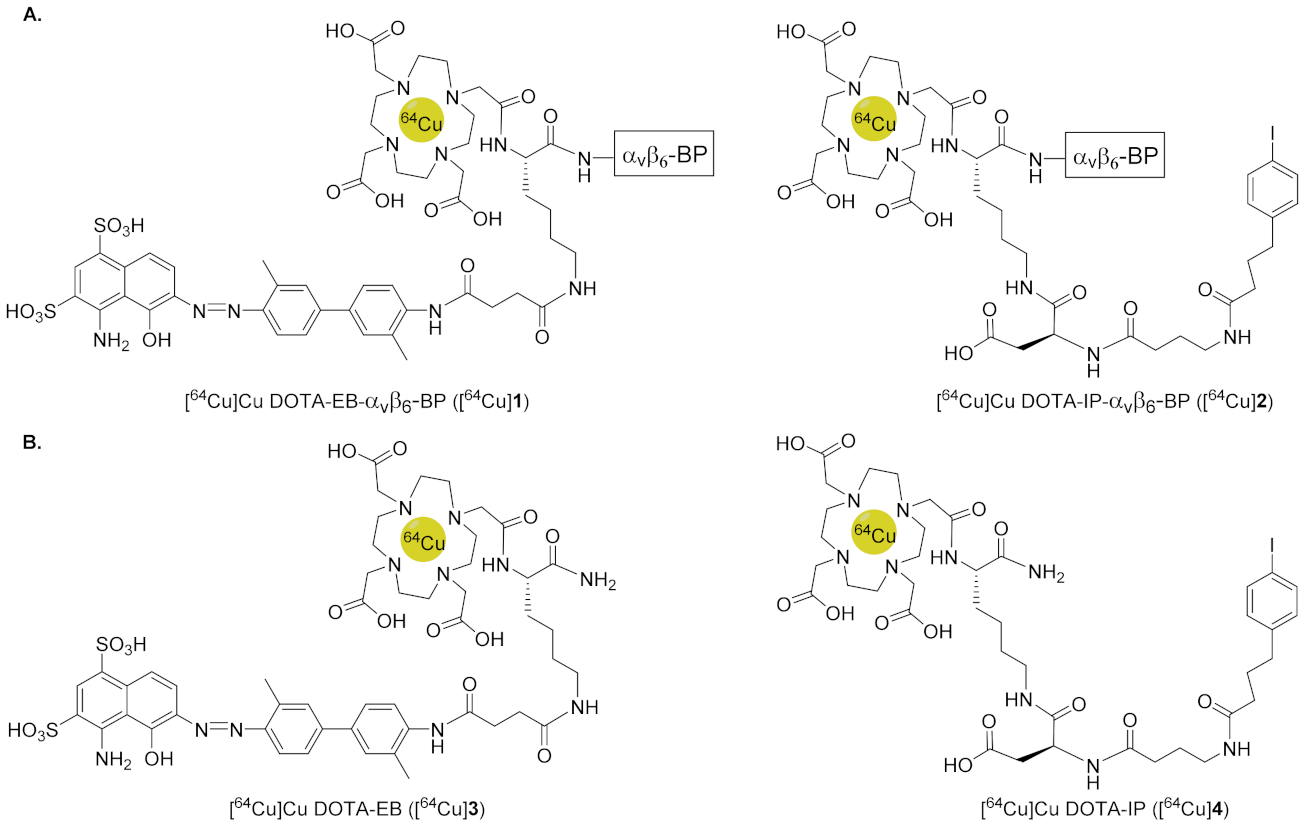
2.3. Synthesis of DOTA-ABM-αvβ6-BPs 1 and 2
2.4. Synthesis of Non-Targeting ABMs 3 and 4
2.5. Radiochemical Synthesis of [64Cu]1–4
2.6. Integrin αvβ6 Affinity ELISA
2.7. Cell Binding and Internalization Assay
2.8. Human and Mouse Serum Binding Assay and Stability Assay
2.9. Biodistribution
2.10. Blocking Biodistribution
2.11. PET-Imaging
2.12. Statistical Analysis
3. Results
3.1. Synthesis of EB-ABM 8
3.2. Synthesis and Radiochemical Synthesis of [64Cu]1–4
3.3. Integrin αvβ6 Affinity ELISA
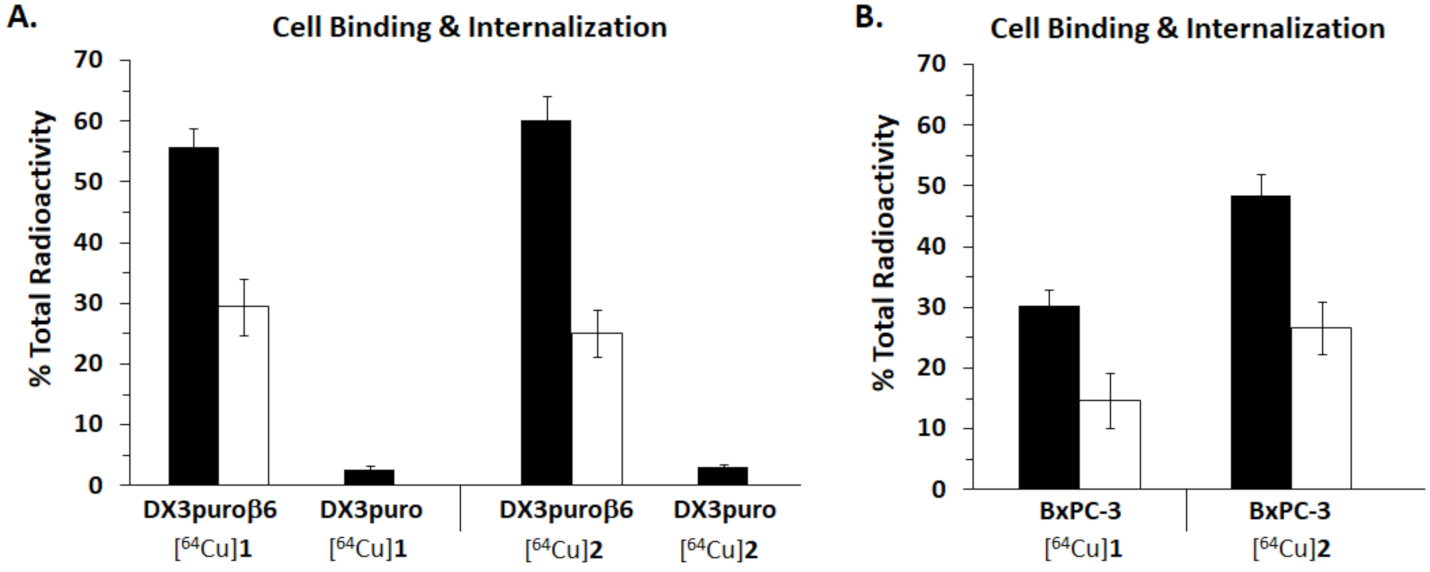
3.4. Cell Binding and Internalization Assay
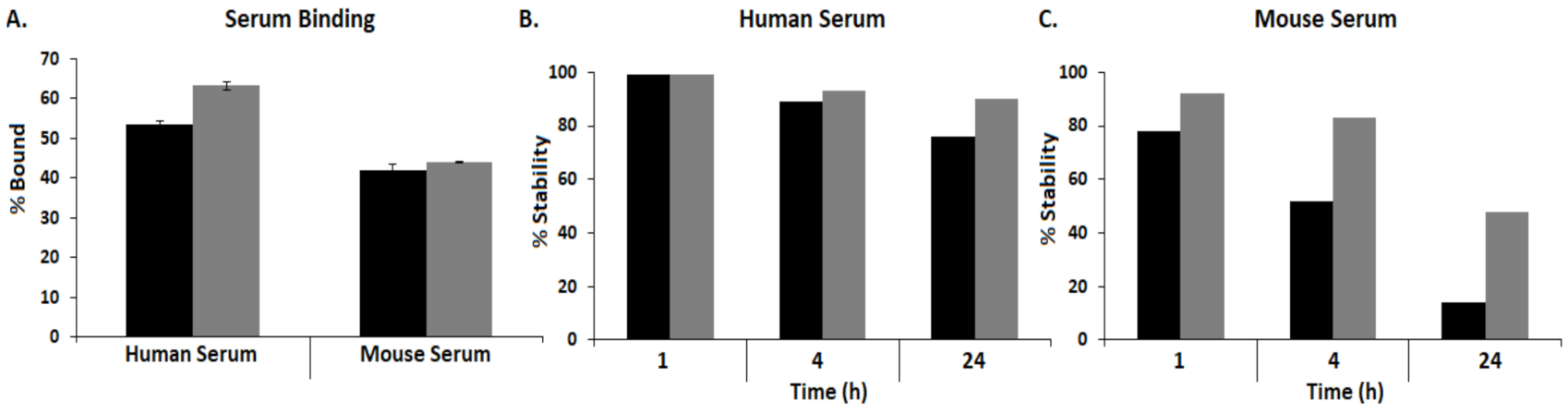
3.5. Human and Mouse Serum Binding Assay and Stability Assay
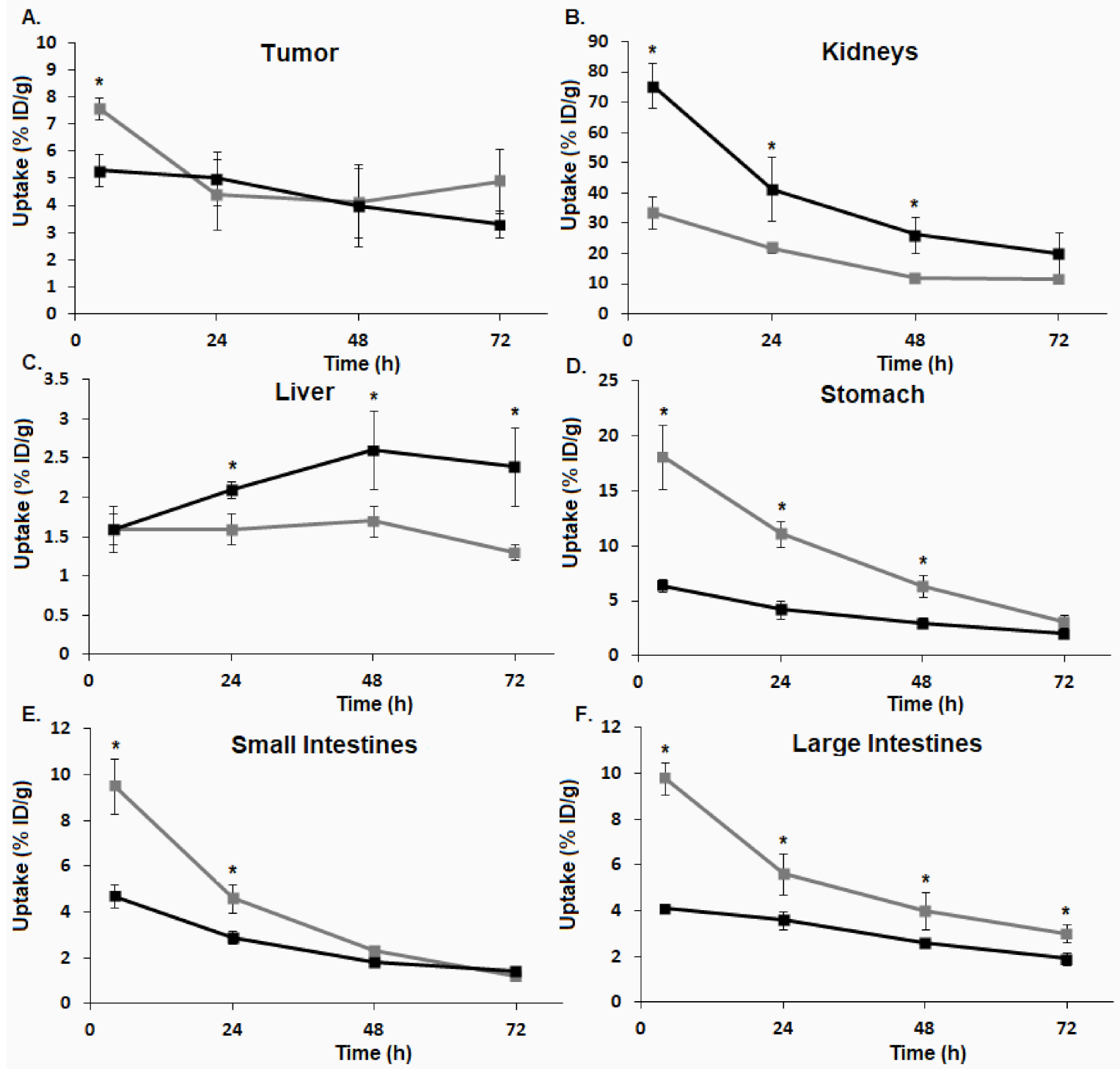
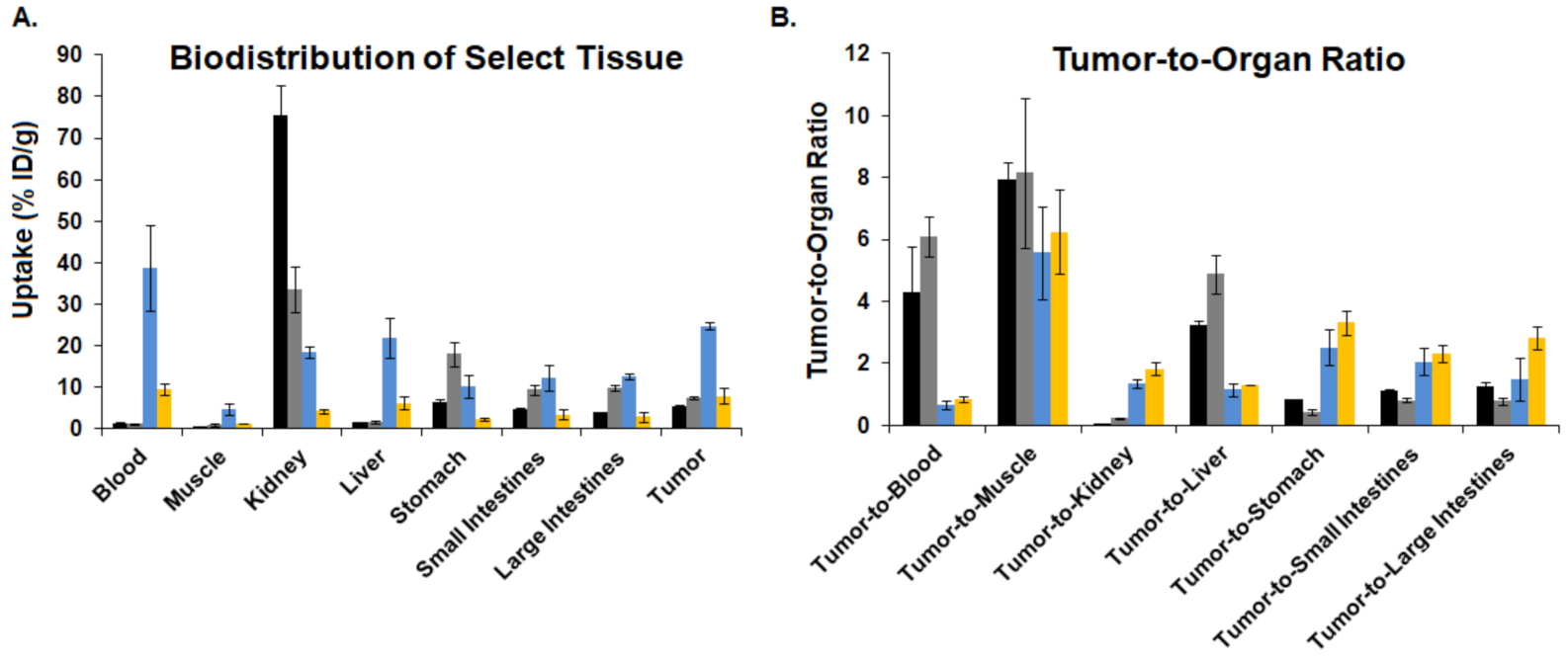
3.6. Biodistribution
3.7. Blocking Biodistribution
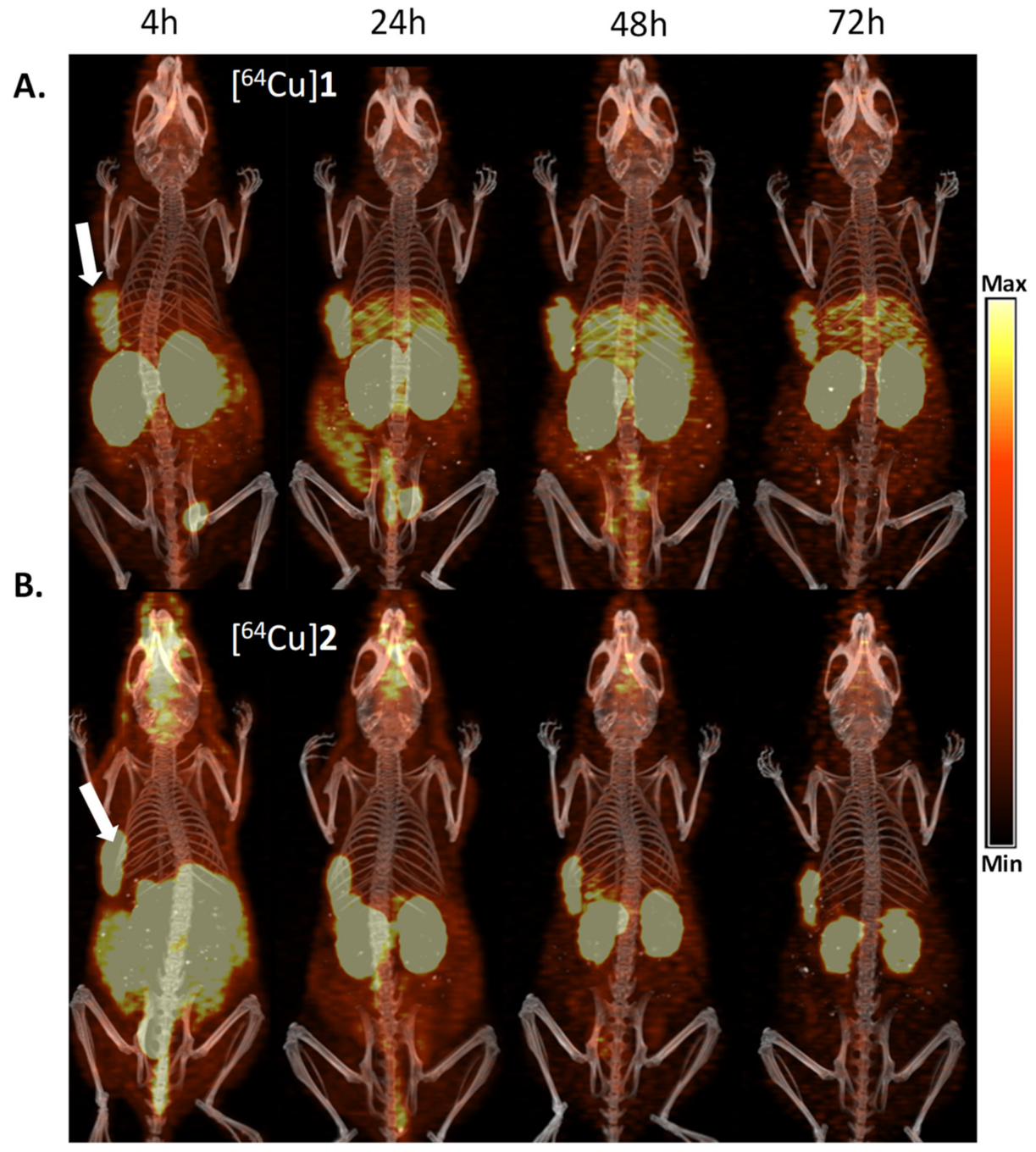
3.8. PET Imaging
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Davis, R.A.; Hausner, S.H.; Sutcliffe, J.L. Peptides as radiopharmaceutical vectors. In Radiopharmaceutical Chemistry; Lewis, J.S., Windhorst, A.D., Zeglis, B.M., Eds.; Springer International Publishing: Cham, Switzerland, 2019; pp. 137–162. [Google Scholar]
- Trier, N.; Hansen, P.; Houen, G. Peptides, antibodies, peptide antibodies and more. Int. J. Mol. Sci. 2019, 20, 6289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kręcisz, P.; Czarnecka, K.; Królicki, L.; Mikiciuk-Olasik, E.; Szymański, P. Radiolabeled peptides and antibodies in medicine. Bioconjugate Chem. 2021, 32, 25–42. [Google Scholar] [CrossRef] [PubMed]
- Zorzi, A.; Linciano, S.; Angelini, A. Non-covalent albumin-binding ligands for extending the circulating half-life of small biotherapeutics. Med. Chem. Commun. 2019, 10, 1068–1081. [Google Scholar] [CrossRef] [PubMed]
- Zorzi, A.; Middendorp, S.J.; Wilbs, J.; Deyle, K.; Heinis, C. Acylated heptapeptide binds albumin with high affinity and application as tag furnishes long-acting peptides. Nat. Commun. 2017, 8, 16092. [Google Scholar] [CrossRef] [PubMed]
- Tian, R.; Jacobson, O.; Niu, G.; Kiesewetter, D.O.; Wang, Z.; Zhu, G.; Ma, Y.; Liu, G.; Chen, X. Evans blue attachment enhances somatostatin receptor subtype-2 imaging and radiotherapy. Theranostics 2018, 8, 735–745. [Google Scholar] [CrossRef]
- Chen, H.; Wang, G.; Lang, L.; Jacobson, O.; Kiesewetter, D.O.; Liu, Y.; Ma, Y.; Zhang, X.; Wu, H.; Zhu, L.; et al. Chemical conjugation of Evans blue derivative: A strategy to develop long-acting therapeutics through albumin binding. Theranostics 2016, 6, 243–253. [Google Scholar] [CrossRef]
- Dennis, M.S.; Zhang, M.; Meng, G.Y.; Kadkhodayan, M.; Kirchhofer, D.; Combs, D.; Damico, L.A. Albumin binding as a general strategy for improving the pharmacokinetics of proteins. J. Biol. Chem. 2002, 277, 35035–35043. [Google Scholar] [CrossRef] [Green Version]
- Jacobson, O.; Kiesewetter, D.O.; Chen, X. Albumin-binding Evans blue derivatives for diagnostic imaging and production of long-acting therapeutics. Bioconjugate Chem. 2016, 27, 2239–2247. [Google Scholar] [CrossRef]
- Lau, J.; Jacobson, O.; Niu, G.; Lin, K.-S.; Bénard, F.; Chen, X. Bench to bedside: Albumin binders for improved cancer radioligand therapies. Bioconjugate Chem. 2019, 30, 487–502. [Google Scholar] [CrossRef]
- Brandt, M.; Cardinale, J.; Giammei, C.; Guarrochena, X.; Happl, B.; Jouini, N.; Mindt, T.L. Mini-review: Targeted radiopharmaceuticals incorporating reversible, low molecular weight albumin binders. Nucl. Med. Biol. 2019, 70, 46–52. [Google Scholar] [CrossRef]
- Gao, H.; Luo, C.; Yang, G.; Du, S.; Li, X.; Zhao, H.; Shi, J.; Wang, F. Improved in vivo targeting capability and pharmacokinetics of 99mTc-Labeled isoDGR by dimerization and albumin-binding for glioma imaging. Bioconjugate Chem. 2019, 30, 2038–2048. [Google Scholar] [CrossRef] [PubMed]
- Cheng, T.-L.; Chuang, K.-H.; Chen, B.-M.; Roffler, S.R. Analytical measurement of PEGylated molecules. Bioconjugate Chem. 2012, 23, 881–899. [Google Scholar] [CrossRef] [PubMed]
- Hausner, S.H.; Bauer, N.; Hu, L.Y.; Knight, L.M.; Sutcliffe, J.L. The effect of bi-terminal PEGylation of an integrin αvβ6–targeted 18F peptide on pharmacokinetics and tumor uptake. J. Nucl. Med. 2015, 56, 784–790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Müller, C.; Farkas, R.; Borgna, F.; Schmid, R.M.; Benešová, M.; Schibli, R. Synthesis, radiolabeling, and characterization of plasma protein-binding ligands: Potential tools for modulation of the pharmacokinetic properties of (radio) pharmaceuticals. Bioconjugate Chem. 2017, 28, 2372–2383. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Chen, X. Simple bioconjugate chemistry serves great clinical advances: Albumin as a versatile platform for diagnosis and precision therapy. Chem. Soc. Rev. 2016, 45, 1432–1456. [Google Scholar] [CrossRef] [Green Version]
- Niu, G.; Lang, L.; Kiesewetter, D.O.; Ma, Y.; Sun, Z.; Guo, N.; Guo, J.; Wu, C.; Chen, X. In vivo labeling of serum albumin for PET. J. Nucl. Med. 2014, 55, 1150–1156. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Jacobson, O.; Niu, G.; Weiss, I.D.; Kiesewetter, D.O.; Liu, Y.; Ma, Y.; Wu, H.; Chen, X. Novel “add-on” molecule based on Evans blue confers superior pharmacokinetics and transforms drugs to theranostic agents. J. Nucl. Med. 2017, 58, 590–597. [Google Scholar] [CrossRef] [Green Version]
- Ehlerding, E.B.; Lan, X.; Cai, W. Albumin hitchhiking” with an Evans blue analog for cancer theranostics. Theranostics 2018, 8, 812–814. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Wang, G.; Zhang, H.; Ma, Y.; Lang, L.; Jacobson, O.; Kiesewetter, D.O.; Zhu, L.; Gao, S.; Ma, Q.; et al. Stable Evans blue derived exendin-4 peptide for type 2 diabetes treatment. Bioconjugate Chem. 2016, 27, 54–58. [Google Scholar] [CrossRef] [Green Version]
- Saunders, N.R.; Dziegielewska, K.M.; Møllgård, K.; Habgood, M.D. Markers for blood-brain barrier integrity: How appropriate is Evans blue in the twenty-first century and what are the alternatives? Front. Neurosci. 2015, 9, 385. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, T.; Ikuta, K.; Oi, K.; Abe, K.; Uwatoku, T.; Murata, M.; Shigetani, N.; Yoshimitsu, K.; Shimokawa, H.; Katayama, Y. First functionalized MRI contrast agent recognizing vascular lesions. Anal. Sci. 2004, 20, 5–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Lang, L.; Zhu, Z.; Li, F.; Niu, G.; Chen, X. Clinical translation of an albumin-binding PET radiotracer 68Ga-NEB. J. Nucl. Med. 2015, 56, 1609–1614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, F.; Xue, J.; Shao, J.; Jin, L. Compilation of 222 drugs’ plasma protein binding data and guidance for study designs. Drug Discov. Today 2012, 17, 475–485. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Lang, L.; Huang, P.; Wang, Z.; Jacobson, O.; Kiesewetter, D.O.; Ali, I.U.; Teng, G.; Niu, G.; Chen, X. In vivo albumin labeling and lymphatic imaging. Proc. Natl. Acad. Sci. USA 2015, 112, 208–213. [Google Scholar] [CrossRef] [Green Version]
- Yao, L.; Xue, X.; Yu, P.; Ni, Y.; Chen, F. Evans blue dye: A revisit of its applications in biomedicine. Contrast Media Mol. Imaging. 2018, 2018, 10. [Google Scholar] [CrossRef] [Green Version]
- Dumelin, C.E.; Trüssel, S.; Buller, F.; Trachsel, E.; Bootz, F.; Zhang, Y.; Mannocci, L.; Beck, S.C.; Drumea-Mirancea, M.; Seeliger, M.W.; et al. A portable albumin binder from a DNA-encoded chemical library. Angew. Chem. Int. Ed. 2008, 47, 3196–3201. [Google Scholar] [CrossRef] [PubMed]
- Umbricht, C.A.; Benešová, M.; Schibli, R.; Müller, C. Preclinical development of novel PSMA-targeting radioligands: Modulation of albumin-binding properties to improve prostate cancer therapy. Mol. Pharm. 2018, 15, 2297–2306. [Google Scholar] [CrossRef]
- Rousseau, E.; Lau, J.; Zhang, Z.; Uribe, C.F.; Kuo, H.-T.; Zhang, C.; Zeisler, J.; Colpo, N.; Lin, K.-S.; Bénard, F. Effects of adding an albumin binder chain on [177Lu]Lu-DOTATATE. Nucl. Med. Biol. 2018, 66, 10–17. [Google Scholar] [CrossRef]
- Hausner, S.H.; Bold, R.J.; Cheuy, L.Y.; Chew, H.K.; Daly, M.E.; Davis, R.A.; Foster, C.C.; Kim, E.J.; Sutcliffe, J.L. Preclinical development and first-in human imaging of integrin αvβ6-binding peptide in metastatic carcinoma. Clinic. Cancer Res. 2019, 25, 1206–1215. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Wang, W.; Niu, W.; Liu, E.; Liu, X.; Wang, J.; Peng, C.; Liu, S.; Xu, L.; Wang, L.; et al. SDF-1/CXCR4 axis promotes directional migration of colorectal cancer cells through upregulation of integrin αvβ6. Carcinogenesis 2013, 35, 282–291. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Lin, P.; Gao, C.; Peng, C.; Liu, S.; Gao, H.; Wang, B.; Wang, J.; Niu, J.; Niu, W. Integrin β6 acts as an unfavorable prognostic indicator and promotes cellular malignant behaviors via ERK-ETS1 pathway in pancreatic ductal adenocarcinoma (PDAC). Tumor. Biol. 2016, 37, 5117–5131. [Google Scholar] [CrossRef] [PubMed]
- Izabela, Ł.; Jacek, M. Integrins as a new target for cancer treatment. Anti-Cancer Agents Med. Chem. 2019, 19, 580–586. [Google Scholar]
- Bandyopadhyay, A.; Raghavan, S. Defining the role of integrin avb6 in cancer. Curr. Drug Targets. 2009, 10, 645–652. [Google Scholar] [CrossRef]
- Ahmed, N.; Pansino, F.; Clyde, R.; Murthi, P.; Quinn, M.A.; Rice, G.E.; Agrez, M.V.; Mok, S.; Baker, M.S. Overexpression of αvβ6 integrin in serous epithelial ovarian cancer regulates extracellular matrix degradation via the plasminogen activation cascade. Carcinogenesis 2002, 23, 237–244. [Google Scholar] [CrossRef]
- Elayadi, A.N.; Samli, K.N.; Prudkin, L.; Liu, Y.-H.; Bian, A.; Xie, X.-J.; Wistuba, I.I.; Roth, J.A.; McGuire, M.J.; Brown, K.C. A peptide selected by biopanning identifies the integrin αvβ6 as a prognostic biomarker for nonsmall cell lung cancer. Cancer Res. 2007, 67, 5889–5895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore, K.M.; Thomas, G.J.; Duffy, S.W.; Warwick, J.; Gabe, R.; Chou, P.; Ellis, I.O.; Green, A.R.; Haider, S.; Brouilette, K.; et al. Therapeutic targeting of integrin αvβ6 in breast cancer. J. Natl. Cancer Inst. 2014, 106, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.Y.; Xu, K.S.; Wang, J.S.; Yang, G.Y.; Wang, W.; Wang, J.Y.; Niu, W.B.; Liu, E.Y.; Mi, Y.T.; Niu, J. Integrin αvβ6 acts as a prognostic indicator in gastric carcinoma. Clin. Oncol. 2008, 20, 61–66. [Google Scholar] [CrossRef]
- Hsiao, J.-R.; Chang, Y.; Chen, Y.-L.; Hsieh, S.-H.; Hsu, K.-F.; Wang, C.-F.; Tsai, S.-T.; Jin, Y.-T. Cyclic αvβ6-targeting peptide selected from biopanning with clinical potential for head and neck squamous cell carcinoma. Head Neck. 2010, 32, 160–172. [Google Scholar] [PubMed]
- Bates, R.C. The αvβ6 integrin as a novel molecular target for colorectal cancer. Future Oncol. 2005, 1, 821–828. [Google Scholar] [CrossRef]
- Berghoff, A.S.; Kovanda, A.K.; Melchardt, T.; Bartsch, R.; Hainfellner, J.A.; Sipos, B.; Schittenhelm, J.; Zielinski, C.C.; Widhalm, G.; Dieckmann, K.; et al. αvβ3, αvβ5 and αvβ6 integrins in brain metastases of lung cancer. Clin. Exper. Met. 2014, 31, 841–851. [Google Scholar] [CrossRef]
- Hausner, S.H.; Bauer, N.; Davis, R.A.; Ganguly, T.; Tang, S.Y.C.; Sutcliffe, J.L. The effects of an albumin binding moiety on the targeting and pharmacokinetics of an integrin αvβ6-selective peptide labeled with aluminum [18F]fluoride. Mol. Imaging Biol. 2020, 22, 1543–1552. [Google Scholar] [CrossRef] [PubMed]
- Hausner, S.H.; Bauer, N.; Sutcliffe, J.L. In vitro and in vivo evaluation of the effects of aluminum [18F]fluoride radiolabeling on an integrin αvβ6-specific peptide. Nucl. Med. Biol. 2014, 41, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Ganguly, T.; Bauer, N.; Davis, R.A.; Hausner, S.H.; Tang, S.Y.; Sutcliffe, J.L. Evaluation of copper-64-labeled αvβ6-targeting peptides: Addition of an albumin binding moiety to improve pharmacokinetics. Mol. Pharm. 2021, 18, 4437–4447. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Zhu, G.; Jacobson, O.; Liu, Y.; Chen, K.; Yu, G.; Ni, Q.; Fan, J.; Yang, Z.; Xu, F.; et al. Transformative nanomedicine of an amphiphilic camptothecin prodrug for long circulation and high tumor uptake in cancer therapy. ACS Nano. 2017, 11, 8838–8848. [Google Scholar] [CrossRef]
- Favre-Besse, F.-C.; Poirel, O.; Bersot, T.; Kim-Grellier, E.; Daumas, S.; El Mestikawy, S.; Acher, F.C.; Pietrancosta, N. Design, synthesis and biological evaluation of small-azo-dyes as potent vesicular glutamate transporters inhibitors. Eur. J. Med. Chem. 2014, 78, 236–247. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.C.; Davis, R.A.; Ganguly, T.; Sutcliffe, J.L. Identification, characterization, and optimization of integrin αvβ6-targeting peptides from a one-bead one-compound (OBOC) library: Towards the development of positron emission tomography (PET) imaging agents. Molecules 2019, 24, 309. [Google Scholar] [CrossRef] [Green Version]
- Padma, V.V. An overview of targeted cancer therapy. BioMedicine 2015, 5, e46. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [Green Version]
- Willemieke, T.S.; Farina-Sarasquota, A.; Boonstra, M.C.; Prevoo, H.A.; Sier, C.F.; Mieog, J.S.; Morreau, J.; van Eijck, C.H.; Kuppen, P.J.; van de Velde, C.J.; et al. Selection of optimal molecular targets for tumor-specific imaging in pancreatic ductal adenocarcinoma. Oncotarget 2017, 8, 56816–56828. [Google Scholar]
- Färber, S.F.; Wurzer, A.; Reichart, F.; Beck, R.; Kessler, H.; Wester, H.-J.; Notni, J. Therapeutic radiopharmaceuticals targeting integrin αvβ6. ACS Omega. 2018, 3, 2428–2436. [Google Scholar] [CrossRef] [Green Version]
- Müller, C.; Struthers, H.; Winiger, C.; Zhernosekov, K.; Schibli, R. DOTA conjugate with an albumin-binding entity enables the first folic acid–targeted 177Lu-radionuclide tumor therapy in mice. J. Nucl. Med. 2013, 54, 124–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Jacobson, O.; Tian, R.; Mease, R.C.; Kiesewetter, D.O.; Niu, G.; Pomper, M.G.; Chen, X. Radioligand therapy of prostate cancer with a long-lasting prostate-specific membrane antigen targeting agent 90Y-DOTA-EB-MCG. Bioconjugate Chem. 2018, 29, 2309–2315. [Google Scholar] [CrossRef] [PubMed]
- Choy, C.J.; Ling, X.; Geruntho, J.J.; Beyer, S.K.; Latoche, J.D.; Langton-Webster, B.; Anderson, C.J.; Berkman, C.E. 177Lu-labeled phosphoramidate-based PSMA inhibitors: The effect of an albumin binder on biodistribution and therapeutic efficacy in prostate tumor-bearing mice. Theranostics 2017, 7, 1928–1939. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Tian, R.; Niu, G.; Ma, Y.; Lang, L.; Szajek, L.P.; Kiesewetter, D.O.; Jacobson, O.; Chen, X. Single low-dose injection of Evans blue modified PSMA-617 radioligand therapy eliminates prostate-specific membrane antigen positive tumors. Bioconjugate Chem. 2018, 29, 3213–3221. [Google Scholar] [CrossRef] [PubMed]
- Kuo, H.-T.; Lin, K.-S.; Zhang, Z.; Uribe, C.F.; Merkens, H.; Zhang, C.; Bénard, F. 177Lu-labeled albumin-binder–conjugated PSMA-targeting agents with extremely high tumor uptake and enhanced tumor-to-kidney absorbed dose ratio. J. Nucl. Med. 2021, 62, 521–527. [Google Scholar] [CrossRef] [PubMed]
- Benešová, M.; Umbricht, C.A.; Schibli, R.; Müller, C. Albumin-binding PSMA ligands: Optimization of the tissue distribution profile. Mol. Pharm. 2018, 15, 934–946. [Google Scholar] [CrossRef]
- Siwowska, K.; Haller, S.; Bortoli, F.; Benešová, M.; Broehn, V.; Bernhardt, P.; Schibli, R.; Müller, C. Preclinical comparison of albumin-binding radiofolates: Impact of linker entities on the in vitro and in vivo properties. Mol. Pharm. 2017, 14, 523–532. [Google Scholar] [CrossRef]
- Kaeppeli, S.A.M.; Jodal, A.; Gotthardt, M.; Schibli, R.; Béhé, M. Exendin-4 derivatives with an albumin-binding moiety show decreased renal retention and improved GLP-1 receptor targeting. Mol. Pharm. 2019, 16, 3760–3769. [Google Scholar] [CrossRef]
- Bandara, N.; Jacobson, O.; Mpoy, C.; Chen, X.; Rogers, B.E. Novel structural modification based on Evans blue dye to improve pharmacokinetics of a somastostatin-receptor-based theranostic agent. Bioconjugate Chem. 2018, 29, 2448–2454. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, H.; Jacobson, O.; Cheng, Y.; Niu, G.; Li, F.; Bai, C.; Zhu, Z.; Chen, X. Safety, pharmacokinetics, and dosimetry of a long-acting radiolabeled somatostatin analog 177Lu-DOTA-EB-TATE in patients with advanced metastatic neuroendocrine tumors. J. Nucl. Med. 2018, 59, 1699–1705. [Google Scholar] [CrossRef] [Green Version]
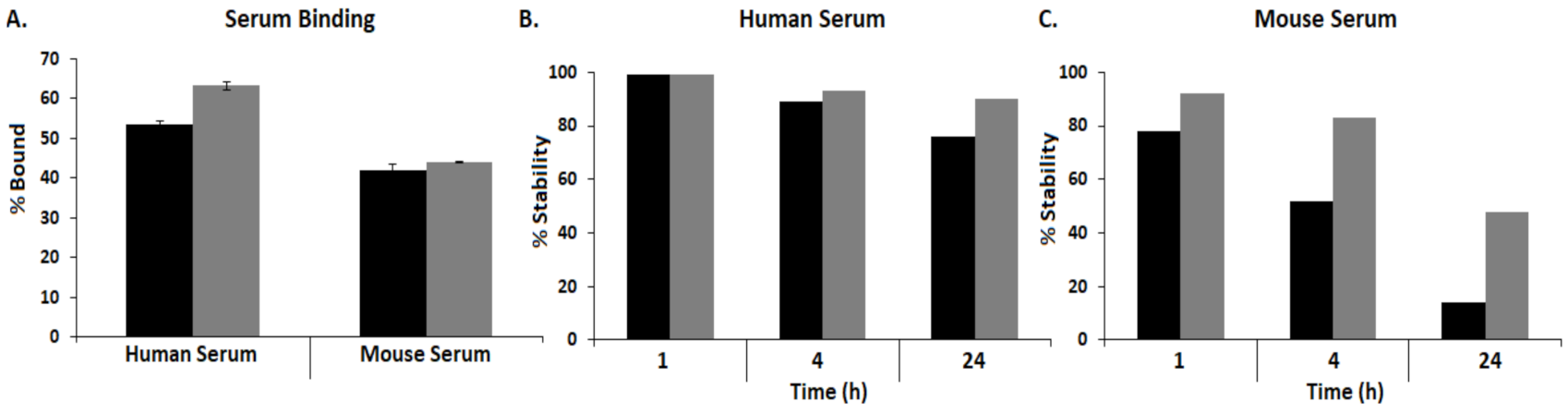
 ).
).
).
). ). (A) BxPC-3 tumors. (B) kidneys. (C) liver. (D) stomach. (E) small intestines. (F) large intestines (* p ≤ 0.05).
). (A) BxPC-3 tumors. (B) kidneys. (C) liver. (D) stomach. (E) small intestines. (F) large intestines (* p ≤ 0.05).
). (A) BxPC-3 tumors. (B) kidneys. (C) liver. (D) stomach. (E) small intestines. (F) large intestines (* p ≤ 0.05).
). (A) BxPC-3 tumors. (B) kidneys. (C) liver. (D) stomach. (E) small intestines. (F) large intestines (* p ≤ 0.05).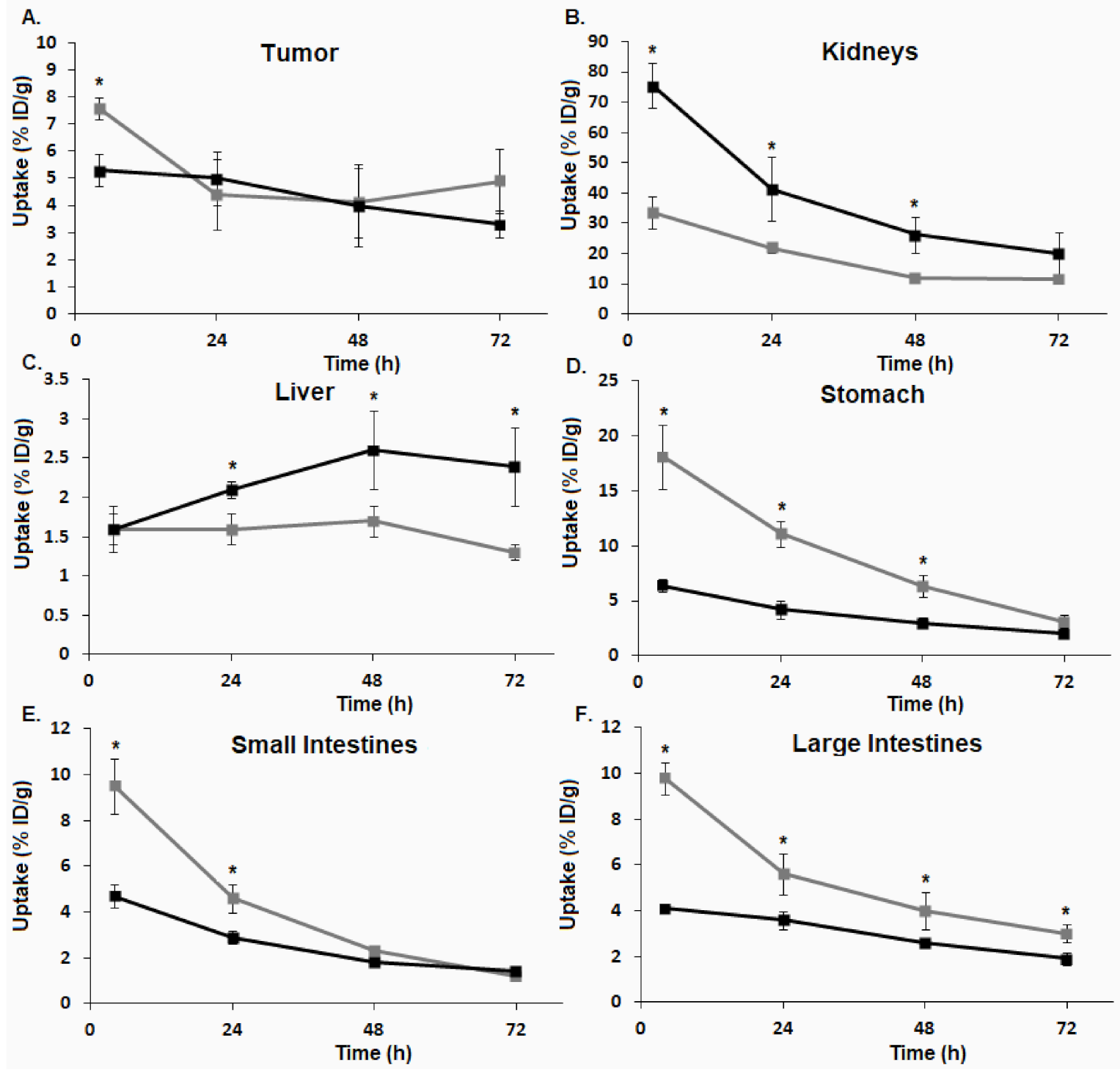 ), [64Cu]3 (
), [64Cu]3 (  ), and [64Cu]4 (
), and [64Cu]4 (  ).
), [64Cu]3 ( ), and [64Cu]4 ( ).
).
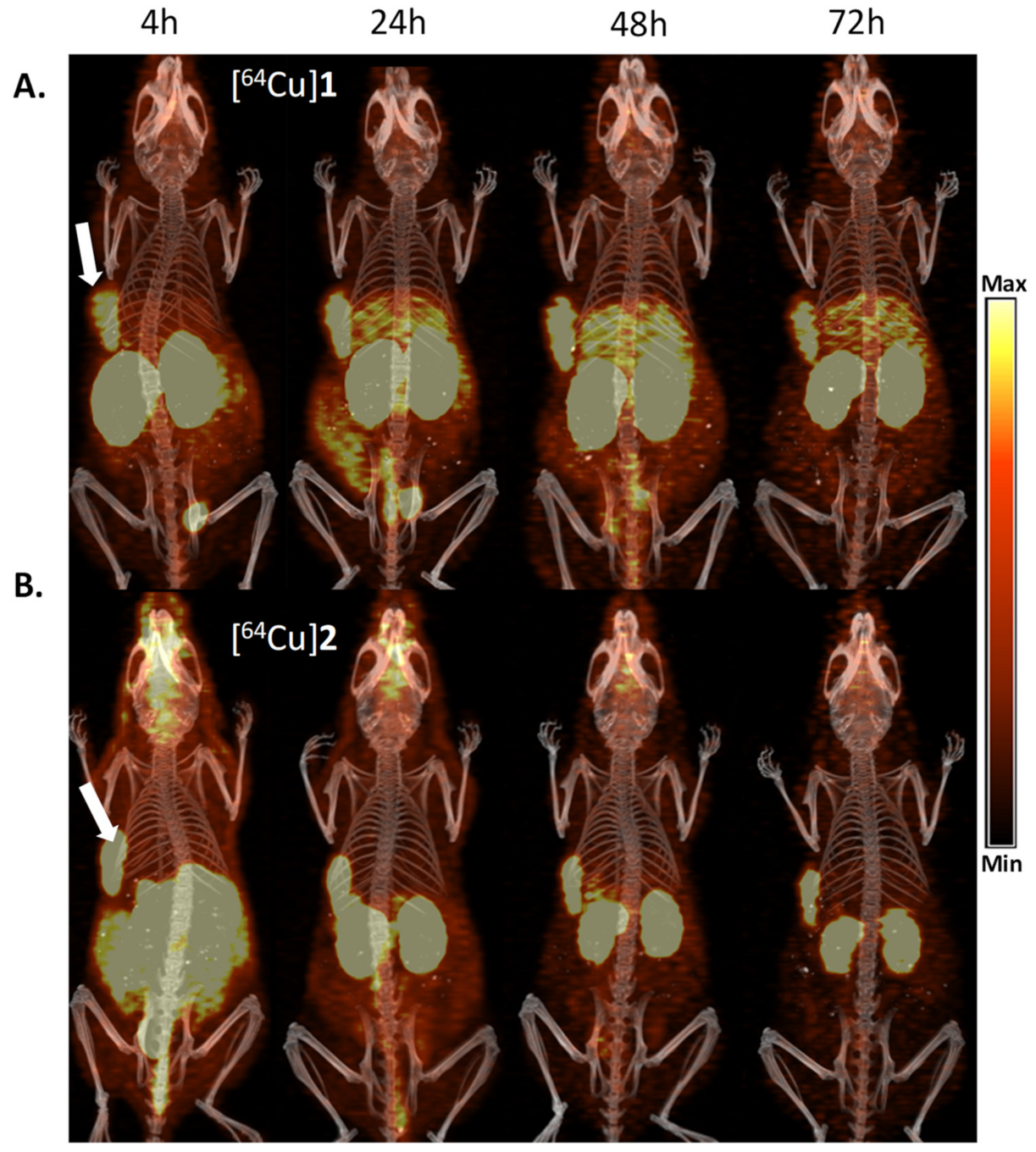
), [64Cu]3 ( ), and [64Cu]4 ( ).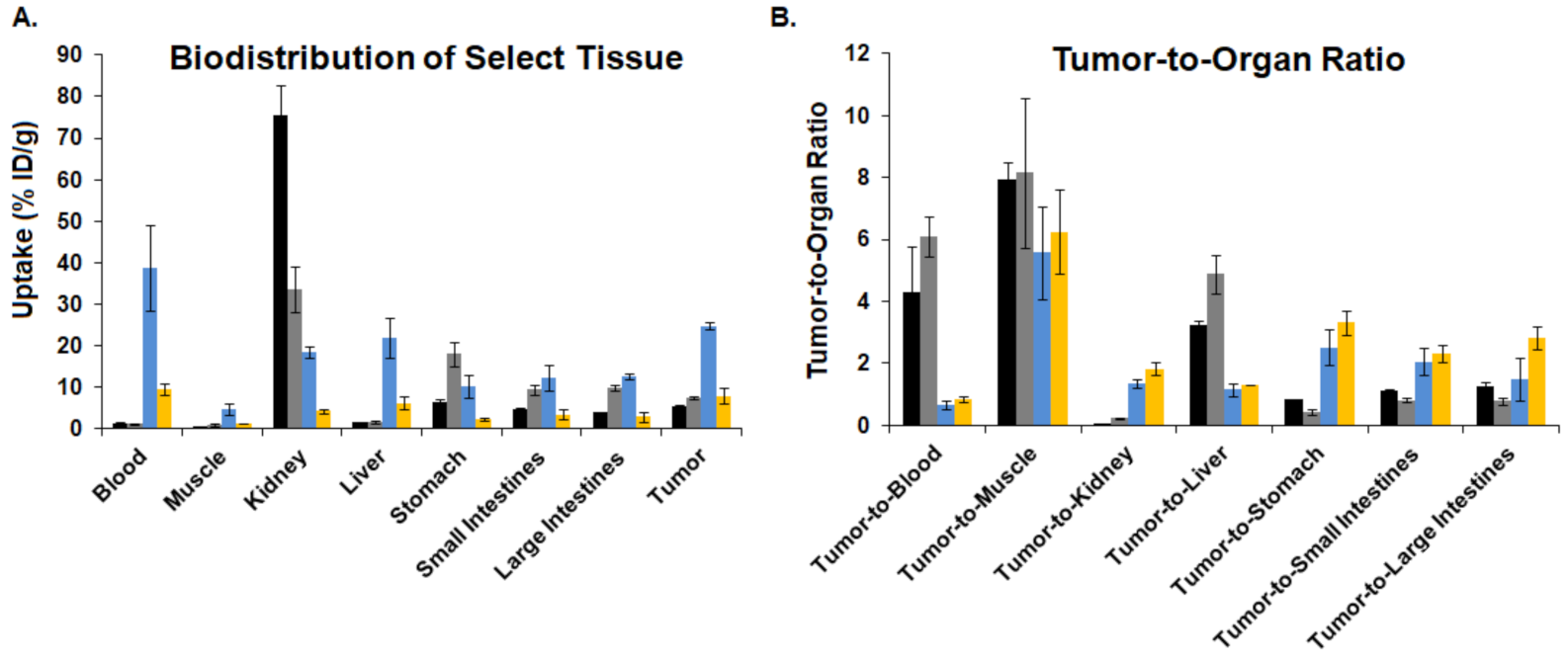
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Davis, R.A.; Hausner, S.H.; Harris, R.; Sutcliffe, J.L. A Comparison of Evans Blue and 4-(p-Iodophenyl)butyryl Albumin Binding Moieties on an Integrin αvβ6 Binding Peptide. Pharmaceutics 2022, 14, 745. https://doi.org/10.3390/pharmaceutics14040745
Davis RA, Hausner SH, Harris R, Sutcliffe JL. A Comparison of Evans Blue and 4-(p-Iodophenyl)butyryl Albumin Binding Moieties on an Integrin αvβ6 Binding Peptide. Pharmaceutics. 2022; 14(4):745. https://doi.org/10.3390/pharmaceutics14040745
Chicago/Turabian StyleDavis, Ryan A., Sven H. Hausner, Rebecca Harris, and Julie L. Sutcliffe. 2022. "A Comparison of Evans Blue and 4-(p-Iodophenyl)butyryl Albumin Binding Moieties on an Integrin αvβ6 Binding Peptide" Pharmaceutics 14, no. 4: 745. https://doi.org/10.3390/pharmaceutics14040745
APA StyleDavis, R. A., Hausner, S. H., Harris, R., & Sutcliffe, J. L. (2022). A Comparison of Evans Blue and 4-(p-Iodophenyl)butyryl Albumin Binding Moieties on an Integrin αvβ6 Binding Peptide. Pharmaceutics, 14(4), 745. https://doi.org/10.3390/pharmaceutics14040745