
Nanotechnology Driven Cancer Chemoradiation: Exploiting the Full Potential of Radiotherapy with a Unique Combination of Gold Nanoparticles and Bleomycin

 , ,
, ,  , ,
, ,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cells and Culture Conditions
2.2. GNP Synthesis, Modification, and Characterization
2.3. Quantification of GNP Uptake Using Inductively Coulples Surface Plasmon Mass Spectrocopy (ICPMS)
2.4. Darkfield and Hyperspectral Imaging (HSI)
2.5. Live-Cell Confocal Imaging
2.6. Cell Cycle Analysis
2.7. Proliferation Assay (Concentration-Dependent)
2.8. Proliferation Radiation Experiment
2.9. DNA Double-Strand Breaks Assay
2.10. Polarization Modulation Infrared Reflection Absorption Spectroscopy (PM-IRRAS) Measurements
2.11. Statistical Analysis
3. Results and Discussion
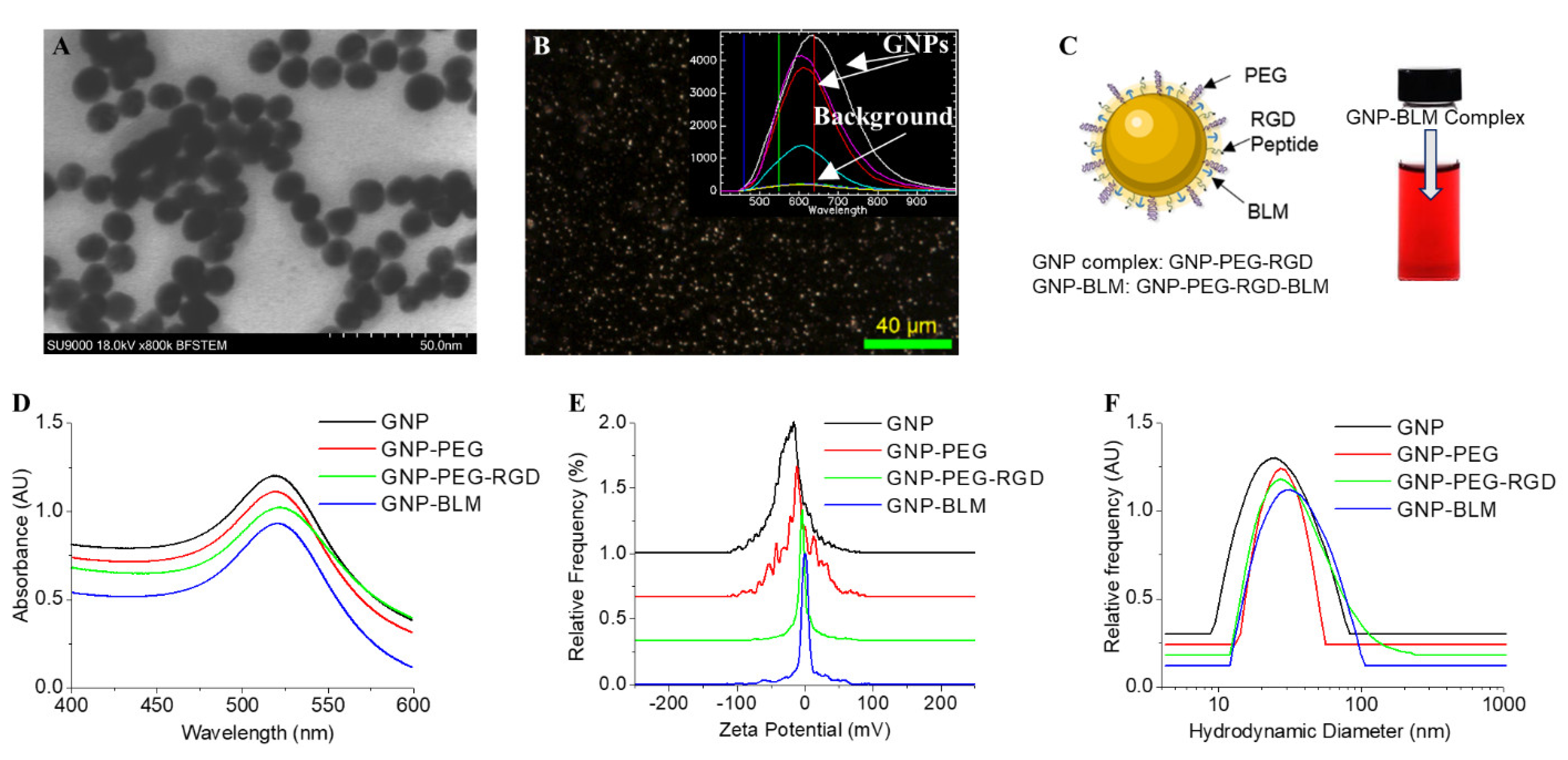
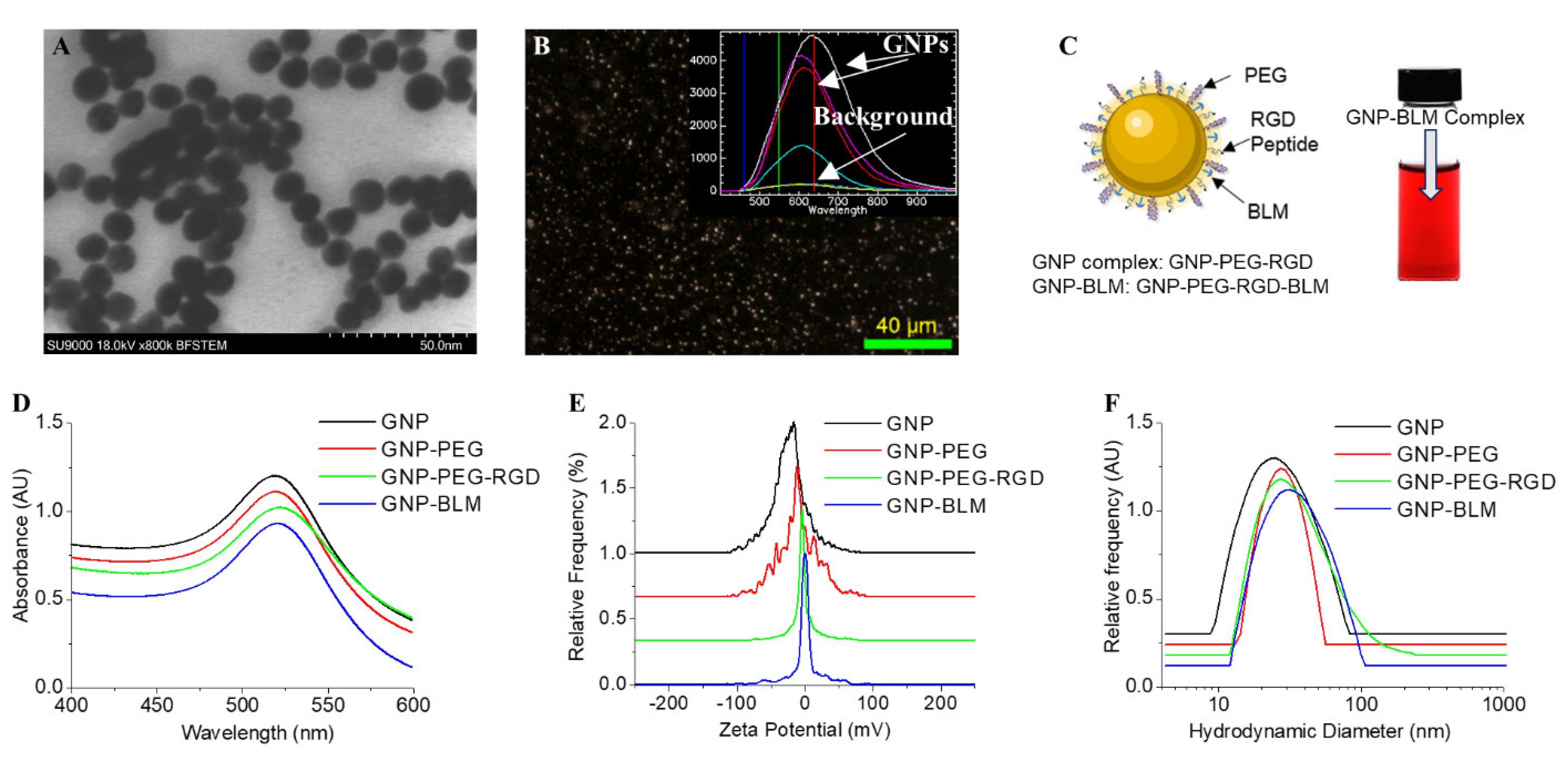
3.1. Characterisation of Nanoparticle Complexes
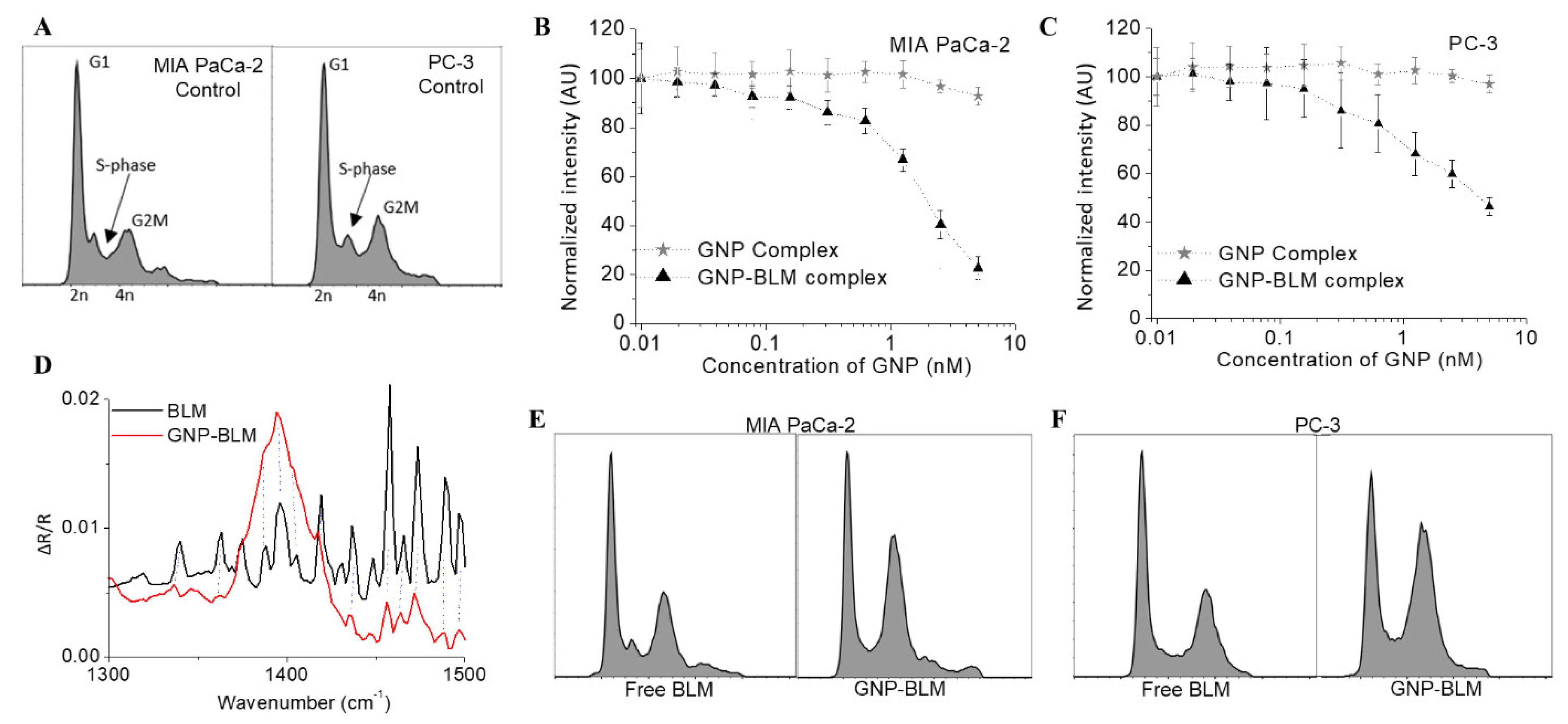
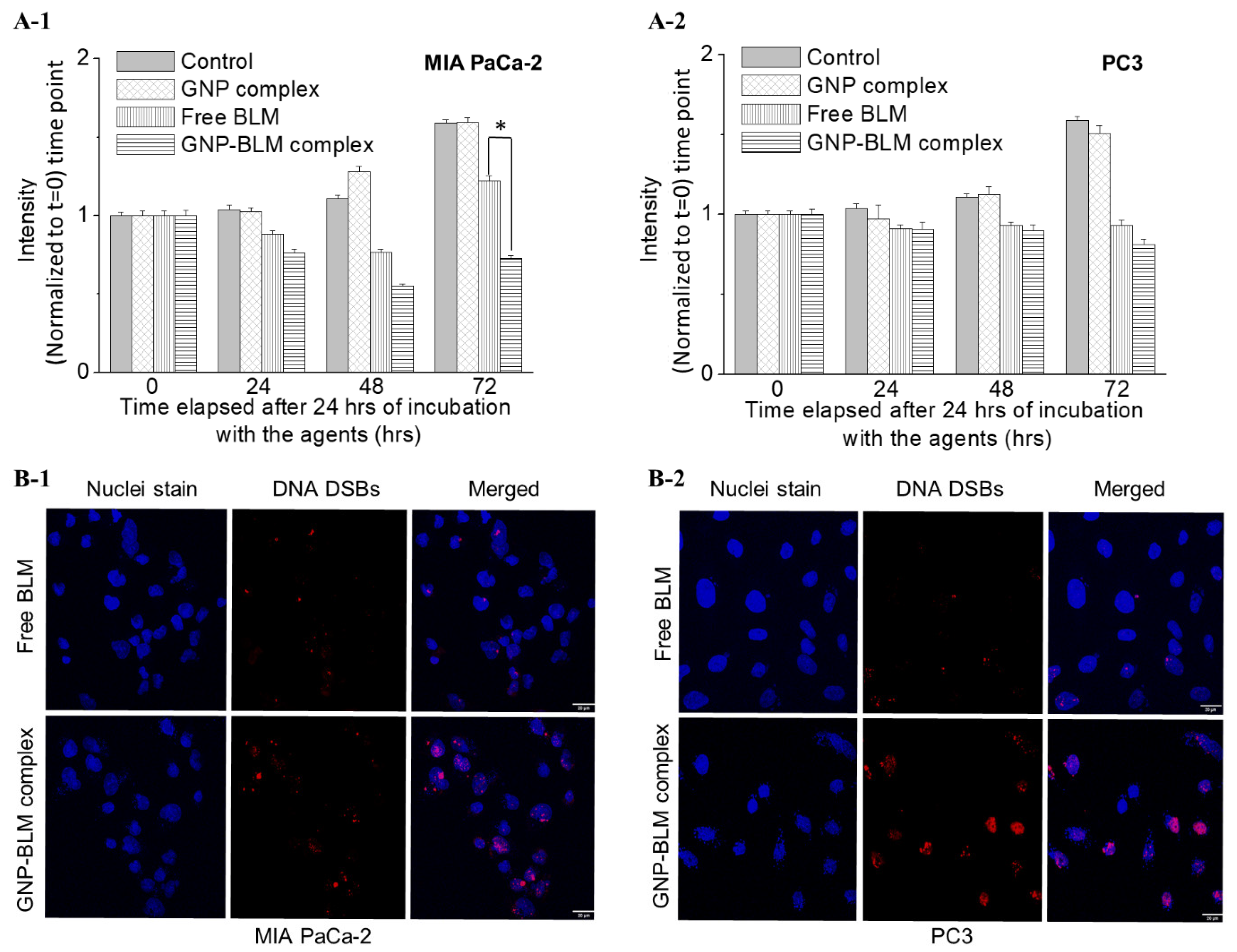
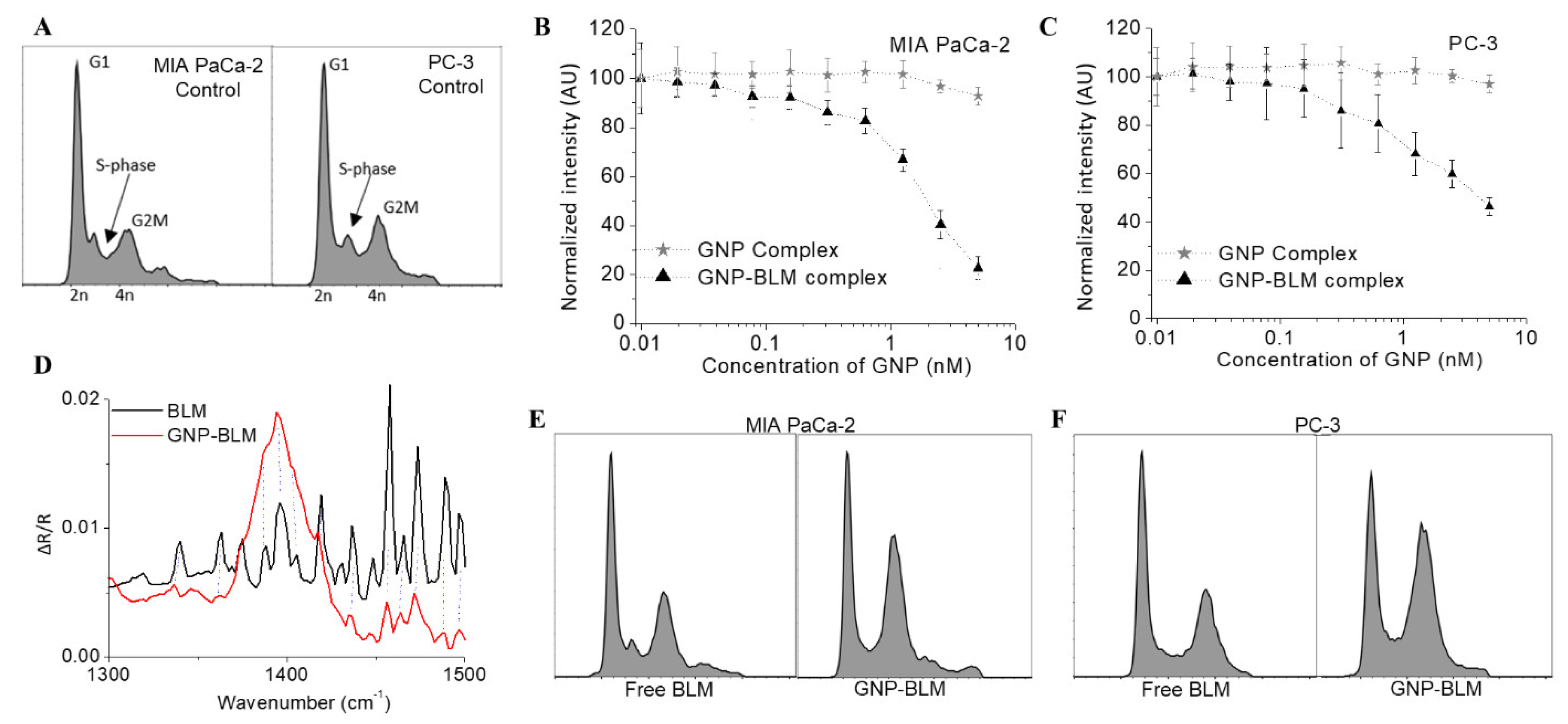
3.2. Action of GNP-BLM Complex vs. Free Drug
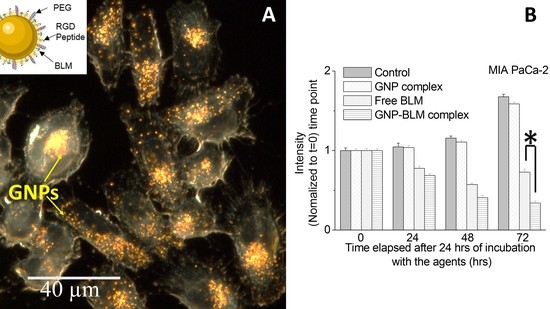
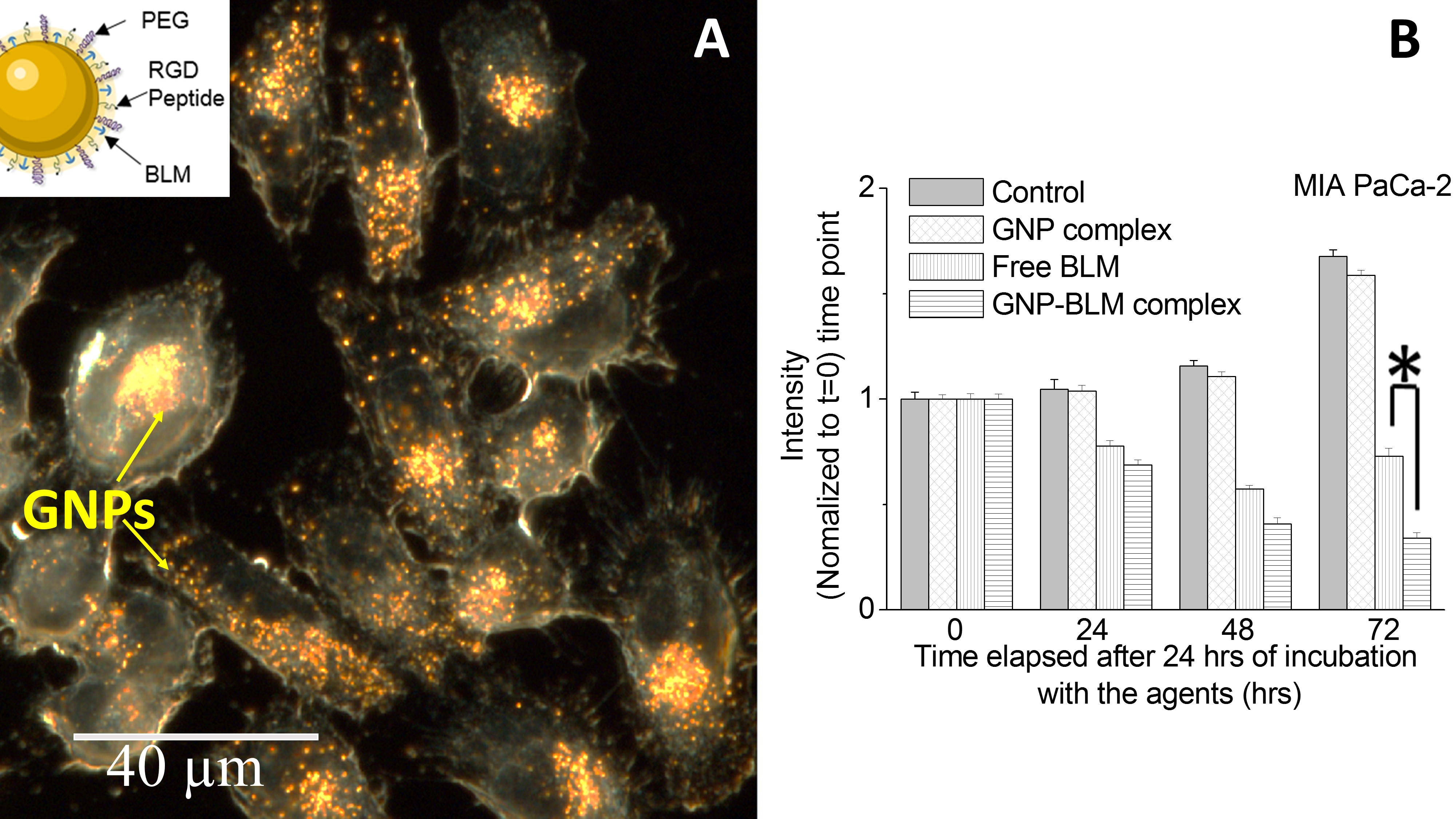
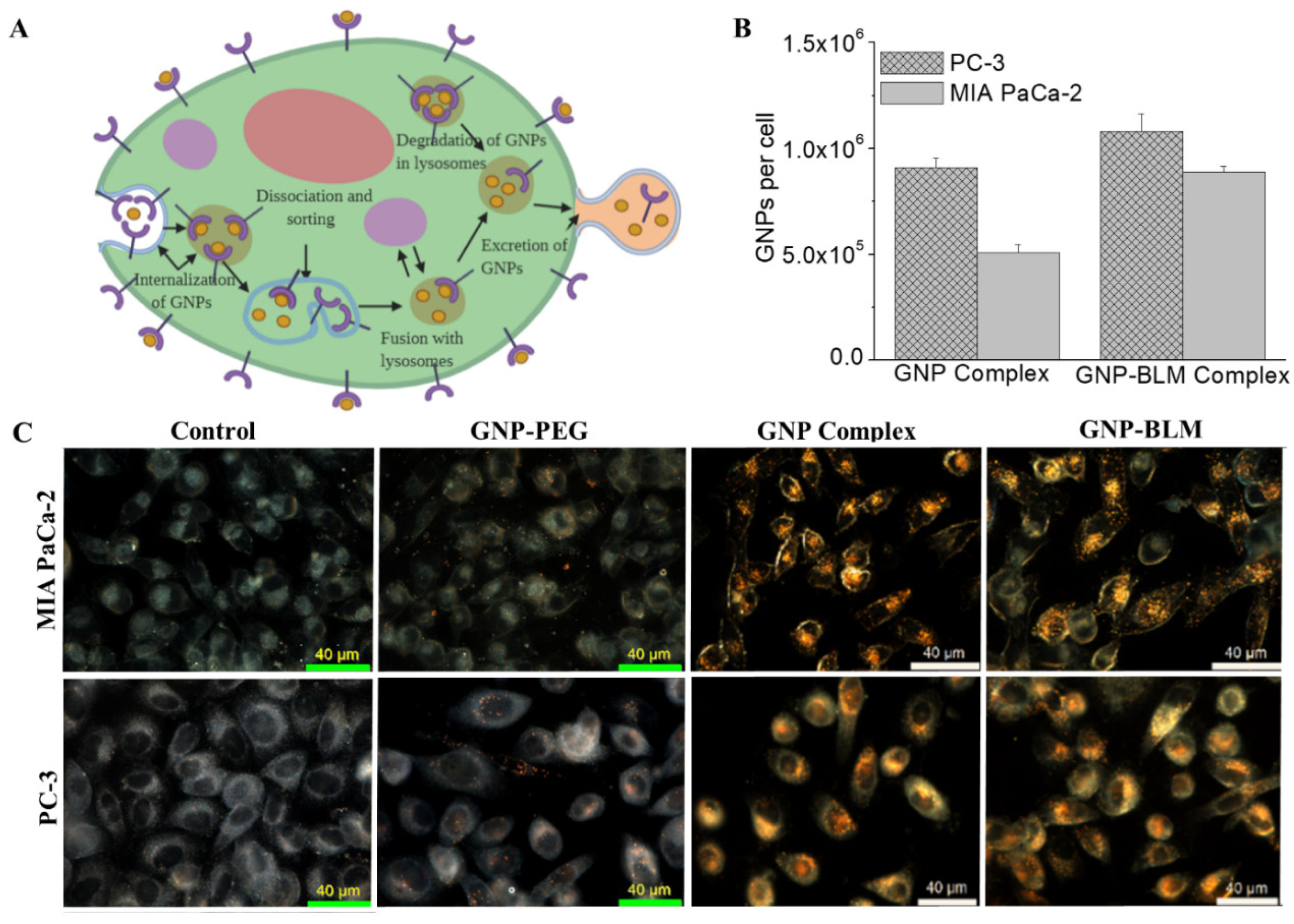
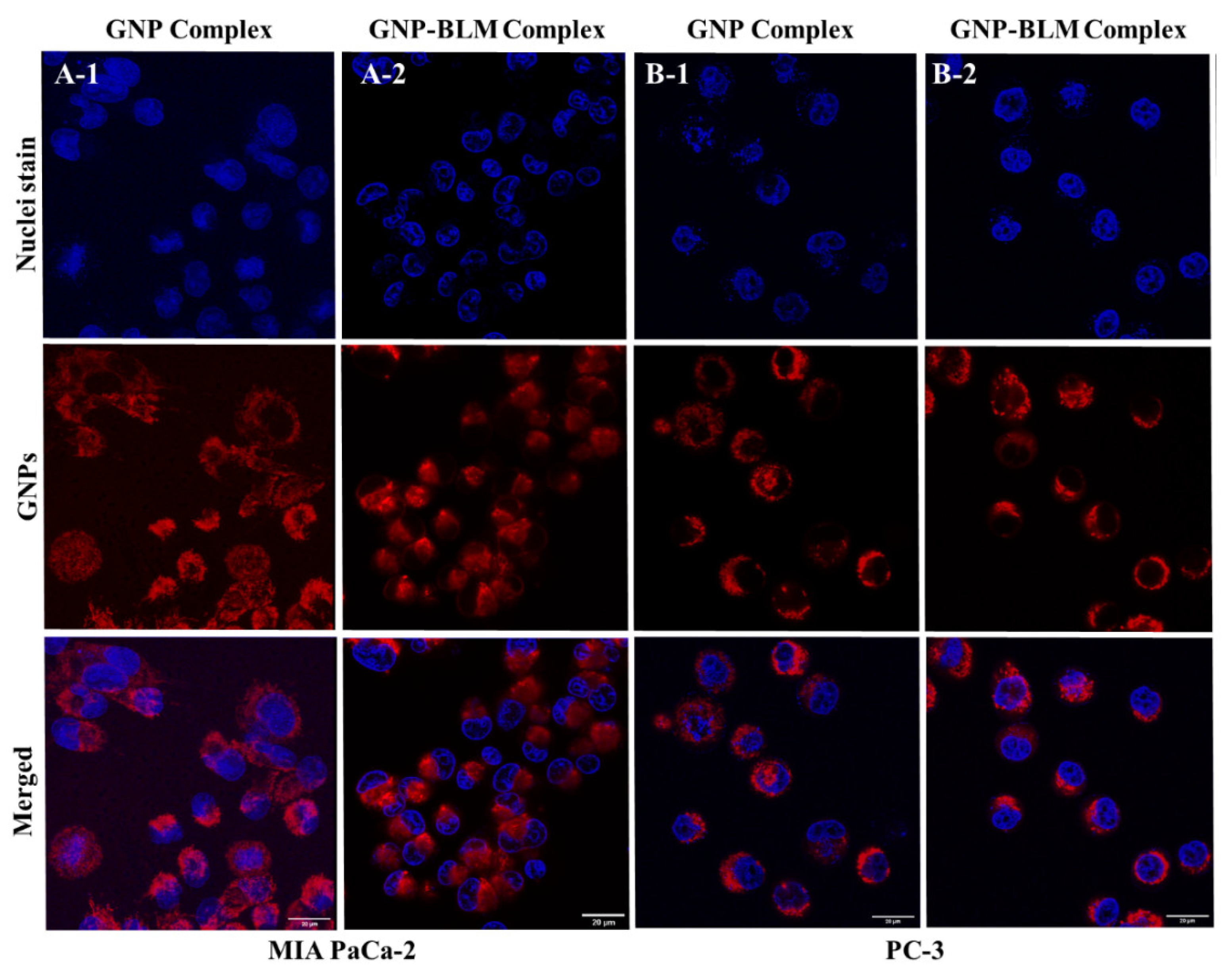
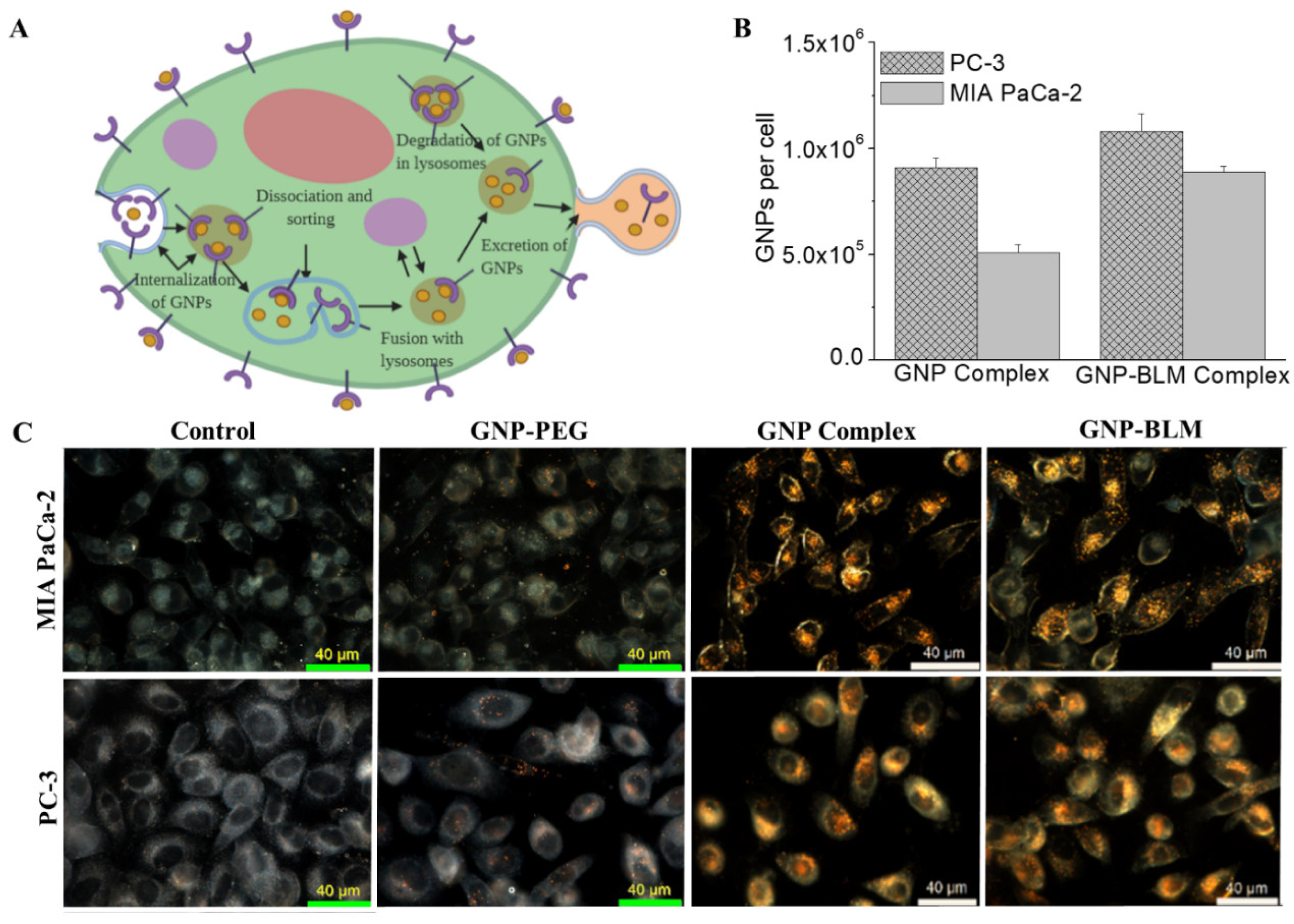
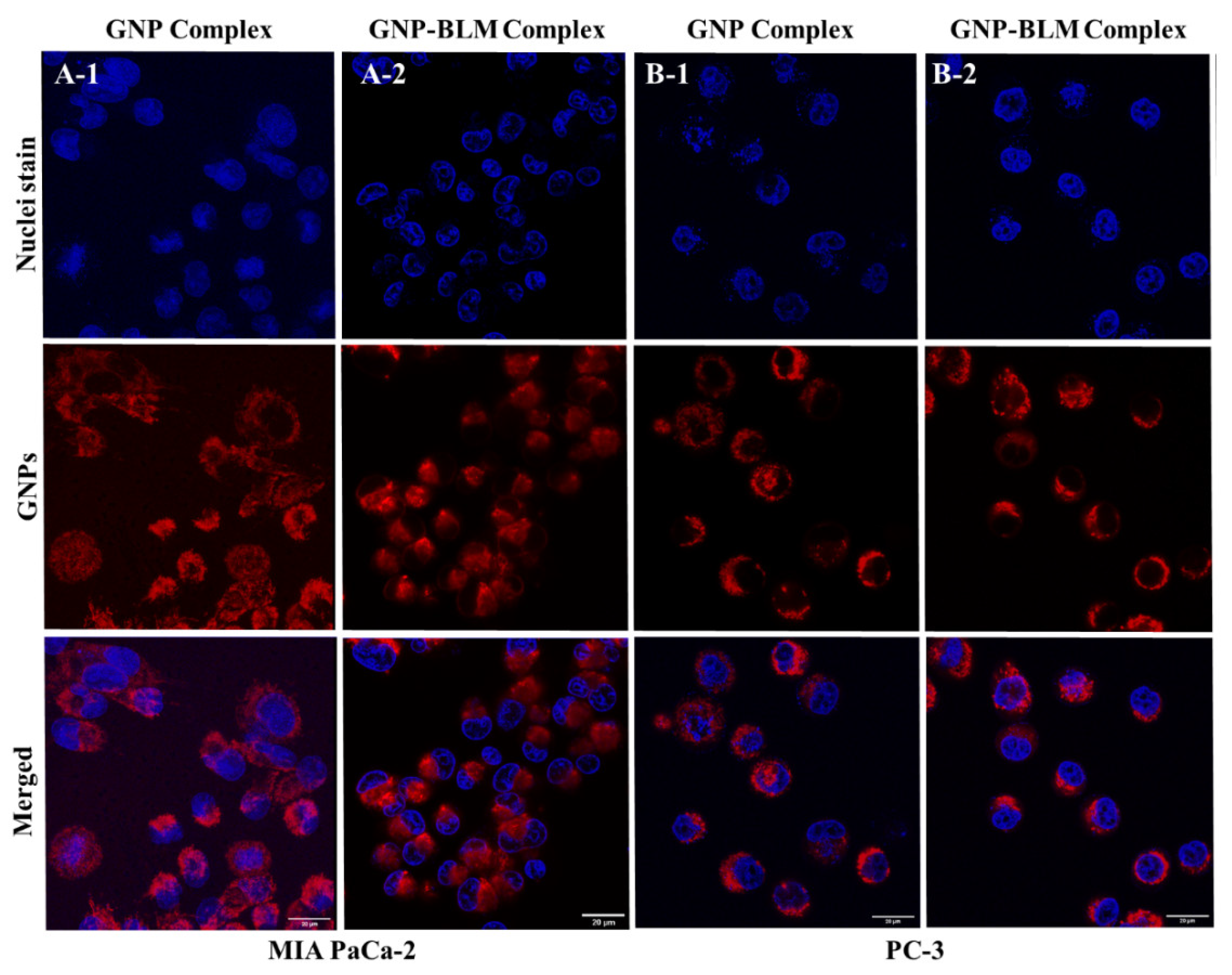
3.3. Intracellular Uptake and Distribution of GNP-BLM and GNP Complexes
3.4. Evaluation of GNP Mediated Drug Delivery
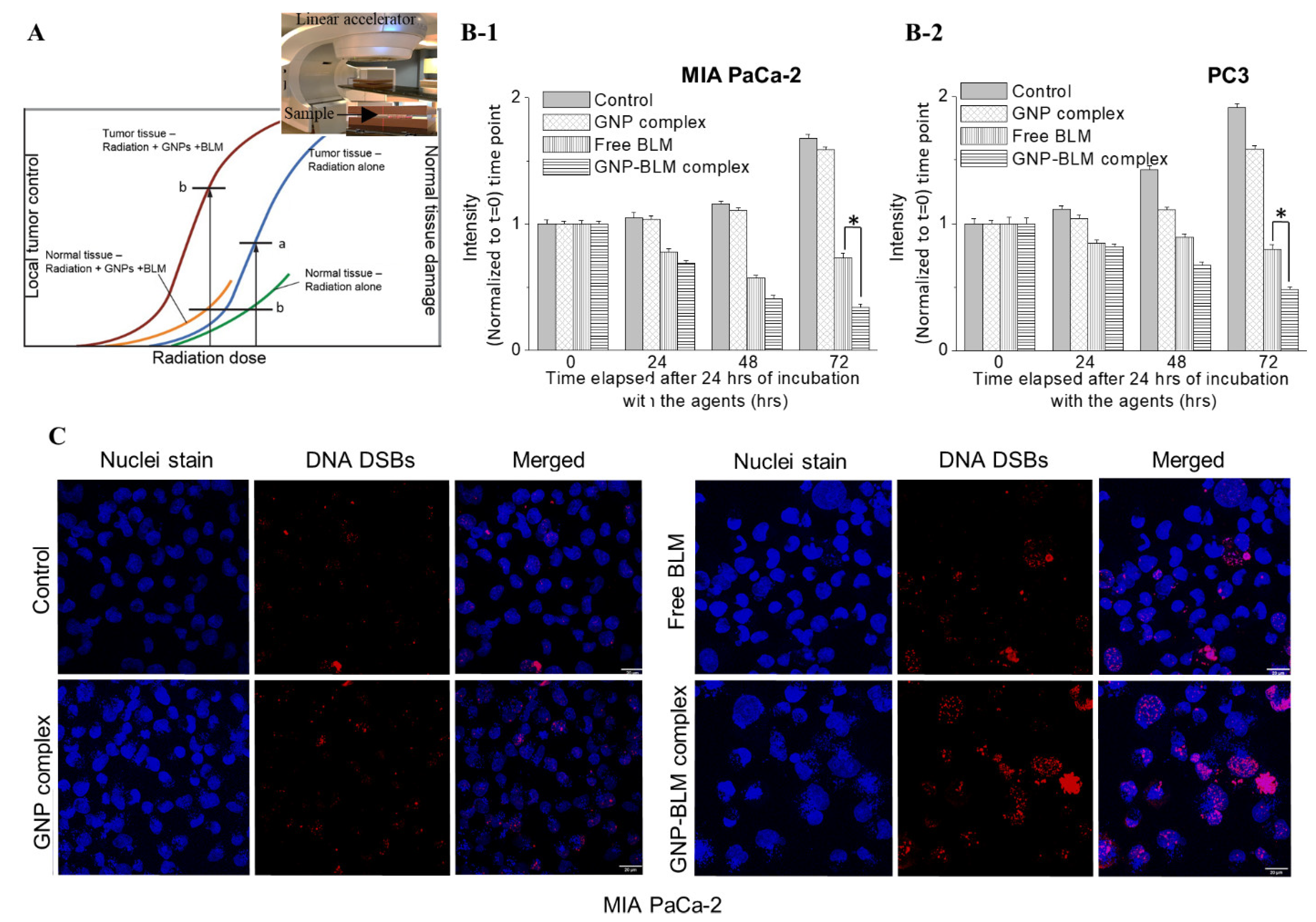
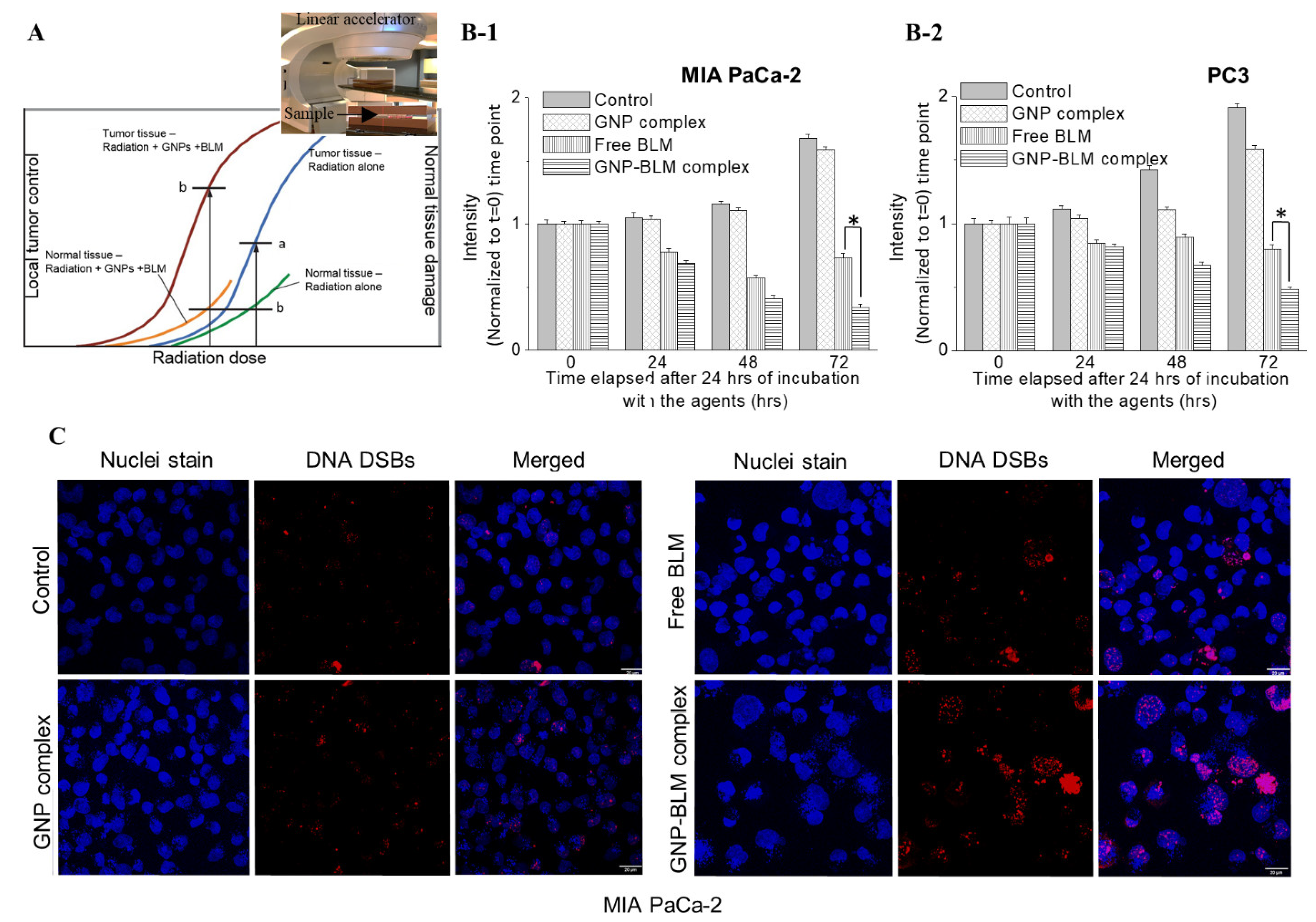
3.5. Combined Approach of GNPs, BLMs, and Radiation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gong, L.; Zhang, Y.; Liu, C.; Zhang, M.; Han, S. Application of radiosensitizers in cancer radiotherapy. Int. J. Nanomed. 2021, 16, 1083–1102. [Google Scholar] [CrossRef]
- Martin, O.A.; Martin, R.F. Cancer radiotherapy: Understanding the price of tumor eradication. Front. Cell Dev. Biol. 2020, 8, 261. [Google Scholar] [CrossRef]
- Rubin, P.; Carter, S.K. Combination radiation therapy and chemotherapy: A logical basis for their clinical use. CA Cancer J. Clin. 1976, 26, 274–292. [Google Scholar] [CrossRef]
- Yapp, D.T.; Lloyd, D.K.; Zhu, J.; Lehnert, S.M. Tumor treatment by sustained intratumoral release of cisplatin: Effects of drug alone and combined with radiation. Int. J. Radiat. Oncol. Biol. Phys. 1997, 39, 497–504. [Google Scholar] [CrossRef]
- Long, W.; Wang, J.; Xu, F.; Wu, H.; Mu, X.; Wang, J.; Sun, Y.; Zhang, X.-D. Catalytic ptpd bimetal nanocrystals with high-index facets for radiation injury repair. Chin. Chem. Lett. 2020, 31, 269–274. [Google Scholar] [CrossRef]
- Chithrani, B.D.; Jelveh, S.; Jalali, F.; van Prooijen, M.; Allen, C.; Bristow, R.G.; Hill, R.P.; Jaffray, D.A. Gold nanoparticles as radiation sensitizers in cancer therapy. Radiat. Res. 2010, 173, 719–728. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P. A new paradigm for the treatment of high-risk prostate cancer: Radiosensitization with docetaxel. Rev. Urol. 2003, 5 (Suppl. 3), S71–S77. [Google Scholar] [PubMed]
- Kumar, P.; Weiss, R. Radiosensitization with docetaxel and 3-D CRT. Results of a completed phase I trial. Proc. Am. Soc. Clin. Oncol. 2003, 22, 404. [Google Scholar]
- Fu, Z.-Z.; Li, K.; Peng, Y.; Zheng, Y.; Cao, L.-Y.; Zhang, Y.-J.; Sun, Y.-M. Efficacy and toxicity of different concurrent chemoradiotherapy regimens in the treatment of advanced cervical cancer: A network meta-analysis. Medicine 2017, 96, e5853. [Google Scholar] [CrossRef]
- Hurwitz, M.D. The docetaxel debate: Impact of chemotherapy in high-risk non-metastatic prostate cancer. Transl. Androl. Urol. 2019, 8, S303–S306. [Google Scholar] [CrossRef]
- Szturz, P.; Wouters, K.; Kiyota, N.; Tahara, M.; Prabhash, K.; Noronha, V.; Adelstein, D.; Van Gestel, D.; Vermorken, J.B. Low-dose vs. High-dose cisplatin: Lessons learned from 59 chemoradiotherapy trials in head and neck cancer. Front. Oncol. 2019, 9, 86. [Google Scholar] [PubMed] [Green Version]
- Ma, H.; Yuelin, W.; Zhang, W.; Zhang, H.; Miao, Z.; Zhuang, C. Radiosensitization of human pancreatic cancer by piperlongumine analogues. Chin. Chem. Lett. 2021, 32, 1197–1201. [Google Scholar] [CrossRef]
- McMahon, S.J.; Hyland, W.B.; Muir, M.F.; Coulter, J.A.; Jain, S.; Butterworth, K.T.; Schettino, G.; Dickson, G.R.; Hounsell, A.R.; O’Sullivan, J.M.; et al. Biological consequences of nanoscale energy deposition near irradiated heavy atom nanoparticles. Sci. Rep. 2011, 1, 18. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.-J.; Wang, C.-H.; Chen, S.-T.; Chen, H.-H.; Leng, W.-H.; Chien, C.-C.; Wang, C.-L.; Kempson, I.M.; Hwu, Y.; Lai, T.-C. Enhancement of cell radiation sensitivity by pegylated gold nanoparticles. Phys. Med. Biol. 2010, 55, 931. [Google Scholar] [CrossRef] [PubMed]
- Kwatra, D.; Venugopal, A.; Anant, S. Nanoparticles in radiation therapy: A summary of various approaches to enhance radiosensitization in cancer. Transl. Cancer Res. 2013, 2, 330–342. [Google Scholar]
- Bromma, K.; Chithrani, D.B. Advances in gold nanoparticle-based combined cancer therapy. Nanomaterials 2020, 10, 1671. [Google Scholar] [CrossRef]
- Schuemann, J.; Bagley, A.F.; Berbeco, R.; Bromma, K.; Butterworth, K.T.; Byrne, H.L.; Chithrani, B.D.; Cho, S.H.; Cook, J.R.; Favaudon, V. Roadmap for metal nanoparticles in radiation therapy: Current status, translational challenges, and future directions. Phys. Med. Biol. 2020, 65, 21RM02. [Google Scholar] [CrossRef]
- Abolfazli, M.K.; Mahdavi, S.; Ataei, G. Studying effects of gold nanoparticle on dose enhancement in megavoltage radiation. J. Biomed. Phys. Eng. 2015, 5, 185. [Google Scholar]
- Wolfe, T.; Chatterjee, D.; Lee, J.; Grant, J.D.; Bhattarai, S.; Tailor, R.; Goodrich, G.; Nicolucci, P.; Krishnan, S. Targeted gold nanoparticles enhance sensitization of prostate tumors to megavoltage radiation therapy in vivo. Nanomed. Nanotechnol. Biol. Med. 2015, 11, 1277–1283. [Google Scholar] [CrossRef] [Green Version]
- Rosa, S.; Connolly, C.; Schettino, G.; Butterworth, K.T.; Prise, K.M. Biological mechanisms of gold nanoparticle radiosensitization. Cancer Nanotechnol. 2017, 8, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Penninckx, S.; Heuskin, A.-C.; Michiels, C.; Lucas, S. Gold nanoparticles as a potent radiosensitizer: A transdisciplinary approach from physics to patient. Cancers 2020, 12, 2021. [Google Scholar] [CrossRef]
- Bilynsky, C.; Millot, N.; Papa, A.-L. Radiation nanosensitizers in cancer therapy—From preclinical discoveries to the outcomes of early clinical trials. Bioeng. Transl. Med. 2021, e10256. [Google Scholar] [CrossRef]
- Umezawa, Y.; Morishima, H.; Saito, S.; Takita, T.; Umezawa, H.; Kobayashi, S.; Otsuka, M.; Narita, M.; Ohno, M. Synthesis of the pyrimidine moiety of bleomycin. J. Am. Chem. Soc. 1980, 102, 6630–6631. [Google Scholar] [CrossRef]
- Hecht, S.M. The chemistry of activated bleomycin. Acc. Chem. Res. 1986, 19, 383–391. [Google Scholar] [CrossRef]
- Georgelin, T.; Bombard, S.; Siaugue, J.M.; Cabuil, V. Nanoparticle-mediated delivery of bleomycin. Angew. Chem. Int. Ed. 2010, 49, 8897–8901. [Google Scholar] [CrossRef] [PubMed]
- Emerich, D.F.; Thanos, C.G. Targeted nanoparticle-based drug delivery and diagnosis. J. Drug Target. 2007, 15, 163–183. [Google Scholar] [CrossRef]
- Groneberg, D.A.; Giersig, M.; Welte, T.; Pison, U. Nanoparticle-based diagnosis and therapy. Curr. Drug Targets 2006, 7, 643–648. [Google Scholar] [CrossRef]
- Davis, M.E. Nanoparticle therapeutics: An emerging treatment modality for cancer. Nat. Rev. Drug Discov. 2008, 7, 771–782. [Google Scholar] [CrossRef]
- Duncan, B.; Kim, C.; Rotello, V.M. Gold nanoparticle platforms as drug and biomacromolecule delivery systems. J. Control. Release 2010, 148, 122–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrari, M. Cancer nanotechnology: Opportunities and challenges. Nat. Rev. Cancer 2005, 5, 161–171. [Google Scholar] [CrossRef]
- Lavan, D.A.; McGuire, T.; Langer, R. Small-scale systems for in vivo drug delivery. Nat. Biotechnol. 2003, 21, 1184–1191. [Google Scholar] [CrossRef]
- Homayoni, H.; Menon, J.U.; Nguyen, K.T. Chitosan-based nanoparticles for drug delivery. Rev. Nanosci. Nanotechnol. 2014, 3, 133–148. [Google Scholar] [CrossRef]
- Bindhani, B.K.; Parida, U.K.; Biswal, S.K.; Panigrahi, A.K.; Nayak, P.L. Gold nanoparticles and their biomedical applications. Rev. Nanosci. Nanotechnol. 2013, 2, 247–260. [Google Scholar] [CrossRef]
- Ghosh, P.; Han, G.; De, M.; Kim, C.K.; Rotello, V.M. Gold nanoparticles in delivery applications. Adv. Drug Deliv. Rev. 2008, 60, 1307–1315. [Google Scholar] [CrossRef] [PubMed]
- Libutti, S.K.; Paciotti, G.F.; Byrnes, A.A.; Alexander, H.R., Jr.; Gannon, W.E.; Walker, M.; Seidel, G.D.; Yuldasheva, N.; Tamarkin, L. Phase i and pharmacokinetic studies of cyt-6091, a novel pegylated colloidal gold-rhtnf nanomedicine. Clin. Cancer Res. 2010, 16, 6139–6149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paciotti, G.F.; Zhao, J.; Cao, S.; Brodie, P.J.; Tamarkin, L.; Huhta, M.; Myer, L.D.; Friedman, J.; Kingston, D.G.I. Synthesis and evaluation of paclitaxel-loaded gold nanoparticles for tumor-targeted drug delivery. Bioconjug. Chem. 2016, 27, 2646–2657. [Google Scholar] [CrossRef] [PubMed]
- Kumthekar, P. Nu-0129 in Treating Patients with Recurrent Glioblastoma or Gliosarcoma Undergoing Surgery; US National Library of Medicine: Chicago, IL, USA, 2019.
- George, A.; Canfield, S.; Jue, J.; Lewis, S.; Davenport, M.; Tammisetti, V.; Maruf, M.; Borregalaes, L.; Schwartz, J.; Halas, N. Mp46-08 a multi-institutional study of mri/ultrasound fusion imaging and biopsy in combination with nanoparticle directed focal therapy for ablation of prostate tissue. J. Urol. 2021, 206, e816. [Google Scholar] [CrossRef]
- Schuemann, J.; Berbeco, R.; Chithrani, D.B.; Cho, S.H.; Kumar, R.; McMahon, S.J.; Sridhar, S.; Krishnan, S. Roadmap to clinical use of gold nanoparticles for radiation sensitization. Int. J. Radiat. Oncol. Biol. Phys. 2016, 94, 189–205. [Google Scholar] [CrossRef] [Green Version]
- Connor, E.E.; Mwamuka, J.; Gole, A.; Murphy, C.J.; Wyatt, M.D. Gold nanoparticles are taken up by human cells but do not cause acute cytotoxicity. Small 2005, 1, 325–327. [Google Scholar] [CrossRef]
- Male, K.B.; Lachance, B.; Hrapovic, S.; Sunahara, G.; Luong, J.H.T. Assessment of cytotoxicity of quantum dots and gold nanoparticles using cell-based impedance spectroscopy. Anal. Chem. 2008, 80, 5487–5493. [Google Scholar] [CrossRef] [Green Version]
- Kadhim, R.J.; Karsh, E.H.; Taqi, Z.J.; Jabir, M.S. Biocompatibility of gold nanoparticles: In-vitro and in-vivo study. Mater. Today Proc. 2021, 42, 3041–3045. [Google Scholar] [CrossRef]
- Yang, C.; Uertz, J.; Chithrani, D.B. Colloidal gold-mediated delivery of bleomycin for improved outcome in chemotherapy. Nanomaterials 2016, 6, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schulze, G.; Jirasek, A.; Yu, M.M.L.; Lim, A.; Turner, R.F.B.; Blades, M.W. Investigation of selected baseline removal techniques as candidates for automated implementation. Appl. Spectrosc. 2005, 59, 545–574. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Bromma, K.; Chithrani, B.D. Peptide mediated in vivo tumor targeting of nanoparticles through optimization in single and multilayer in vitro cell models. Cancers 2018, 10, 84. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.; Uertz, J.; Yohan, D.; Chithrani, B.D. Peptide modified gold nanoparticles for improved cellular uptake, nuclear transport, and intracellular retention. Nanoscale 2014, 6, 12026–12033. [Google Scholar] [CrossRef] [PubMed]
- Cruje, C.; Yang, C.J.; Uertz, J.; Prooijen, M.V.; Chithrani, B.D. Optimization of peg coated nanoscale gold particles for enhanced radiation therapy. RSC Adv. 2015, 5, 101525–101532. [Google Scholar] [CrossRef]
- Thambiraj, S.; Vijayalakshmi, R.; Shankaran, D.R. An effective strategy for development of docetaxel encapsulated gold nanoformulations for treatment of prostate cancer. Sci. Rep. 2021, 11, 2808. [Google Scholar] [CrossRef]
- Chen, J.; Stubbe, J. Bleomycins: Towards better therapeutics. Nat. Rev. Cancer 2005, 5, 102–112. [Google Scholar] [CrossRef] [PubMed]
- Bannister, A.H.; Bromma, K.; Sung, W.; Monica, M.; Cicon, L.; Howard, P.; Chow, R.L.; Schuemann, J.; Chithrani, D.B. Modulation of nanoparticle uptake, intracellular distribution, and retention with docetaxel to enhance radiotherapy. Br. J. Radiol. 2020, 93, 20190742. [Google Scholar] [CrossRef]
- Alhussan, A.; Bromma, K.; Bozdoğan, E.P.; Metcalfe, A.; Karasinska, J.; Beckham, W.; Alexander, A.S.; Renouf, D.J.; Schaeffer, D.F.; Chithrani, D.B. Investigation of nano-bio interactions within a pancreatic tumor microenvironment for the advancement of nanomedicine in cancer treatment. Curr. Oncol. 2021, 28, 1962–1979. [Google Scholar] [CrossRef] [PubMed]
- Mahmoudi, M.; Azadmanesh, K.; Shokrgozar, M.A.; Journeay, W.S.; Laurent, S. Effect of nanoparticles on the cell life cycle. Chem. Rev. 2011, 111, 3407–3432. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Huang, C.; Liu, L.; Hu, R.; Qu, J. Effect of surface coating of gold nanoparticles on cytotoxicity and cell cycle progression. Nanomaterials 2018, 8, 1063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanžić, N.; Horvat, A.; Bibić, J.; Unfried, K.; Jurkin, T.; Dražić, G.; Marijanović, I.; Slade, N.; Gotić, M. Syntheses of gold nanoparticles and their impact on the cell cycle in breast cancer cells subjected to megavoltage x-ray irradiation. Mater. Sci. Eng. C 2018, 91, 486–495. [Google Scholar] [CrossRef] [PubMed]
- Cho, E.C.; Xie, J.; Wurm, P.A.; Xia, Y. Understanding the role of surface charges in cellular adsorption versus internalization by selectively removing gold nanoparticles on the cell surface with a i2/ki etchant. Nano Lett. 2009, 9, 1080–1084. [Google Scholar] [CrossRef] [PubMed]
- Chithrani, B.D. Intracellular uptake, transport, and processing of gold nanostructures. Mol. Membr. Biol. 2010, 27, 299–311. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Shi, W.; Freund, L.B. Mechanics of receptor-mediated endocytosis. Proc. Natl. Acad. Sci. USA 2005, 102, 9469–9474. [Google Scholar] [CrossRef] [Green Version]
- Chithrani, B.D.; Chan, W.C.W. Elucidating the mechanism of cellular uptake and removal of protein-coated gold nanoparticles of different sizes and shapes. Nano Lett. 2007, 7, 1542–1550. [Google Scholar] [CrossRef] [PubMed]
- Yuba, E.; Osaki, T.; Ono, M.; Park, S.; Harada, A.; Yamashita, M.; Azuma, K.; Tsuka, T.; Ito, N.; Imagawa, T.; et al. Bleomycin-loaded ph-sensitive polymer–lipid-incorporated liposomes for cancer chemotherapy. Polymers 2018, 10, 74. [Google Scholar] [CrossRef] [Green Version]
- Chi-Jen, L.; Chang-Hai, W.; Chia-Chi, C.; Tsung-Yeh, Y.; Shin-Tai, C.; Wei-Hua, L.; Cheng-Feng, L.; Kuen-Ho, L.; Hwu, Y.; Yao-Chang, L.; et al. Enhanced X-ray irradiation-induced cancer cell damage by gold nanoparticles treated by a new synthesis method of polyethylene glycol modification. Nanotechnology 2008, 19, 295104. [Google Scholar]
- Barton, M.B.; Jacob, S.; Shafiq, J.; Wong, K.; Thompson, S.R.; Hanna, T.P.; Delaney, G.P. Estimating the demand for radiotherapy from the evidence: A review of changes from 2003 to 2012. Radiother. Oncol. J. Eur. Soc. Ther. Radiol. Oncol. 2014, 112, 140–144. [Google Scholar] [CrossRef]
- Gelband, H.; Jha, P.; Sankaranarayanan, R.; Horton, S. (Eds.) Cancer: Disease Control Priorities, 3rd ed.; The International Bank for Reconstruction and Development/The World Bank: Washington, DC, USA, 2015; Volume 3. [Google Scholar]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Han, O.; Bromma, K.; Palmerley, N.; Bido, A.T.; Monica, M.; Alhussan, A.; Howard, P.L.; Brolo, A.G.; Beckham, W.; Alexander, A.S.; et al. Nanotechnology Driven Cancer Chemoradiation: Exploiting the Full Potential of Radiotherapy with a Unique Combination of Gold Nanoparticles and Bleomycin. Pharmaceutics 2022, 14, 233. https://doi.org/10.3390/pharmaceutics14020233
Han O, Bromma K, Palmerley N, Bido AT, Monica M, Alhussan A, Howard PL, Brolo AG, Beckham W, Alexander AS, et al. Nanotechnology Driven Cancer Chemoradiation: Exploiting the Full Potential of Radiotherapy with a Unique Combination of Gold Nanoparticles and Bleomycin. Pharmaceutics. 2022; 14(2):233. https://doi.org/10.3390/pharmaceutics14020233
Chicago/Turabian StyleHan, Ocean, Kyle Bromma, Nicholas Palmerley, Ariadne T. Bido, Mesa Monica, Abdulaziz Alhussan, Perry L. Howard, Alexandre G. Brolo, Wayne Beckham, Abraham S. Alexander, and et al. 2022. "Nanotechnology Driven Cancer Chemoradiation: Exploiting the Full Potential of Radiotherapy with a Unique Combination of Gold Nanoparticles and Bleomycin" Pharmaceutics 14, no. 2: 233. https://doi.org/10.3390/pharmaceutics14020233
APA StyleHan, O., Bromma, K., Palmerley, N., Bido, A. T., Monica, M., Alhussan, A., Howard, P. L., Brolo, A. G., Beckham, W., Alexander, A. S., & Chithrani, D. B. (2022). Nanotechnology Driven Cancer Chemoradiation: Exploiting the Full Potential of Radiotherapy with a Unique Combination of Gold Nanoparticles and Bleomycin. Pharmaceutics, 14(2), 233. https://doi.org/10.3390/pharmaceutics14020233