
Post-Injury Buprenorphine Administration Is Associated with Long-Term Region-Specific Glial Alterations in Rats
 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Surgical Preparation, Injury Induction, and Drug Administration
2.3. Physiologic Assessment
2.4. Whisker Nuisance Task
2.5. Tissue Processing
2.6. Assessment of Cell Damage/Loss
2.7. Assessment of Neuronal Membrane Disruption
2.8. Immunohistochemistry
2.9. Assessment of Microglial Morphology
2.10. Assessment of Astrocyte Morphology
2.11. Assessment of Myelin Integrity
2.12. Quantification of Protein Expression
2.13. Statistical Analysis
3. Results
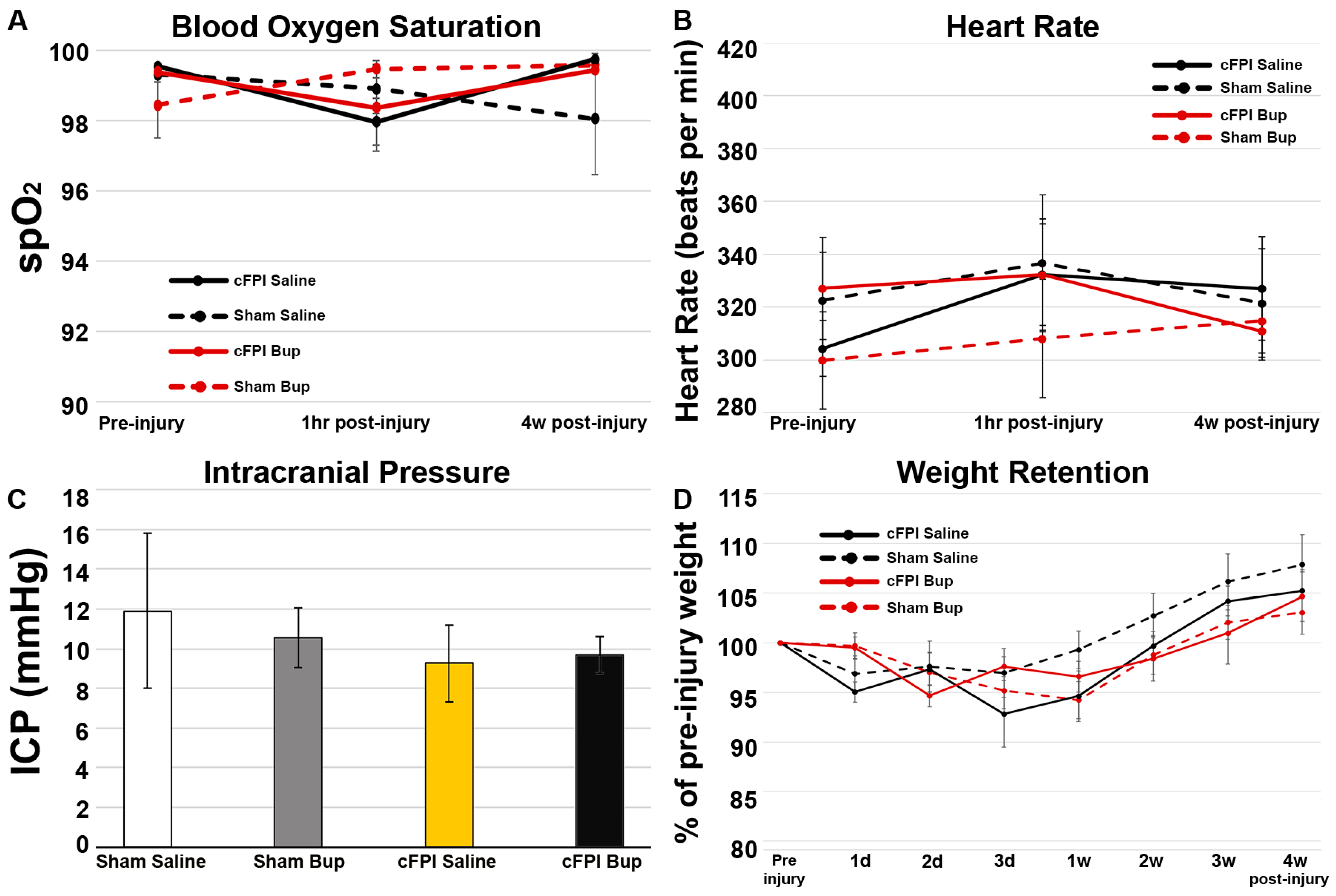
3.1. Physiology Was Not Impacted by Bup-SR-Lab Treatment
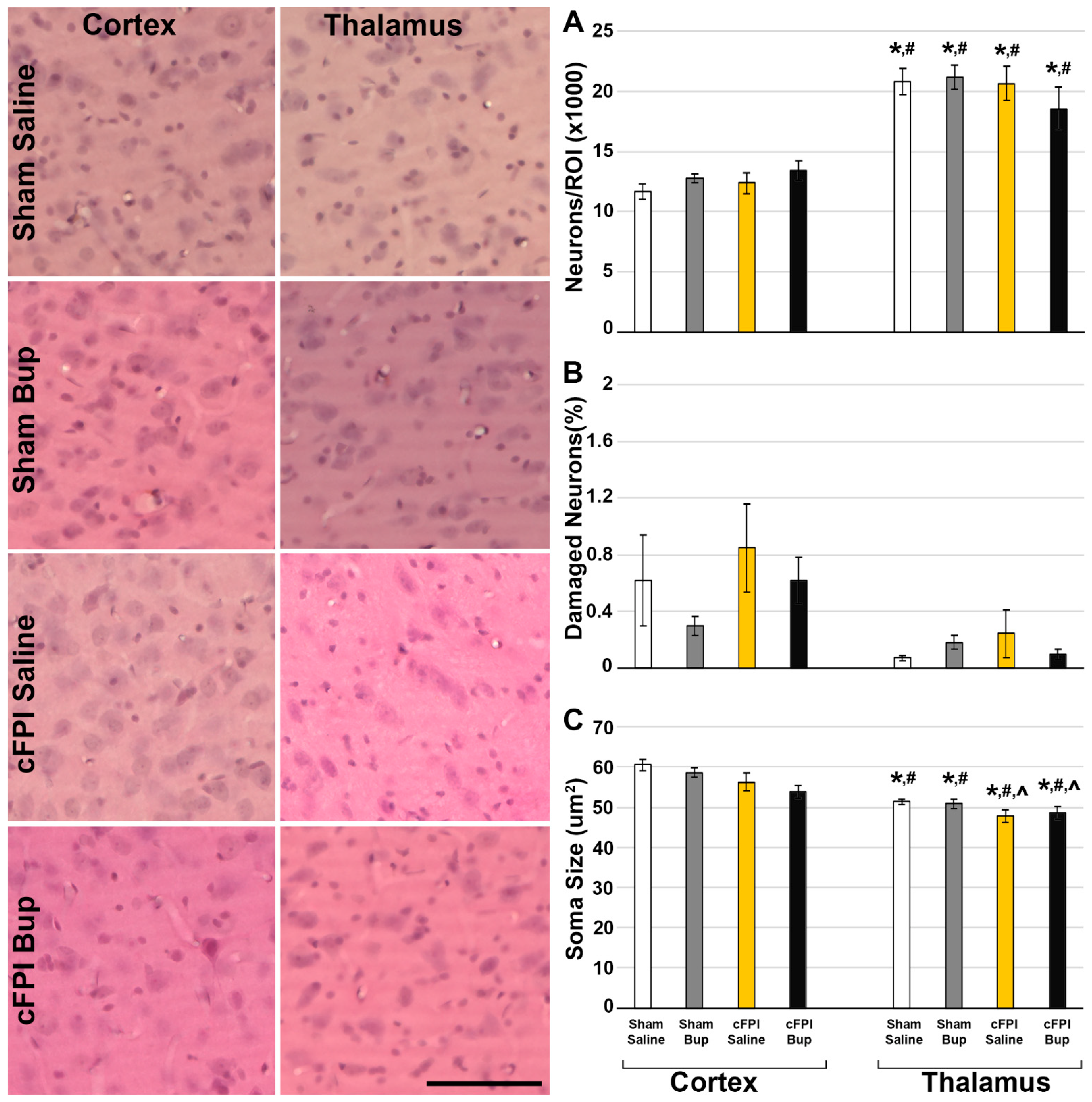
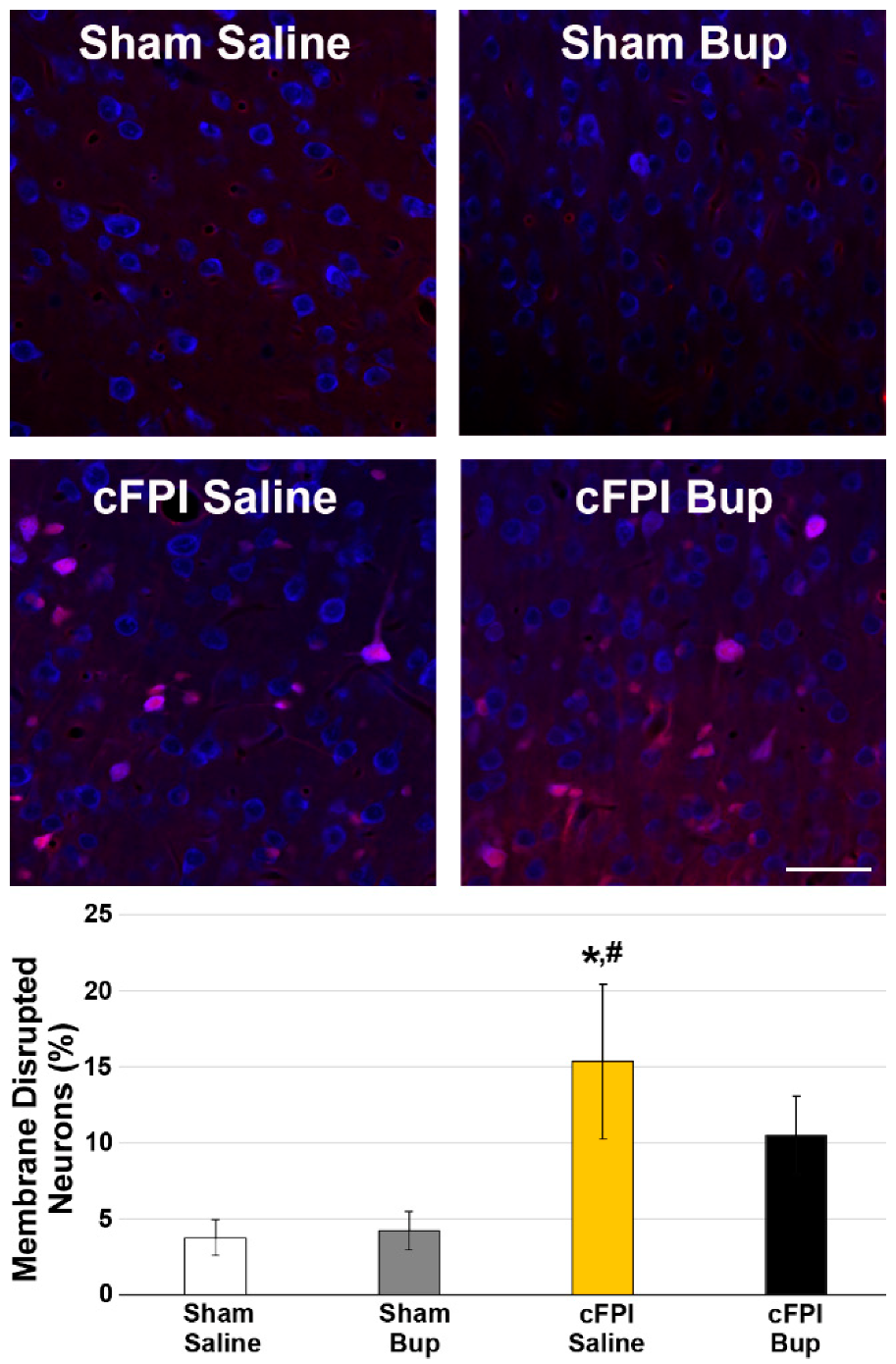
3.2. Neuronal Damage
3.3. Cytokine Changes Chronically following Bup-SR-Lab Administration
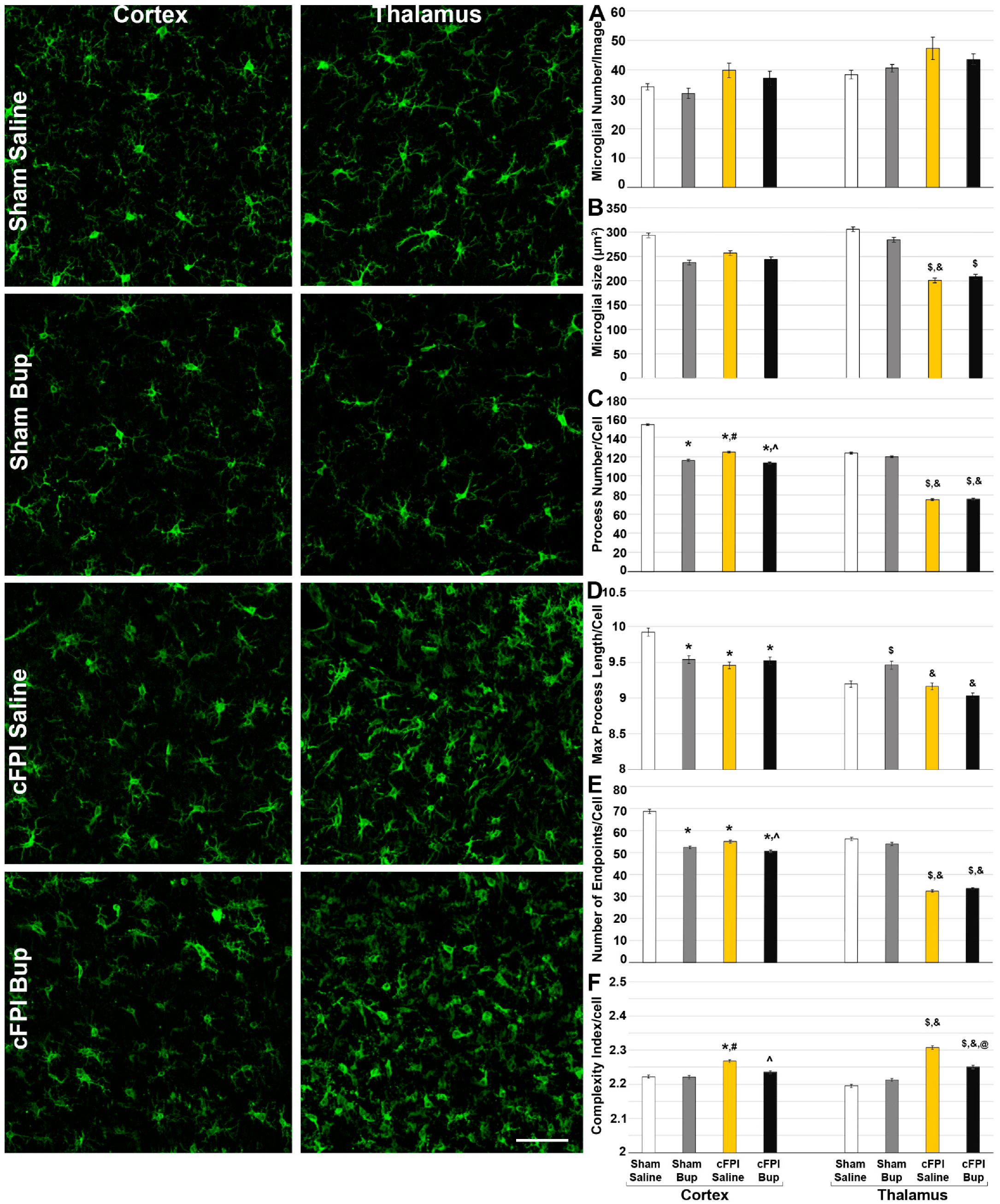
3.4. Microglial Morphology Is Altered Chronically following Bup-SR-Lab Administration
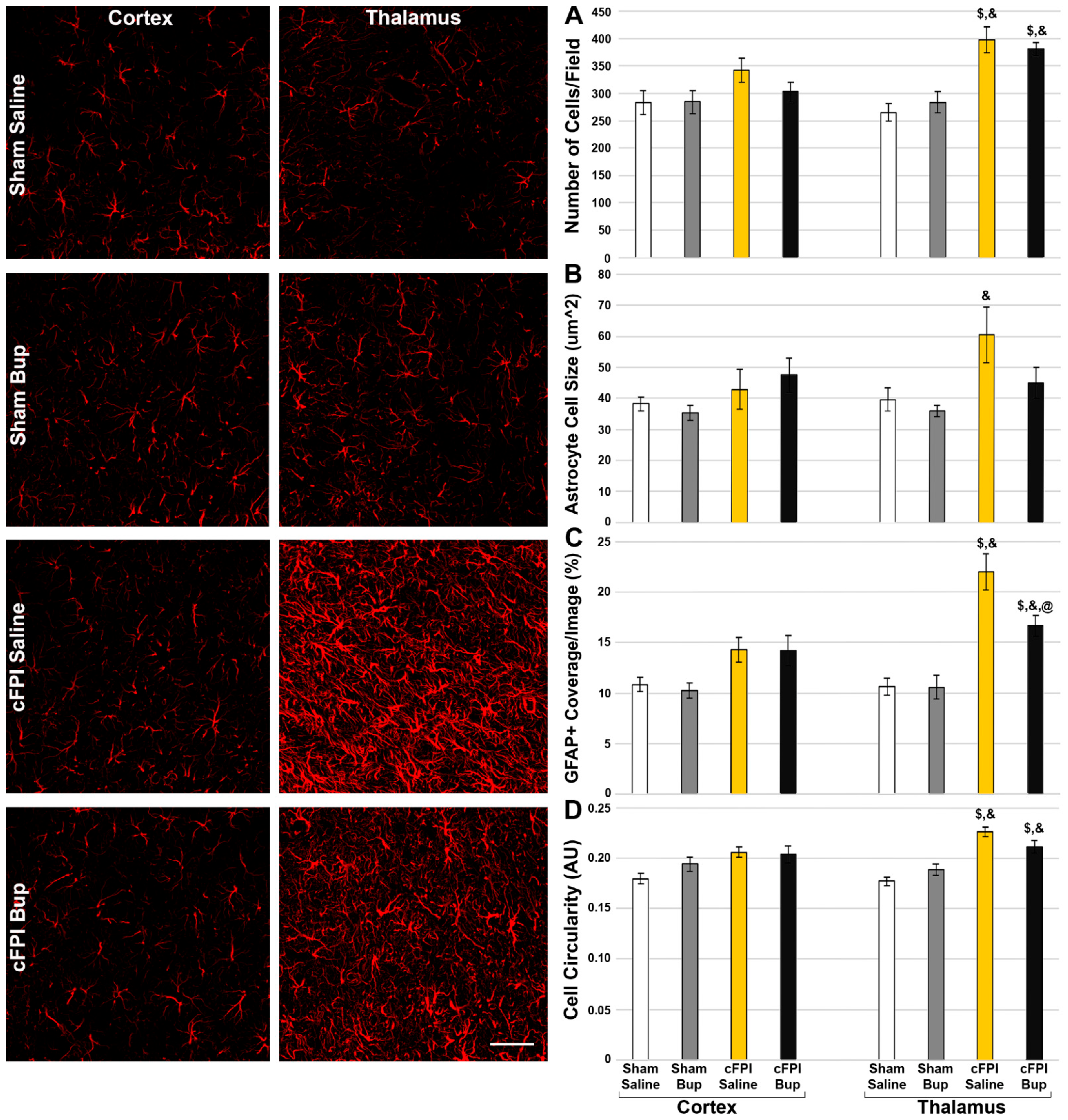
3.5. Astrocyte Changes Chronically following Bup-SR-Lab Administration
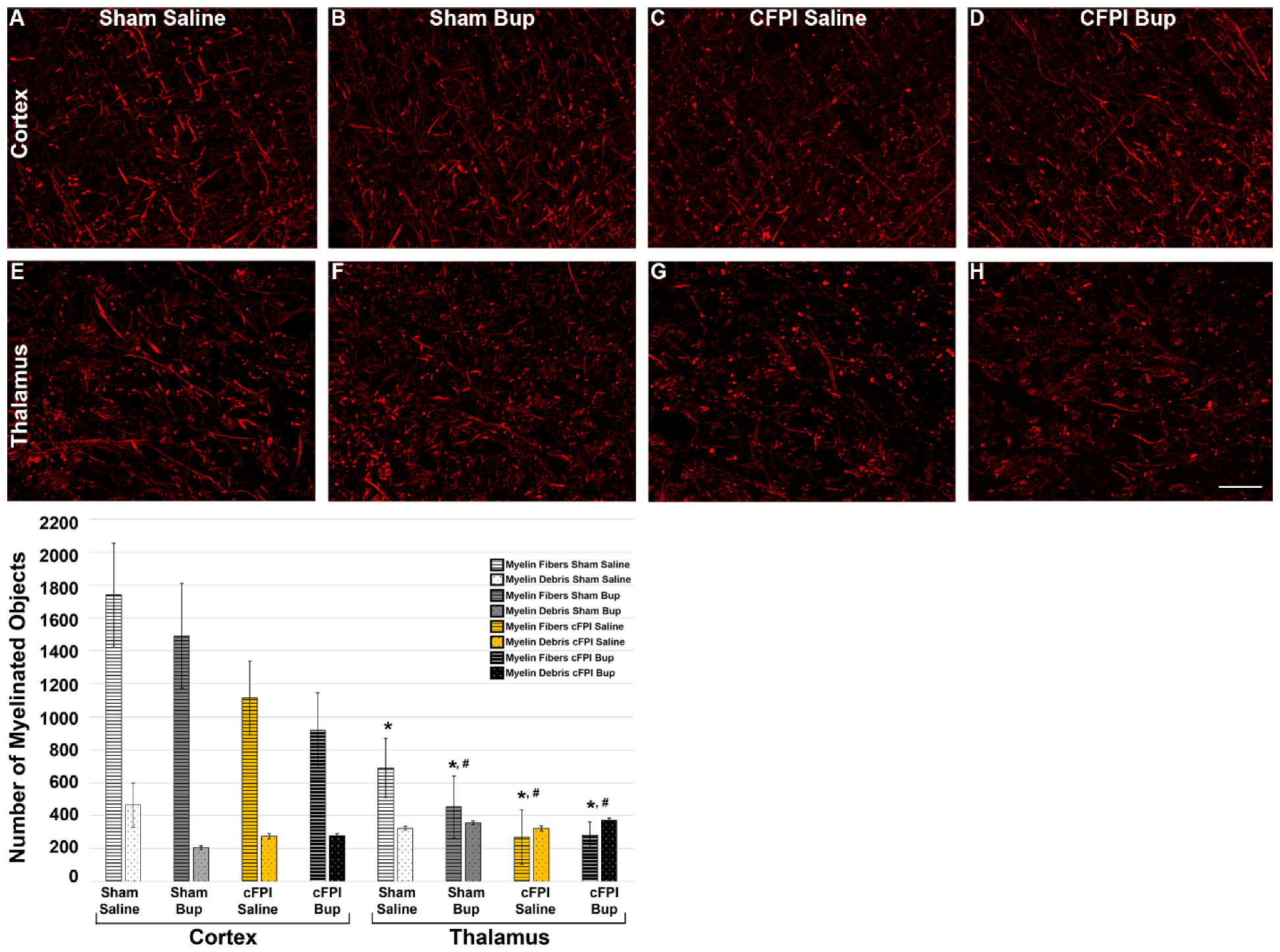
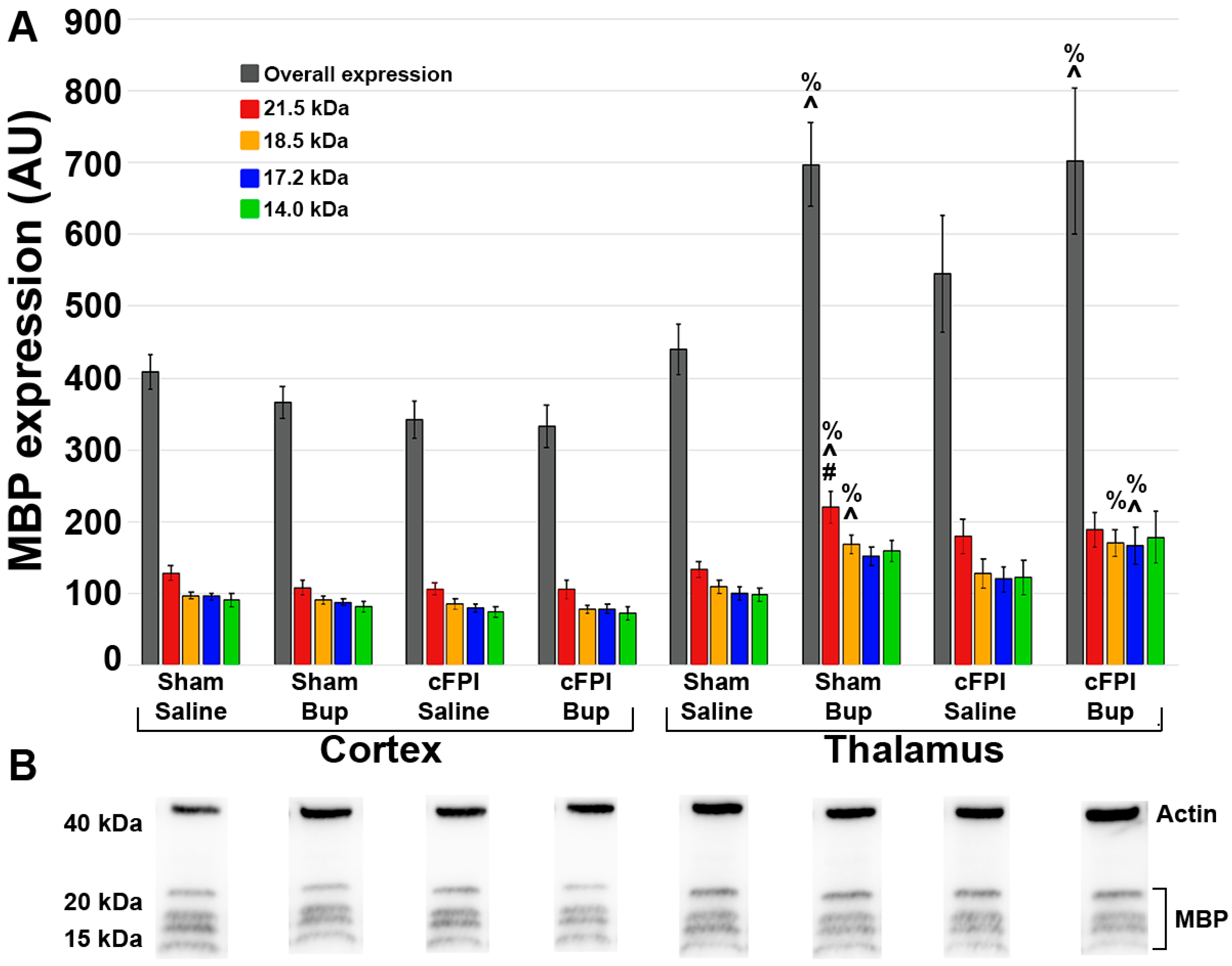
3.6. Treatment with Bup-SR-Lab Altered Myelin Fibers in an Injury and Region-Specific Manner at 4 Weeks Post-Injury
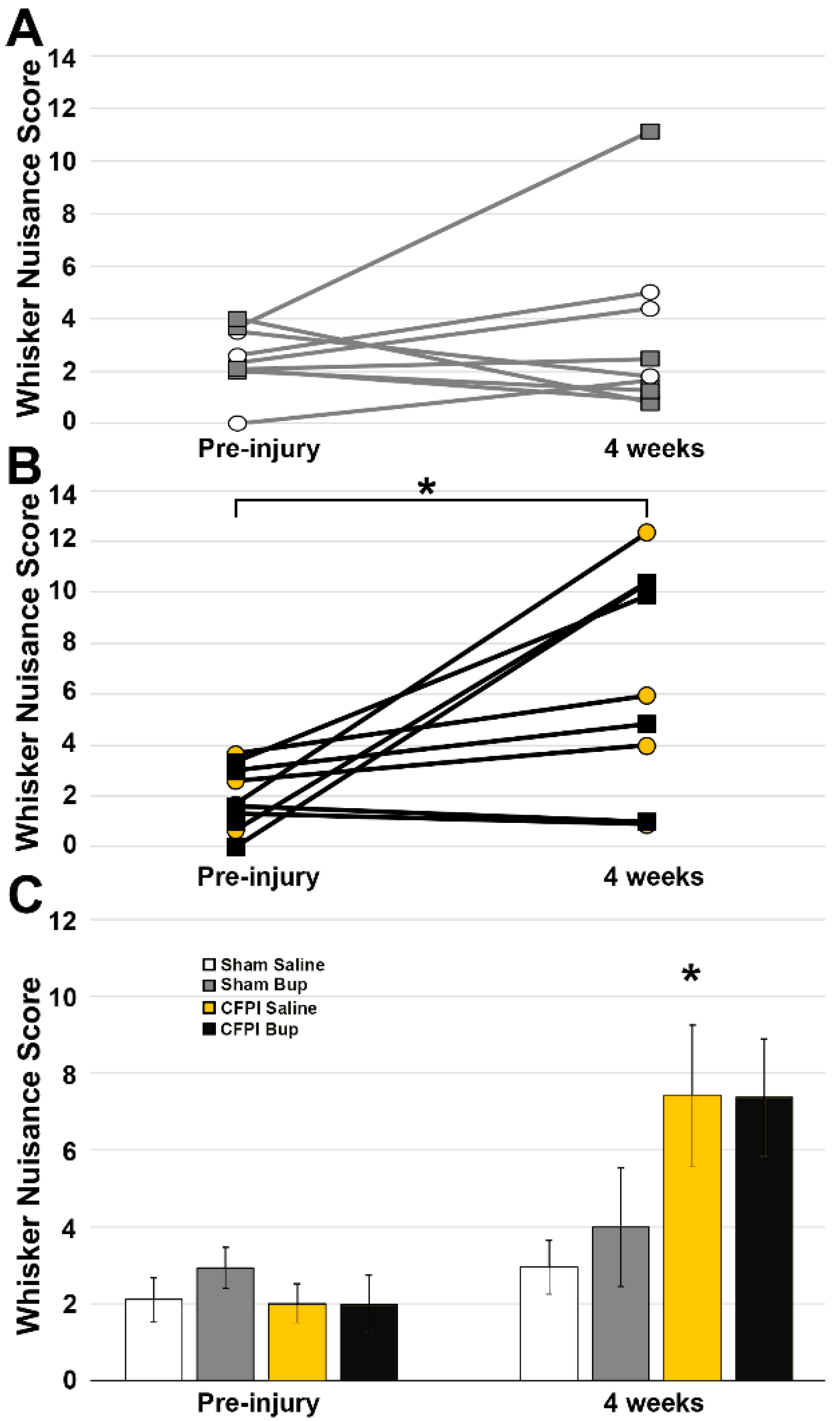
3.7. Post-Injury Somatosensory Hypersensitivity Is Not Altered by Bup-SR-Lab Treatment
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Coronado, V.G.; Xu, L.; Basavaraju, S.V.; McGuire, L.C.; Wald, M.M.; Faul, M.; Hemphill, J.D. Surveillance for traumatic brain injury-related deaths—United States, 1997–2007. Morb. Mortal. Wkly. Rep. Surveill. Summ. 2011, 60, 1–32. [Google Scholar]
- Bramlett, H.M.; Dietrich, W.D. Pathophysiology of cerebral ischemia and brain trauma: Similarities and differences. J. Cereb. Blood Flow Metab. 2004, 24, 133–150. [Google Scholar] [CrossRef] [PubMed]
- Taylor, C.A.; Bell, J.M.; Breiding, M.J.; Xu, L. Traumatic Brain Injury–Related Emergency Department Visits, Hospitalizations, and Deaths—United States, 2007 and 2013. MMWR. Surveill. Summ. 2017, 66, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Brau, R.H.; Acevedo-Salas, Y.; Giovannetti, K. Epidemiological trends of traumatic brain and spinal cord injury in Puerto Rico from November 10th, 2006, through May 24th, 2011. Puerto. Rico. Health Sci. J. 2018, 37, 67–77. [Google Scholar]
- Center-TBI. Traumatic Brain Injury Fact sheFets and Policy Brief. Available online: https://www.center-tbi.eu/files/news/21571f81-20b8-4860-a3dd-1f6e27d02b3d.pdf (accessed on 10 August 2022).
- Gardner, R.C.; Yaffe, K. Epidemiology of mild traumatic brain injury and neurodegenerative disease. Mol. Cell. Neurosci. 2015, 66, 75–80. [Google Scholar] [CrossRef]
- Ortiz-Prado, E.; Mascialino, G.; Paz, C.; Rodriguez-Lorenzana, A.; Gómez-Barreno, L.; Simbaña-Rivera, K.; Diaz, A.M.; Coral-Almeida, M.; Espinosa, P.S. A Nationwide Study of Incidence and Mortality Due to Traumatic Brain Injury in Ecuador (2004–2016). Neuroepidemiology 2020, 54, 33–44. [Google Scholar] [CrossRef]
- Koskinen, S.; Alaranta, H. Traumatic brain injury in Finland 1991-2005: A nationwide register study of hospitalized and fatal TBI. Brain Inj. 2008, 22, 205–214. [Google Scholar] [CrossRef]
- Bramlett, H.M.; Dietrich, W.D. Progressive damage after brain and spinal cord injury: Pathomechanisms and treatment strategies. Prog. Brain Res. 2007, 161, 125–141. [Google Scholar]
- Grossman, E.J.; Inglese, M. The Role of Thalamic Damage in Mild Traumatic Brain Injury. J. Neurotrauma 2016, 33, 163–167. [Google Scholar] [CrossRef]
- Little, D.M.; Kraus, M.F.; Joseph, J.; Geary, E.K.; Susmaras, T.; Zhou, X.J.; Pliskin, N.; Gorelick, P.B. Thalamic integrity underlies executive dysfunction in traumatic brain injury. Neurology 2010, 74, 558–564. [Google Scholar] [CrossRef]
- National Research Council. Guide Laboratory Animals for the Care and Use of Eighth Edition Committee for the Update of the Guide for the Care and Use of Laboratory Animals Institute for Laboratory Animal Research Division on Earth and Life Studies; National Research Council of the National Academies; The National Academies Press: Washington, DC, USA, 2011. [Google Scholar]
- Murphy, A.; Barbaro, J.; Martínez-Aguado, P.; Chilunda, V.; Jaureguiberry-Bravo, M.; Berman, J.W. The Effects of Opioids on HIV Neuropathogenesis. Front. Immunol. 2019, 10, 2445. [Google Scholar] [CrossRef] [PubMed]
- Villiger, J.W.; Taylor, K.M. Buprenorphine: Characteristics of binding sites in the rat central nervous system. Life Sci. 1981, 29, 2699–2708. [Google Scholar] [CrossRef]
- Boas, R.A.; Villiger, J.W. Clinical Actions of Fentanyl and Buprenorphine the Significance of Receptor Binding. Br. J. Anaesth. 1985, 57, 192–196. [Google Scholar] [CrossRef] [PubMed]
- Rosenblum, A.; Marsch, L.A.; Joseph, H.; Portenoy, R.K. Opioids and the Treatment of Chronic Pain: Controversies, Current Status, and Future Directions. Exp. Clin. Psychopharmacol. 2008, 16, 405–416. [Google Scholar] [CrossRef] [PubMed]
- Alamaw, E.D.; Franco, B.D.; Jampachaisri, K.; Huss, M.K.; Pacharinsak, C. Extended-release Buprenorphine, an FDA-indexed Analgesic, Attenuates Mechanical Hypersensitivity in Rats (Rattus norvegicus). J. Am. Assoc. Lab. Anim. Sci. 2022, 61, 81–88. [Google Scholar] [CrossRef]
- Blaney, A.; Jampachaisri, K.; Huss, M.K.; Pacharinsak, C. Sustained release buprenorphine effectively attenuates postoperative hypersensitivity in an incisional pain model in neonatal rats (Rattus norvegicus). PLoS ONE 2021, 16, e0246213. [Google Scholar] [CrossRef]
- Clark, T.S.; Clark, D.D.; Hoyt, R.F. Pharmacokinetic comparison of sustained-release and standard buprenorphine in mice. J. Am. Assoc. Lab. Anim. Sci. 2014, 53, 387–391. [Google Scholar]
- Chum, H.H.; Jampachairsri, K.; McKeon, G.P.; Yeomans, D.C.; Pacharinsak, C.; Felt, S.A. Antinociceptive effects of sustained-release buprenorphine in a model of incisional pain in rats (Rattus norvegicus). J. Am. Assoc. Lab. Anim. Sci. 2014, 53, 193–197. [Google Scholar]
- Dooley, S.B.; Aarnes, T.K.; Lakritz, J.; Lerche, P.; Bednarski, R.M.; Hubbell, J.A.E. Pharmacokinetics and pharmacodynamics of buprenorphine and sustained-release buprenorphine after administration to adult alpacas. Am. J. Vet. Res. 2017, 78, 321–329. [Google Scholar] [CrossRef]
- Ryu, J.; Stone, P.; Lee, S.; Payne, B.; Gorse, K.; Lafrenaye, A. Buprenorphine alters microglia and astrocytes acutely following diffuse traumatic brain injury. Sci. Rep. 2021, 11, 8620. [Google Scholar] [CrossRef]
- Percie Du Sertid, N.; Hurst, V.; Ahluwaliaid, A.; Almid, S.; Aveyid, M.T.; Baker, M.; Browneid, W.J.; Clarkid, A.; Cuthillid, I.C.; Dirnaglid, U.; et al. The ARRIVE Guidelines 2.0: Updated Guidelines for Reporting Animal Research. PLoS Biol. 2020, 49, 1769–1777. [Google Scholar] [CrossRef]
- Lafrenaye, A.D.; Krahe, T.E.; Povlishock, J.T. Moderately elevated intracranial pressure after diffuse traumatic brain injury is associated with exacerbated neuronal pathology and behavioral morbidity in the rat. J. Cereb. Blood Flow Metab. Off. J. Int. Soc. Cereb. Blood Flow Metab. 2014, 34, 1628–1636. [Google Scholar] [CrossRef]
- Hernandez, M.L.; Marone, M.; Gorse, K.M.; Lafrenaye, A.D. Cathepsin B Relocalization in Late Membrane Disrupted Neurons Following Diffuse Brain Injury in Rats. ASN Neuro 2022, 14, 175909142210991. [Google Scholar] [CrossRef] [PubMed]
- Dixon, C.E.; Dixon, C.E.; Lyeth, B.G.; Povlishock, G.T.; Findling, R.L.; Hamm, R.J.; Marmarou, A.; Young, H.F.; Hayes, R.L. A fluid percussion model of experimental brain injury in the rat. J. Neurosurg. 1987, 67, 110–119. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, M.L.; Chatlos, T.; Gorse, K.M.; Lafrenaye, A.D. Neuronal Membrane Disruption Occurs Late Following Diffuse Brain Trauma in Rats and Involves a Subpopulation of NeuN Negative Cortical Neurons. Front. Neurol. 2019, 10, 1238. [Google Scholar] [CrossRef]
- McNamara, K.C.; Lisembee, A.M.; Lifshitz, J. The whisker nuisance task identifies a late-onset, persistent sensory sensitivity in diffuse brain-injured rats. J. Neurotrauma 2010, 27, 695–706. [Google Scholar] [CrossRef]
- Learoyd, A.E.; Lifshitz, J. Comparison of rat sensory behavioral tasks to detect somatosensory morbidity after diffuse brain-injury. Behav. Brain Res. 2012, 226, 197–204. [Google Scholar] [CrossRef]
- Thomas, T.C.; Hinzman, J.M.; Gerhardt, G.A.; Lifshitz, J. Hypersensitive glutamate signaling correlates with the development of late-onset behavioral morbidity in diffuse brain-injured circuitry. J. Neurotrauma 2012, 29, 187–200. [Google Scholar] [CrossRef]
- Krishna, G.; Bromberg, C.; Connell1, E.C.; Mian, E.; Hu, C.; Lifshitz, J.; Adelson, P.D.; Thomas, T.C. Traumatic Brain Injury-Induced Sex-Dependent Changes in Late-Onset Sensory Hypersensitivity and Glutamate Neurotransmission. Front. Neurol. 2020, 11, 749. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef]
- Simon, C.M.; Sharif, S.; Tan, R.P.; LaPlaca, M.C. Spinal cord contusion causes acute plasma membrane damage. J. Neurotrauma 2009, 26, 563–574. [Google Scholar] [CrossRef] [PubMed]
- Gorse, K.M.; Lafrenaye, A.D. The importance of inter-species variation in traumatic brain injury-induced alterations of microglial-axonal interactions. Front. Neurol. 2018, 9, 778. [Google Scholar] [CrossRef] [PubMed]
- Lafrenaye, A.D.; Todani, M.; Walker, S.A.; Povlishock, J.T. Microglia processes associate with diffusely injured axons following mild traumatic brain injury in the micro pig. J. Neuro inflamm. 2015, 12, 186. [Google Scholar] [CrossRef] [PubMed]
- Kelley, B.J.; Lifshitz, J.; Povlishock, J.T. Neuroinflammatory responses after experimental diffuse traumatic brain injury. J. Neuropathol. Exp. Neurol. 2007, 66, 989–1001. [Google Scholar] [CrossRef] [PubMed]
- Lifshitz, J.; Kelley, B.J.; Povlishock, J.T. Perisomatic thalamic axotomy after diffuse traumatic brain injury is associated with atrophy rather than cell death. J. Neuropathol. Exp. Neurol. 2007, 66, 218–229. [Google Scholar] [CrossRef] [PubMed]
- Witcher, K.G.; Bray, C.E.; Dziabis, J.E.; McKim, D.B.; Benner, B.N.; Rowe, R.K.; Kokiko-Cochran, Q.N.; Popovich, P.G.; Lifshitz, J.; Eiferman, D.S.; et al. Traumatic brain injury-induced neuronal damage in the somatosensory cortex causes formation of rod-shaped microglia that promote astrogliosis and persistent neuroinflammation. Glia 2018, 66, 2719–2736. [Google Scholar] [CrossRef] [PubMed]
- Lafrenaye, A.D.; McGinn, M.J.; Povlishock, J.T. Increased intracranial pressure after diffuse traumatic brain injury exacerbates neuronal somatic membrane poration but not axonal injury: Evidence for primary intracranial pressure-induced neuronal perturbation. J. Cereb. Blood Flow Metab. 2012, 32, 1919–1932. [Google Scholar] [CrossRef]
- Byrnes, K.R.; Loane, D.J.; Stoica, B.A.; Zhang, J.; Faden, A.I. Delayed mGluR5 activation limits neuroinflammation and neurodegeneration after traumatic brain injury. J. Neuroinflamm. 2012, 9, 43. [Google Scholar] [CrossRef]
- Navarro, K.; Jampachaisri, K.; Huss, M.; Pacharinsak, C. Lipid bound extended release buprenorphine (high and low doses) and sustained release buprenorphine effectively attenuate post-operative hypersensitivity in an incisional pain model in mice (Mus musculus). Anim. Model Exp. Med. 2021, 4, 129–137. [Google Scholar] [CrossRef]
- Hamood, Y.; Abdullah, M.; Ghoul, H.E.; Saad, N.; Dysko, R.C.; Zhang, Z. Sex specific effects of buprenorphine on behavior, astrocytic opioid receptor expression and neuroinflammation after pediatric traumatic brain injury in mice. Brain Behav. Immun. Health 2022, 22, 100469. [Google Scholar] [CrossRef]
- LaPlaca, M.C.; Lessinga, M.C.; Pradoa, G.R.; Zhoub, R.; Tatea, C.C.; Geddes-Kleinc, D.; Meaneyc, D.F.; Zhangb, L. Mechanoporation is a potential indicator of tissue strain and subsequent degeneration following experimental traumatic brain injury. Clin. Biomech. 2019, 64, 2–13. [Google Scholar] [CrossRef] [PubMed]
- Wofford, K.L.; Harrisac, J.P.; Browneac, K.D.; Brownac, D.P.; Grovolaac, M.R.; Mietusc, C.J.; Wolfac, J.A.; Dudaad, J.E.; Putte, M.E.; Spillerb, K.L.; et al. Rapid neuroinflammatory response localized to injured neurons after diffuse traumatic brain injury in swine. Exp. Neurol. 2017, 290, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Greer, J.E.; McGinn, M.J.; Povlishock, J.T. Diffuse traumatic axonal injury in the mouse induces atrophy, c-Jun activation, and axonal outgrowth in the axotomized neuronal population. J. Neurosci. Off. J. Soc. Neurosci. 2011, 31, 5089–5105. [Google Scholar] [CrossRef] [PubMed]
- García-Juárez, M.; Camacho-Morales, A. Defining the Role of Anti- and Pro-inflammatory Outcomes of Interleukin-6 in Mental Health. Neuroscience 2022, 492, 32–46. [Google Scholar] [CrossRef] [PubMed]
- Zhu, T.; Yao, Z.; Yuan, H.; Lu, B.; Yang, S. Changes of interleukin-1 beta, tumor necrosis factor alpha and interleukin-6 in brain and plasma after brain injury in rats. Chin. J. Traumatol. 2004, 7, 32–35. [Google Scholar]
- Rowe, R.K.; Ellis, G.I.; Harrison, J.L.; Bachstetter, A.D.; Corder, G.F.; Eldik, L.J.V.; Taylor, B.K.; Marti, F.; Lifshitz, J. Diffuse traumatic brain injury induces prolonged immune dysregulation and potentiates hyperalgesia following a peripheral immune challenge. Mol. Pain 2016, 12, 1–12. [Google Scholar] [CrossRef]
- Curvello, V.; Hekierski, H.; Riley, J.; Vavilala, M.; Armstead, W.M. Sex and age differences in phenylephrine mechanisms and outcomes after piglet brain injury. Pediatr. Res. 2017, 82, 108–113. [Google Scholar] [CrossRef]
- Chen, L.; Zhu, L.; Lu, D.; Wu, Z.; Han, Y.; Xu, P.; Chang, L.; Wu, Q. Interleukin 4 Affects Epilepsy by Regulating Glial Cells: Potential and Possible Mechanism. Front. Mol. Neurosci. 2020, 13, 554547. [Google Scholar] [CrossRef]
- Gadani, S.P.; Cronk, J.C.; Norris, G.T.; Kipnis, J. Interleukin-4: A Cytokine to Remember. J. Immunol. 2012, 189, 4213–4219. [Google Scholar] [CrossRef]
- Sun, J.; Guo, W.; Du, X. Buprenorphine differentially affects M1- and M2-polarized macrophages from human umbilical cord blood. Eur. Cytokine Netw. 2017, 28, 85–92. [Google Scholar] [CrossRef]
- Jiang, X.; Yi, S.; Liu, Q.; Zhang, J. The secretome of microglia induced by IL-4 of IFN-γ differently regulate proliferation, differentiation and survival of adult neural stem/progenitor cell by targeting the PI3K-Akt pathway. Cytotechnology 2022, 74, 407–420. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Zhi, F.; Peng, Y.; Mao, J.; Balboni, G.; Yang, Y.; Xia, Y. A Critical Role of δ-Opioid Receptor in Anti-microglial Activation Under Stress. Front. Aging Neurosci. 2022, 14, 847386. [Google Scholar] [CrossRef] [PubMed]
- Vicario, N.; Denaro, S.; Turnaturi, R.; Longhitano, L.; Spitale, F.M.; Spoto, S.; Marrazzo, A.; Zappalà, A.; Tibullo, D.; Volti, G.L.; et al. Mu and Delta Opioid Receptor Targeting Reduces Connexin 43-Based Heterocellular Coupling during Neuropathic Pain. Int. J. Mol. Sci. 2022, 23, 5864. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Bachstetter, A.D.; Cass, W.A.; Lifshitz, J.; Bing, G. Pioglitazone Attenuates Neuroinflammation and Promotes Dopaminergic Neuronal Survival in the Nigrostriatal System of Rats after Diffuse Brain Injury. J. Neurotrauma 2017, 34, 414–422. [Google Scholar] [CrossRef]
- Ziebell, J.M.; Taylor, S.E.; Cao, T.; Harrison, J.L.; Lifshitz, J. Rod microglia: Elongation, alignment, and coupling to form trains across the somatosensory cortex after experimental diffuse brain injury. J. Neuro inflamm. 2012, 9, 247. [Google Scholar] [CrossRef]
- Pestana, F.; Edwards-Faret, G.; Belgard, T.G.; Martirosyan, A.; Holt, M.G. No Longer Underappreciated: The Emerging Concept of Astrocyte Heterogeneity in Neuroscience. Brain Sci. 2020, 10, 168. [Google Scholar] [CrossRef]
- Farhy-Tselnicker, I.; Boisvert, M.M.; Liu, H.; Dowling, C.; Erikson, G.A.; Blanco-Suarez, E.; Farhy, C.; Shokhirev, M.N.; Ecker, J.; Allen, N.J. Activity-Dependent Modulation of Synapse-Regulating Genes in Astrocytes. eLife 2021, 10, e70514. [Google Scholar] [CrossRef]
- Somaiya, R.D.; Somaiya, R.D.; Huebschman, N.A.; Chaunsali, L.; Sabbagh, U.; Carrillo, G.L.; Tewari, B.P.; Fox, M.A. Development of astrocyte morphology and function in mouse visual thalamus. J. Comp. Neurol. 2022, 530, 945–962. [Google Scholar] [CrossRef]
- Bayraktar, O.A.; Bartels, T.; Holmqvist, S.; Kleshchevnikov, V.; Martirosyan, A.; Polioudakis, D.; Haim, L.B.; Young, A.M.H.; Batiuk, M.Y.; Prakash, K.; et al. Astrocyte layers in the mammalian cerebral cortex revealed by a single-cell in situ transcriptomic map. Nat. Neurosci. 2020, 23, 500–509. [Google Scholar] [CrossRef]
- Torres-Platas, S.G.; Nagy, C.; Wakid, M.; Turecki, G.; Mechawar, N. Glial fibrillary acidic protein is differentially expressed across cortical and subcortical regions in healthy brains and downregulated in the thalamus and caudate nucleus of depressed suicides. Mol. Psychiatry 2016, 21, 509–515. [Google Scholar] [CrossRef]
- Kimura, A.; Takekoshi, A.; Yoshikura, N.; Hayashi, Y.; Shimohata, T. Clinical characteristics of autoimmune GFAP astrocytopathy. J. Neuroimmunol. 2019, 332, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.-H.; Tao, P.-L.; Tsay, H.-J.; Chiang, Y.-C.; Chang, W.-T.; Ho, I.K.; Shie, F.-S. Dextromethorphan Dampens Neonatal Astrocyte Activation and Endoplasmic Reticulum Stress Induced by Prenatal Exposure to Buprenorphine. Behav. Neurol. 2021, 2021, 6301458. [Google Scholar] [CrossRef] [PubMed]
- Üçeyler, N.; Topuzoǧlu, T.; Schießer, P.; Hahnenkamp, S.; Sommer, C. IL-4 deficiency is associated with mechanical hypersensitivity in mice. PLoS ONE 2011, 6, e28205. [Google Scholar] [CrossRef] [PubMed]
- Reiss, D.; Maurin, H.; Audouard, E.; Martínez-Navarro, M.; Xue, Y.; Herault, Y.; Maldonado, R.; Cabañero, D.; Gaveriaux-Ruff, C. Delta Opioid Receptor in Astrocytes Contributes to Neuropathic Cold Pain and Analgesic Tolerance in Female Mice. Front. Cell Neurosci. 2021, 15, 745178. [Google Scholar] [CrossRef]
- Lampron, A.; Laflamme, N.; Préfontaine, P.; Plante, M.-M.; Sánchez, M.G.; Yong, Y.V.W.; Stys, P.K.; Tremblay, M.-E. Serge Rivest Inefficient clearance of myelin debris by microglia impairs remyelinating processes. J. Exp. Med. 2015, 212, 481–495. [Google Scholar] [CrossRef] [PubMed]
- Mierzwa, A.J.; Marion, C.M.; Sullivan, G.M.; Mcdaniel, D.P.; Armstrong, R.C. Components of Myelin Damage and Repair in the Progression of White Matter Pathology After Mild Traumatic. Brain Inj. 2015, 74, 218–232. [Google Scholar] [CrossRef]
- Wang, F.; Mei, F. Kappa opioid receptor and oligodendrocyte remyelination. Vitam. Horm. 2019, 111, 281–297. [Google Scholar]
- Liu, M.C.; Akle, V.; Zheng, W.; Kitlen, J.; O’Steen, B.; Larner, S.F.; Dave, J.R.; Tortella, F.C.; Hayes, R.L.; Wang, K.K.W. Extensive degradation of myelin basic protein isoforms by calpain following traumatic brain injury. J. Neurochem. 2006, 98, 700–712. [Google Scholar] [CrossRef]
- Harauz, G.; Boggs, J.M. Myelin management by the 18.5–kDa and 21.5–kDa classic myelin basic protein isoforms. J. Neurochem. 2013, 125, 334–361. [Google Scholar] [CrossRef]
- Flygt, J.; Gumucio, A.; Ingelsson, M.; Skoglund, K.; Holm, J.; Alafuzoff, I.; Marklund, N. Human Traumatic Brain Injury Results in Oligodendrocyte Death and Increases the Number of Oligodendrocyte Progenitor Cells. J. Neuropathol. Exp. Neurol. 2016, 75, 503–515. [Google Scholar] [CrossRef]
- Dent, K.A.; Christie, K.J.; Bye, N.; Basrai, H.S.; Turbic, A.; Habgood, M.; Cate, H.S.; Turnley, A.M. Oligodendrocyte Birth and Death following Traumatic Brain Injury in Adult Mice. PLoS ONE 2015, 10, e0121541. [Google Scholar] [CrossRef] [PubMed]
- Myers, P.H.; Goulding, D.R.; Wiltshire, R.A.; McGee, C.A.; Dickerson, A.B.; Comins, M.M.; Shi, M.; Kissling, G.E.; Lih, F.B.; Deterding, L.J.; et al. Serum Buprenorphine Concentrations and Behavioral Activity in Mice After a Single Subcutaneous Injection of Simbadol, Buprenorphine SR-LAB, or Standard Buprenorphine. J. Am. Assoc. Lab. Anim. Sci. 2021, 60, 661–666. [Google Scholar] [CrossRef] [PubMed]
- Becerra, L.; Upadhyay, J.; Chang, P.-C.; Bishop, J.; Anderson, J.; Baumgartner, R.; Schwarz, A.J.; Coimbra, A.; Wallin, D.; Nutile, L.; et al. Parallel Buprenorphine phMRI Responses in Conscious Rodents and Healthy Human Subjects. J. Pharmacol. Exp. Ther. 2013, 345, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Erbs, E.; Faget, L.; Scherrer, G.; Matifas, A.; Filliol, D.; Vonesch, J.-L.; Koch, M.; Kessler, P.; Hentsch, D.; Birling, M.-C.; et al. A mu–delta opioid receptor brain atlas reveals neuronal co-occurrence in subcortical networks. Brain Struct. Funct. 2015, 220, 677–702. [Google Scholar] [CrossRef] [PubMed]
- Shafiei, F.; Afarinesh, M.R.; Golshan, F.; Haghpanah, T.; Sabzalizadeh, M.; Zangiabadi, I.; Sheibani, V. Comparison of pre-pulse inhibition, tactile discrimination learning and barrel cortical neural response in adult male rats following chronic exposure to morphine, methadone and buprenorphine. Physiol. Behav. 2019, 212, 112694. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sham Saline | Sham Bup | cFPI Saline | cFPI Bup | |
|---|---|---|---|---|
| Pre-injury Weight (g) | 482 (62) | 489 (71) | 498 (87) | 513 (89) |
| Injury Intensity (atm) | Sham injury | Sham injury | 2.08 (0.04) | 2.05 (0.05) |
| Injury duration (ms) | Sham injury | Sham injury | 20.97 (0.56) | 21.43 (0.50) |
| Recovery Time (min) | 62.67 (43.43) | 48.33 (6.47) | 60.83 (29.21) | 72.50 (36.75) |
| IL-1a | IL-1b | IL-4 | IL-6 | IL-10 | IL-12 | IFNy | TNFa | |
|---|---|---|---|---|---|---|---|---|
| Sham Saline cortex | 17.36 (6.01) | 7.23 (1.56) | 14.43 (2.37) | 119.80 (29.20) | 26.05 (1.62) | 269.88 (21.10) | 47.03 (8.09) | 28.68 (6.95) |
| Sham Bup Cortex | 22.73 (6.26) | 9.51 (1.14) | 28.02 (7.00) | 158.28 (34.62) @ | 27.84 (1.87) | 324.02 (32.19) | 63.58 (12.54) | 38.11 (4.35) |
| TBI Saline Cortex | 17.08 (4.79) | 9.22 (1.72) | 18.50 (2.34) | 111.41 (20.73) | 22.81 (3.46) | 291.84 (22.75) | 56.51 (8.12) | 42.77 (4.94) @ |
| TBI Bup Cortex | 14.13 (3.89) | 7.95 (1.53) | 19.44 (6.77) | 83.14 (27.02) | 21.16 (5.31) | 276.89 (24.79) | 36.04 (8.02) # | 36.46 (5.12) |
| Sham Saline Thalamus | 23.64 (4.52) | 12.08 (3.92) | 16.49 (7.06) | 164.31 (44.03) @ | 26.57 (3.24) | 330.17 (39.74) | 54.74 (10.63) | 37.51 (9.70) |
| Sham Bup Thalamus | 27.91 (4.84) | 11.27 (2.52) | 22.54 (3.18) | 165.38 (36.41) @ | 20.76 (2.77) | 259.37 (25.48) | 49.37 (4.95) | 47.33 (5.86) *,@ |
| TBI Saline Thalamus | 25.14 (4.18) | 9.85 (2.27) | 15.45 (4.53) | 28.48 (17.46) | 19.44 (3.04) | 314.33 (30.86) | 45.86 (5.44) | 23.97 (5.02) |
| TBI Bup Thalamus | 26.28 (6.63) | 14.88 (4.19) * | 29.03 (5.68) | 123.40 (39.34) @ | 17.64 (4.24) # | 293.65 (22.03) | 52.68 (12.08) | 37.41 (7.78) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ryu, J.; Jeizan, P.; Ahmed, S.; Ehsan, S.; Jose, J.; Regan, S.; Gorse, K.; Kelliher, C.; Lafrenaye, A. Post-Injury Buprenorphine Administration Is Associated with Long-Term Region-Specific Glial Alterations in Rats. Pharmaceutics 2022, 14, 2068. https://doi.org/10.3390/pharmaceutics14102068
Ryu J, Jeizan P, Ahmed S, Ehsan S, Jose J, Regan S, Gorse K, Kelliher C, Lafrenaye A. Post-Injury Buprenorphine Administration Is Associated with Long-Term Region-Specific Glial Alterations in Rats. Pharmaceutics. 2022; 14(10):2068. https://doi.org/10.3390/pharmaceutics14102068
Chicago/Turabian StyleRyu, Jane, Pantea Jeizan, Saira Ahmed, Sareena Ehsan, Jefin Jose, Sean Regan, Karen Gorse, Corrina Kelliher, and Audrey Lafrenaye. 2022. "Post-Injury Buprenorphine Administration Is Associated with Long-Term Region-Specific Glial Alterations in Rats" Pharmaceutics 14, no. 10: 2068. https://doi.org/10.3390/pharmaceutics14102068
APA StyleRyu, J., Jeizan, P., Ahmed, S., Ehsan, S., Jose, J., Regan, S., Gorse, K., Kelliher, C., & Lafrenaye, A. (2022). Post-Injury Buprenorphine Administration Is Associated with Long-Term Region-Specific Glial Alterations in Rats. Pharmaceutics, 14(10), 2068. https://doi.org/10.3390/pharmaceutics14102068