
Reliable Prediction of Caco-2 Permeability by Supervised Recursive Machine Learning Approaches

Abstract
1. Introduction
2. Materials and Methods
2.1. Computational Tool
2.2. Permeability Data Collection
2.3. Chemical and Experimental Data Curation
2.4. Physicochemical and Structural Descriptors
2.5. Variable Selection
2.6. Data Cleaning
2.7. Modeling Algorithm
2.8. Model Evaluation
3. Results and Discussion
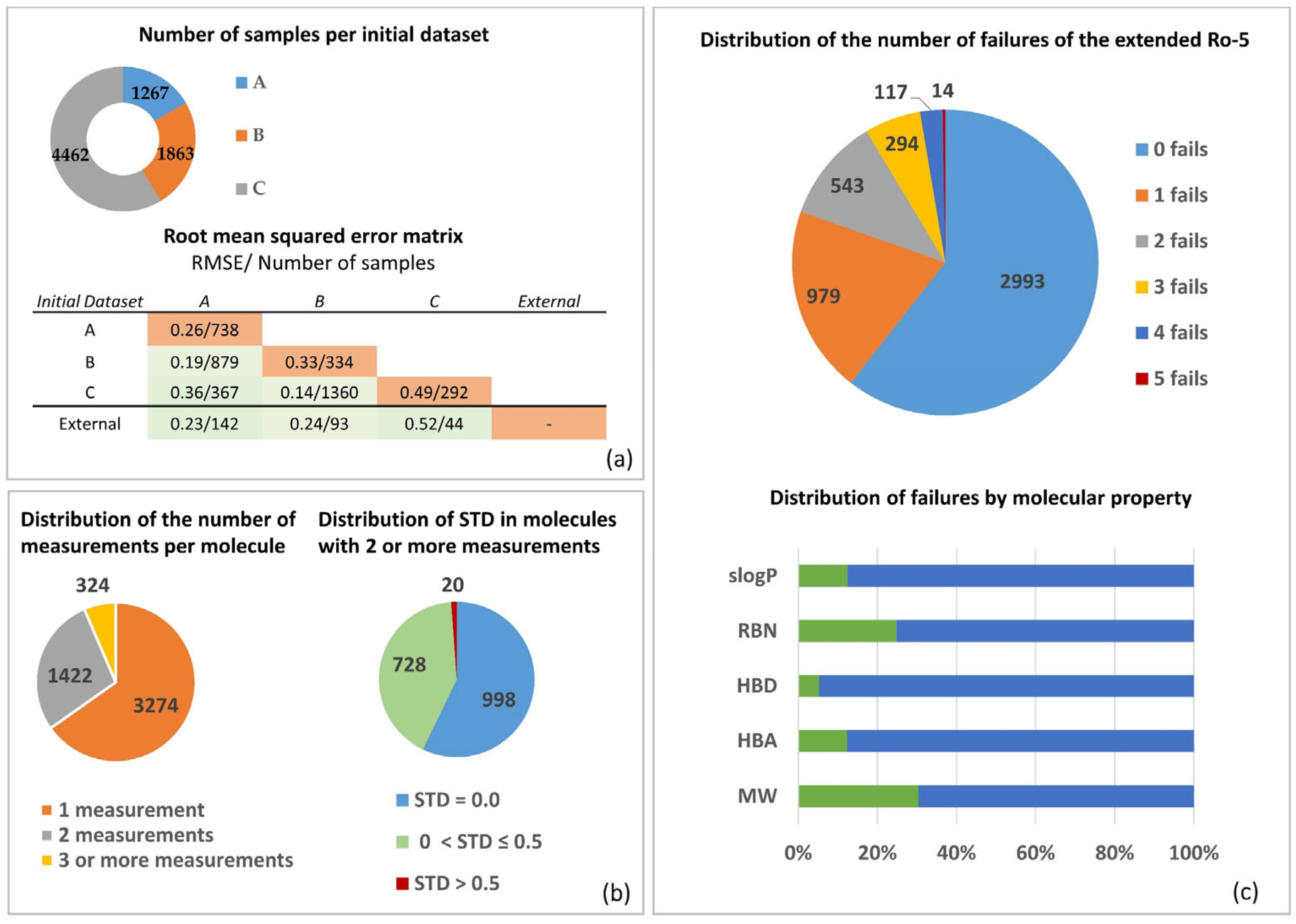
3.1. Permeability Data
3.2. Molecular Properties and Caco-2 MERGED Data
3.3. Model Development
3.4. Applicability Domain
3.5. Model Validation with Reference Drugs and Potential Application to BCS/BDDCS
3.6. Automated Platform for In Silico Caco-2 Permeability Prediction, Concluding Remarks
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A


References
- Alqahtani, M.S.; Kazi, M.; Alsenaidy, M.A.; Ahmad, M.Z. Advances in Oral Drug Delivery. Front. Pharmacol. 2021, 12, 62. [Google Scholar] [CrossRef]
- Lin, L.; Wong, H. Predicting Oral Drug Absorption: Mini Review on Physiologically-Based Pharmacokinetic Models. Pharmaceutics 2017, 9, 41. [Google Scholar] [CrossRef] [PubMed]
- Lanevskij, K.; Didziapetris, R. Physicochemical QSAR Analysis of Passive Permeability Across Caco-2 Monolayers. J. Pharm. Sci. 2019, 108, 78–86. [Google Scholar] [CrossRef]
- Hubatsch, I.; Ragnarsson, E.G.E.; Artursson, P. Determination of Drug Permeability and Prediction of Drug Absorption in Caco-2 Monolayers. Nat. Protoc. 2007, 2, 2111–2119. [Google Scholar] [CrossRef] [PubMed]
- Volpe, D.A. Advances in Cell-Based Permeability Assays to Screen Drugs for Intestinal Absorption. Expert Opin. Drug Discov. 2020, 15, 539–549. [Google Scholar] [CrossRef]
- Sun, H.; Chow, E.C.Y.; Liu, S.; Du, Y.; Pang, K.S. The Caco-2 Cell Monolayer: Usefulness and Limitations. Expert Opin. Drug Metab. Toxicol. 2008, 4, 395–411. [Google Scholar] [CrossRef]
- Skolnik, S.; Lin, X.; Wang, J.; Chen, X.H.; He, T.; Zhang, B. Towards Prediction of in Vivo Intestinal Absorption Using a 96-Well Caco-2 Assay. J. Pharm. Sci. 2010, 99, 3246–3265. [Google Scholar] [CrossRef]
- Bocci, G.; Oprea, T.I.; Benet, L.Z. State of the Art and Uses for the Biopharmaceutics Drug Disposition Classification System (BDDCS): New Additions, Revisions, and Citation References. AAPS J. 2022, 24, 1–17. [Google Scholar] [CrossRef]
- Mostrag-Szlichtyng, A.; Worth, A. Review of QSAR Models and Software Tools for Predicting Biokinetic Properties. JRC Sci. Tech. Rep. 2010, 1–71. [Google Scholar] [CrossRef]
- Ta, G.H.; Jhang, C.S.; Weng, C.F.; Leong, M.K. Development of a Hierarchical Support Vector Regression-Based in Silico Model for Caco-2 Permeability. Pharmaceutics 2021, 13, 174. [Google Scholar] [CrossRef]
- Volpe, D.A. Variability in Caco-2 and MDCK Cell-Based Intestinal Permeability Assays. J. Pharm. Sci. 2008, 97, 712–725. [Google Scholar] [CrossRef] [PubMed]
- Oltra-Noguera, D.; Mangas-Sanjuan, V.; Centelles-Sangüesa, A.; Gonzalez-Garcia, I.; Sanchez-Castaño, G.; Gonzalez-Alvarez, M.; Casabo, V.G.; Merino, V.; Gonzalez-Alvarez, I.; Bermejo, M. Variability of Permeability Estimation from Different Protocols of Subculture and Transport Experiments in Cell Monolayers. J. Pharmacol. Toxicol. Methods 2015, 71, 21–32. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.B.; Zgair, A.; Taha, D.A.; Zang, X.; Kagan, L.; Hwan, T.; Kim, M.G.; Yun, H.y.; Fischer, P.M.; Gershkovich, P.; et al. Quantitative Analysis of Lab-to-Lab Variability in Caco-2 Permeability Assays. Eur. J. Pharm. Biopharm. 2017, 114, 38–42. [Google Scholar] [CrossRef]
- Pham-The, H.; Cabrera-Pérez, M.Á.; Nam, N.-H.; Castillo-Garit, J.A.; Rasulev, B.; Le-Thi-Thu, H.; Casañola-Martin, G.M. In Silico Assessment of ADME Properties: Advances in Caco-2 Cell Monolayer Permeability Modeling. Curr. Top. Med. Chem. 2019, 18, 2209–2229. [Google Scholar] [CrossRef] [PubMed]
- Nordqvist, A.; Nilsson, J.; Lindmark, T.; Eriksson, A.; Garberg, P.; Kihlén, M. A General Model for Prediction of Caco-2 Cell Permeability. QSAR Comb. Sci. 2004, 23, 303–310. [Google Scholar] [CrossRef]
- Castillo-Garit, J.A.; Marrero-Ponce, Y.; Torrens, F.; Garcia-Domenech, R. Estimation of ADME Properties in Drug Discovery: Predicting Caco-2 Cell Permeability Using Atom-Based Stochastic and Non-Stochastic Linear Indices. J. Pharm Sci. 2008, 97, 1946–1976. [Google Scholar] [CrossRef]
- Paixão, P.; Gouveia, L.F.; Morais, J.A.G. Prediction of the in Vitro Permeability Determined in Caco-2 Cells by Using Artificial Neural Networks. Eur. J. Pharm. Sci. 2010, 41, 107–117. [Google Scholar] [CrossRef]
- Wilkinson, M.D.; Dumontier, M.; Aalbersberg, I.J.; Appleton, G.; Axton, M.; Baak, A.; Blomberg, N.; Boiten, J.W.; da Silva Santos, L.B.; Bourne, P.E.; et al. Comment: The FAIR Guiding Principles for Scientific Data Management and Stewardship. Sci. Data 2016, 3, 1–9. [Google Scholar] [CrossRef]
- Wise, J.; De Barron, A.G.; Splendiani, A.; Balali-Mood, B.; Vasant, D.; Little, E.; Mellino, G.; Harrow, I.; Smith, I.; Taubert, J.; et al. Implementation and Relevance of FAIR Data Principles in Biopharmaceutical R&D. Drug Discov. Today 2019, 24, 933–938. [Google Scholar] [CrossRef]
- Merz, K.M.; Amaro, R.; Cournia, Z.; Rarey, M.; Soares, T.; Tropsha, A.; Wahab, H.A.; Wang, R. Editorial: Method and Data Sharing and Reproducibility of Scientific Results. J. Chem. Inf. Model. 2020, 60, 5868–5869. [Google Scholar] [CrossRef]
- KNIME Analytics Platform 4.4.2. Available online: Https://Www.Knime.Com/Download-Previous-Versions. (accessed on 3 March 2022).
- Wang, N.N.; Dong, J.; Deng, Y.H.; Zhu, M.F.; Wen, M.; Yao, Z.J.; Lu, A.-P.P.; Wang, J.B.; Cao, D.-S.S. ADME Properties Evaluation in Drug Discovery: Prediction of Caco-2 Cell Permeability Using a Combination of NSGA-II and Boosting. J. Chem. Inf. Model. 2016, 56, 763–773. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Chen, X. QSPR Model for Caco-2 Cell Permeability Prediction Using a Combination of HQPSO and Dual-RBF Neural Network. RSC Adv. 2020, 10, 42938–42952. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Liu, M.; Zhang, L.; Wang, Y.; Li, Y.; Lu, T. Optimizing Pharmacokinetic Property Prediction Based on Integrated Datasets and a Deep Learning Approach. J. Chem. Inf. Model. 2020, 60, 4603–4613. [Google Scholar] [CrossRef]
- Hou, T.J.; Zhang, W.; Xia, K.; Qiao, X.B.; Xu, X.J. ADME Evaluation in Drug Discovery. 5 Correlation of Caco-2 Permeation with Simple Molecular Properties. J. Chem. Inf. Comput. Sci. 2004, 44, 1585–1600. [Google Scholar] [CrossRef] [PubMed]
- Fourches, D.; Muratov, E.; Tropsha, A. Trust, but Verify II: A Practical Guide to Chemogenomics Data Curation. J. Chem. Inf. Model. 2016, 56, 1243–1252. [Google Scholar] [CrossRef] [PubMed]
- Landrum, G. RDKit: Open-Source Cheminformatics Software; Version 2021.06.4. Available online: http://www.rdkit.org (accessed on 16 September 2022).
- Falcón, G.; Molina, C.; Ángel, M.; Pérez, C. ADME Prediction with KNIME: In Silico Aqueous Solubility Consensus Model Based on Supervised Recursive Random Forest Approaches. ADMET DMPK 2020, 8, 251–273. [Google Scholar]
- Pham The, H.; González-Álvarez, I.; Bermejo, M.; Mangas Sanjuan, V.; Centelles, I.; Garrigues, T.M.; Cabrera-Pérez, M.Á.; The, H.P.; González-Álvarez, I.; Bermejo, M.; et al. In Silico Prediction of Caco-2 Cell Permeability by a Classification QSAR Approach. Mol. Inform. 2011, 30, 376–385. [Google Scholar] [CrossRef]
- Alexander, D.L.J.; Tropsha, A.; Winkler, D.A. Beware of R2: Simple, Unambiguous Assessment of the Prediction Accuracy of QSAR and QSPR Models. J. Chem. Inf. Model. 2015, 55, 1316. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular Properties That Influence the Oral Bioavailability of Drug Candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and Computational Approaches to Estimate Solubility and Permeability in Drug Discovery and Development Settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Di, L.; Artursson, P.; Avdeef, A.; Benet, L.Z.; Houston, J.B.; Kansy, M.; Kerns, E.H.; Lennernäs, H.; Smith, D.A.; Sugano, K. The Critical Role of Passive Permeability in Designing Successful Drugs. ChemMedChem 2020, 15, 1862–1874. [Google Scholar] [CrossRef] [PubMed]
- Jacek, K.; Hanna, P.; Anna, M.; Beata, D.; Bernard, M.K. The Log P Parameter as a Molecular Descriptor in the Computer-Aided Drug Design—An Overview. CMST 2012, 18, 81–88. [Google Scholar] [CrossRef]
- Irvine, J.D.; Takahashi, L.; Lockhart, K.; Cheong, J.; Tolan, J.W.; Selick, H.E.; Grove, J.R. MDCK (Madin-Darby Canine Kidney) Cells: A Tool for Membrane Permeability Screening. J. Pharm. Sci. 1999, 88, 28–33. [Google Scholar] [CrossRef] [PubMed]
- Desai, P.V.; Raub, T.J.; Blanco, M. Bioorganic & Medicinal Chemistry Letters How Hydrogen Bonds Impact P-Glycoprotein Transport and Permeability. Bioorg. Med. Chem. Lett. 2012, 22, 6540–6548. [Google Scholar] [CrossRef]
- Chen, C.; Lee, M.H.; Weng, C.F.; Leong, M.K. Theoretical Prediction of the Complex P-Glycoprotein Substrate Efflux Based on the Novel Hierarchical Support Vector Regression Scheme. Mol. J. Synth. Chem. Nat. Prod. Chem. 2018, 23, 1820. [Google Scholar] [CrossRef]
- Zhu, Q.; Lu, Y.; He, X.; Liu, T.; Chen, H.; Wang, F.; Zheng, D.; Dong, H.; Ma, J. Entropy and Polarity Control the Partition and Transportation of Drug-like Molecules in Biological Membrane. Sci. Rep. 2017, 7, 17749. [Google Scholar] [CrossRef]
- Bennion, B.J.; Be, N.A.; McNerney, M.W.; Lao, V.; Carlson, E.M.; Valdez, C.A.; Malfatti, M.A.; Enright, H.A.; Nguyen, T.H.; Lightstone, F.C.; et al. Predicting a Drug’s Membrane Permeability: A Computational Model Validated with in Vitro Permeability Assay Data. J. Phys. Chem. B 2017, 121, 5228–5237. [Google Scholar] [CrossRef]
- Martin, Y.C.; Abagyan, R.; Ferenczy, G.G.; Gillet, V.J.; Oprea, T.I.; Ulander, J.; Winkler, D.; Zefirov, N.S. Glossary of Terms Used in Computational Drug Design, Part II (IUPAC Recommendations 2015). Pure Appl. Chem. 2016, 88, 239–264. [Google Scholar] [CrossRef]
- Hall, L.H.; Kiert, L.B. The Molecular Connectivity Chi Indexes and Kappa Shape Indexes in Structure-Property Modeling. Rev. Comp. Chem. 1991, 2, 367–422. [Google Scholar]
- Caron, G.; Digiesi, V.; Solaro, S.; Ermondi, G. Flexibility in Early Drug Discovery: Focus on the beyond-Rule-of-5 Chemical Space. Drug Discov. Today 2020, 25, 621–627. [Google Scholar] [CrossRef]
- Sherer, E.C.; Verras, A.; Madeira, M.; Hagmann, W.K.; Sheridan, R.P.; Roberts, D.; Bleasby, K.; Cornell, W.D. QSAR Prediction of Passive Permeability in the LLC-PK1 Cell Line: Trends in Molecular Properties and Cross-Prediction of Caco-2 Permeabilities. Mol. Inform. 2012, 31, 231–245. [Google Scholar] [CrossRef] [PubMed]
- Fredlund, L.; Winiwarter, S.; Hilgendorf, C. In Vitro Intrinsic Permeability: A Transporter-Independent Measure of Caco-2 Cell Permeability in Drug Design and Development. Mol. Pharm. 2017, 14, 1601–1609. [Google Scholar] [CrossRef] [PubMed]
- Refsgaard, H.H.F.; Jensen, B.F.; Brockhoff, P.B.; Padkjær, S.B.; Guldbrandt, M. In Silico Prediction of Membrane Permeability from Calculated Molecular. J. Med. Chem. 2005, 48, 805–811. [Google Scholar] [CrossRef]
- Esaki, T.; Ohashi, R.; Watanabe, R.; Natsume-Kitatani, Y.; Kawashima, H.; Nagao, C.; Komura, H.; Mizuguchi, K. Constructing an In Silico Three-Class Predictor of Human Intestinal Absorption With Caco-2 Permeability and Dried-DMSO Solubility. J. Pharm. Sci. 2019, 108, 3630–3639. [Google Scholar] [CrossRef]
- Committee for Medicinal Products for Human Use ICH M9 Guideline on Biopharmaceutics Classification System-Based Biowaivers ICH M9 on Biopharmaceutics Classification System-Based Biowaivers. 2020. Available online: https://www.ema.europa.eu/en/ich-m9-biopharmaceutics-classification-system-based-biowaivers (accessed on 16 September 2022).
- CDER/FDA Guidance for Industry: Waiver of In Vivo Bioavailability and Bioequivalence Studies for Immediate-Release Solid Oral Dosage Forms Based on a Biopharmaceutics Classification System. 2017. Available online: https://www.gmp-compliance.org/files/guidemgr/UCM070246.pdf (accessed on 16 September 2022).
- Bergström, C.A.S.; Strafford, M.; Lazorova, L.; Avdeef, A.; Luthman, K.; Artursson, P. Absorption Classification of Oral Drugs Based on Molecular Surface Properties. J. Med. Chem. 2003, 46, 558–570. [Google Scholar] [CrossRef]
- Avdeef, A.; Artursson, P.; Neuhoff, S.; Lazorova, L.; Gr, J. Caco-2 Permeability of Weakly Basic Drugs Predicted with the Double-Sink PAMPA PKa (Flux) Method. Eur. J. Pharm. Sci. 2005, 24, 333–349. [Google Scholar] [CrossRef]
- Balimane, P.V.; Han, Y.H.; Chong, S. Current Industrial Practices of Assessing Permeability and P-Glycoprotein Interaction. AAPS J. 2006, 8, E1–E13. [Google Scholar] [CrossRef] [PubMed]
- Pham-the, H.; González-, I.; Bermejo, M.; Garrigues, T.; Le-thi-thu, H. The Use of Rule-Based and QSPR Approaches in ADME Profiling: A Case Study on Caco-2 Permeability Full Paper. Mol. Inform. 2013, 32, 459–479. [Google Scholar] [CrossRef] [PubMed]
- Alsenz, J.; Haenel, E. Development of a 7-Day, 96-Well Caco-2 Permeability Assay with High-Throughput Direct UV Compound Analysis. Pharm. Res. 2003, 20, 1961–1969. [Google Scholar] [CrossRef]
- Teksin, Z.S.; Seo, P.R.; Polli, J.E. Comparison of Drug Permeabilities and BCS Classification: Three Lipid-Component PAMPA System Method versus Caco-2 Monolayers. AAPS J. 2010, 12, 238. [Google Scholar] [CrossRef]
- Kerns, E.H.; Di, L.; Petusky, S.; Farris, M.; Ley, R.; Jupp, P. Combined Application of Parallel Artificial Membrane Permeability Assay and Caco-2 Permeability Assays in Drug Discovery. J. Pharm. Sci. 2004, 93, 1440–1453. [Google Scholar] [CrossRef] [PubMed]
- Benet, L.Z. The Role of BCS (Biopharmaceutics Classification System) and BDDCS (Biopharmaceutics Drug Disposition Classification System) in Drug Development. J. Pharm. Sci. 2013, 102, 2271–2280. [Google Scholar] [CrossRef] [PubMed]
- Benet, L.Z.; Broccatelli, F.; Oprea, T.I. BDDCS Applied to over 900 Drugs. AAPS J. 2011, 13, 519–547. [Google Scholar] [CrossRef]
- Varma, M.V.; Gardner, I.; Steyn, S.J.; Nkansah, P.; Fenner, K.S.; El-Kattan, A.F. PH-Dependent Solubility and Permeability Criteria for Provisional Biopharmaceutics Classification (BCS and BDDCS) in Early Drug Discovery. Mol. Pharm. 2012, 9, 1199–1212. [Google Scholar] [CrossRef] [PubMed]
- Hosey, C.M.; Benet, L.Z. Predicting the Extent of Metabolism Using in Vitro Permeability Rate Measurements and in Silico Permeability Rate Predictions. Mol. Pharm. 2015, 12, 1456–1466. [Google Scholar] [CrossRef]
- Hosey, C.M.; Chan, R.; Benet, L.Z. BDDCS Predictions, Self-Correcting Aspects of BDDCS Assignments, BDDCS Assignment Corrections, and Classification for More than 175 Additional Drugs. AAPS J. 2016, 18, 251–260. [Google Scholar] [CrossRef]
- Lindenberg, M.; Kopp, S.; Dressman, J.B. Classification of Orally Administered Drugs on the World Health Organization Model List of Essential Medicines According to the Biopharmaceutics Classification System. Eur. J. Pharm. Biopharm. 2004, 58, 265–278. [Google Scholar] [CrossRef]
- Dahan, A.; Wolk, O.; Kim, Y.H.; Ramachandran, C.; Crippen, G.M.; Takagi, T.; Bermejo, M.; Amidon, G.L. Purely in Silico BCS Classification: Science Based Quality Standards for the World’s Drugs. Mol. Pharm. 2013, 10, 4378–4390. [Google Scholar] [CrossRef]
- Takagi, T.; Ramachandran, C.; Bermejo, M.; Yamashita, S.; Yu, L.X.; Amidon, G.L. A Provisional Biopharmaceutical Classification of the Top 200 Oral Drug Products in the United States, Great Britain, Spain, and Japan. Mol. Pharm. 2006, 3, 631–643. [Google Scholar] [CrossRef]
- Newby, D.; Freitas, A.; Ghafourian, T. Comparing Multi-Label Classification Methods for Provisional Biopharmaceutics Class Prediction. Mol. Pharm. 2014, 12, 87–102. [Google Scholar] [CrossRef]
- Broccatelli, F.; Cruciani, G.; Benet, L.Z.; Oprea, T.I. BDDCS Class Prediction for New Molecular Entities. Mol. Pharm. 2012, 9, 570–580. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Pham-The, H.; Garrigues, T.; Bermejo, M.; González-Álvarez, I.; Monteagudo, M.C.; Cabrera-Pérez, M.Á. Provisional Classification and in Silico Study of Biopharmaceutical System Based on Caco-2 Cell Permeability and Dose Number. Mol. Pharm. 2013, 10, 2445–2461. [Google Scholar] [CrossRef] [PubMed]
- Golfar, Y.; Shayanfar, A. Prediction of Biopharmaceutical Drug Disposition Classification System (BDDCS) by Structural Parameters. J. Pharm. Pharm. Sci. 2019, 22, 247–269. [Google Scholar] [CrossRef] [PubMed]
- Kasim, N.A.; Whitehouse, M.; Ramachandran, C.; Bermejo Sanz, M.; Lennernäs, H.; Hussain, A.S.; Junginger, H.E.; Stavchansky, S.A.; Midha, K.K.; Shah, V.P.; et al. Molecular Properties of WHO Essential Drugs and Provisional Biopharmaceutical Classification. Mol. Pharm. 2004, 1, 85–96. [Google Scholar] [CrossRef]
- Khandelwal, A.; Bahadduri, P.M.; Chang, C.; Polli, J.E.; Swaan, P.W.; Ekins, S. Computational Models to Assign Biopharmaceutics Drug Disposition Classification from Molecular Structure. Pharm. Res. 2007, 24, 2249–2262. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Validation Set |
N Mean (SD) |
r2 (validation) Mean (SD) |
RMSE (validation) Mean (SD) |
MAE Mean (SD) |
% 0.5 log Mean (SD) |
N (Variables) Mean (SD) |
|---|---|---|---|---|---|---|
| Cross Validation | 4166 (10) | 0.54 (0.01) | 0.51 (0.01) | 0.39 (0.01) | 70 (0.5) | 15 (3) |
| Reliable Set | 728 | 0.61 (0.01) | 0.43 (0.01) | 0.33 (0.01) | 77 (0.6) | 15 (3) |
| External Set | 100 | 0.57 (0 03) | 0.51 (0 03) | 0.40 (0 01) | 69 (3) | 15 (3) |
| Study | Method | Data Size |
Test Set Size | RMSE | NMAE * | r2 | Units | Range |
|---|---|---|---|---|---|---|---|---|
| Wang et al. [24] | MESN | 4464 | 1340 | - | 0.08 | 0.55 | log Papp (10−6 cm/s) | [−2.78; 2.48] |
| Wang et al. [22] | Boosting | 1272 | 255 | 0.31 | - | 0.83 | log Papp | [−7.76; −3.51] |
| Wang and Shen [23] | HQPSO + dual RBF NN | 1863 | 369 | 0.39 | - | 0.77 | log Papp | [−7.9; −3.72] |
| Sherer et al. [43] | RRF | 15,791 | - | 0.2 | - | 0.52 | log Papp | − |
| Fredlund et al. [44] | PLS, RRF, SVM | 2842 | 284 | 0.45 | - | - | log Papp | − |
| Current study | Recursive selection + RRF | 5013 ** | 728 | 0.43 | 0.07 | 0.61 | log Papp | [−8.32; −3.51] |
| 100 | 0.51 | 0.08 | 0.57 |
| ICH Group Permeability | Drug | log Papp obs. | log Papp calc. | Drug Transport | Fabs * (%) | EoM (%) | BDDCS Class | BCS Class | BDDCS Predicted | BCS Predicted |
|---|---|---|---|---|---|---|---|---|---|---|
| High | Antipyrine | −4.55 | −4.58 | Passive diffusion | 100 | 95 | 1 | 1 | 1, 2 | 1, 2 |
| Caffeine | −4.47 | −4.7 | Passive diffusion | 100 | 99 | 1 | 1 | 1, 2 | 1, 2 | |
| Carbamazepine | −4.57 | −4.75 | Passive diffusion | 90 | 98 | 2 | 2 | 1, 2 | 1, 2 | |
| Disopyramide | −5.37 | −5.19 | Passive diffusion | 83 | 3 | 1 | 1, 2 | 3, 4 | ||
| Ketoprofen | −4.55 | −4.58 | Passive diffusion | 100 | 90 | 2 | 1 | 1, 2 | 1, 2 | |
| Metoprolol | −4.65 | −4.73 | Passive diffusion | 95 | 95 | 1 | 1 | 1, 2 | 1, 2 | |
| Minoxidil | −4.75 | −5.27 | Passive diffusion | 95 | 1 | 3, 4 | 3, 4 | |||
| Naproxen | −4.46 | −4.62 | Passive diffusion | 98 | 95 | 2 | 2 | 1, 2 | 1, 2 | |
| Propranolol | −4.65 | −4.64 | Passive diffusion | 90 | 99 | 1 | 1 | 1, 2 | 1, 2 | |
| Theophylline | −5.11 | −5.03 | Passive diffusion | 96 | 90 | 1 | 1 | 1, 2 | 3, 4 | |
| Moderate | Amiloride | −5.68 | −6.23 | Passive diffusion | 50 | 0 | 3 | 1/3 | 3, 4 | 3, 4 |
| Atenolol | −6.27 | −5.58 | Paracellular | 51 | 10 | 3 | 3 | 3, 4 | 3, 4 | |
| Chlorpheniramine | −4.55 | −4.74 | Passive diffusion | 85 | 1 | 1/3 | 1, 2 | 1, 2 | ||
| Enalapril | −5.64 | −5.14 | Passive diffusion | 65 | 1 | 1 | 1, 2 | 3, 4 | ||
| Furosemide | −5.91 | −5.4 | Carrier-mediated | 61 | 10 | 4 | 4 | 3, 4 | 3, 4 | |
| Hydrochlorothiazide | −6.24 | −5.6 | Paracellular | 73 | 0 | 3 | 3 | 3, 4 | 3, 4 | |
| Metformin | −5.37 | −5.64 | Paracellular | 53 | 0 | 3 | 3 | 3, 4 | 3, 4 | |
| Ranitidine | −5.86 | −5.11 | Carrier-mediated | 53 | 30 | 3 | 3 | 1, 2 | 3, 4 | |
| Terbutaline | −5.92 | −5.18 | Paracellular | 67 | 30 | 3 | 1, 2 | 3, 4 | ||
| Low | Acyclovir | −6.38 | −6.16 | Paracellular Carrier-mediated | 25 | 25 | 4 | 1/3 | 3, 4 | 3, 4 |
| Famotidine | −6.21 | −5.59 | Paracellular | 38 | 45 | 3 | 3 | 3, 4 | 3, 4 | |
| Chlorothiazide | −5.88 | −5.44 | Carrier-mediated | 24 | 4 | 4 | 3, 4 | 3, 4 | ||
| Enalaprilat | −5.98 | −6.3 | Carrier-mediated | 17 | 10 | 3 | 3, 4 | 3, 4 | ||
| Foscarnet | −6.19 | −5.35 | Paracellular | 17 | 3 | 3, 4 | 3, 4 | |||
| Lisinopril | −6.42 | −5.67 | Carrier-mediated | 25 | 0 | 3 | 3 | 3, 4 | 3, 4 | |
| Mannitol | −6.09 | −5.59 | Carrier-mediated | 20 | 3 | 3, 4 | 3, 4 | |||
| Nadolol | −5.58 | −5.39 | Paracellular | 31 | 0 | 3 | 3 | 3, 4 | 3, 4 | |
| Sulpiride | −6.23 | −5.68 | Carrier-mediated | 36 | 0 | 3 | 3, 4 | 3, 4 | ||
| Efflux | Digoxin | −6.02 | −6 | Carrier-mediated | 50 | 3 | 1/3 | 3, 4 | 3, 4 | |
| Paclitaxel | −5.81 | −5.48 | Carrier-mediated | 50 | 2 | 4 | 3, 4 | 3, 4 | ||
| Quinidine | −4.56 | −4.65 | Carrier-mediated | 85 | 100 | 1 | 1 | 1, 2 | 1, 2 | |
| Vinblastine | −5.69 | −5.54 | Carrier-mediated | 30 | 2 | 3, 4 | 3, 4 |
| Study | Permeability/Metabolism Prediction Method for BCS/BDDCS | Number of Drugs from the ICH List | Sensitivity | Specificity |
|---|---|---|---|---|
| Golfar et al. (2019) [67] | clogP | 29 | 0.89 | 0.81 |
| Broccatelli et al. (2012) [65] | logD 7.5, logD 7, logD 9, log BB, CP, | 25 | 1 | 0.64 |
| Pham-The et al. (2013) [66] | logP, logD 7.5, TPSA, CACO2 (Volsurf+) | 10 | 0.86 | 0.67 |
| Kassim et al. (2004) [68] | logP, clogP | 15 | 1 | 0.67 |
| In silico log Papp for BCS | 21 | 0.73 | 1 | |
| This study | In silico log Papp for BDDCS | 32 | 0.78 | 0.83 |
| Study | Permeability Prediction Applied on: | Features | Methods | N (Test) | Statistics * | |
|---|---|---|---|---|---|---|
| BCS | BDDCS | |||||
| Broccatelli. (2012) [65] | x | 17 features (Volsurf+ descriptors) | Bayes, SVM | 379 | Se: 0.7 Sp: 0.8 | |
| Golfar (2019) [67] | x | logP ACD | BLR | 99 | Acc: 0.86 | |
| Takagi [63] | x | logP, clogP | Comparison with cut-off value | 29 | Acc: 0.62–0.65 | |
| Pham-The 2013 [66] | x | x | logP, logD 7.5, TPSA, CACO2(Volsurf+) | LDA, BLR, QDA | 675 | Se: 0.73 Sp: 0.68 |
| Khandelwal 2007 [69] | x | HBD, HBA, PSA, clogP, VOLSURF descriptors | RP, RF, SVM | 56 | Se: 0.86 Sp: 0.42 | |
| Dahan 2013 [62] | x | clogP, AlogP, KlogP | Comparison with cut-off value | 29 | Acc: 0.69–0.72 | |
| 14 | Acc: 0.85–0.93 | |||||
| Newby 2014 [64] | x | Volsurf+ descriptors | DT | 127 | Se: 0.63 Sp: 0.62 | |
| This study | x | x | In silico log Papp computed from 15 molecular descriptors using CCM | Comparison with cut-off value | 204 (BDDCS) | Se: 0.8 Sp: 0.7 Acc: 0.77 |
| 32 (BDDCS) | Se: 0.78 Sp: 0.83 Acc: 0.81 | |||||
| 22 (BCS) | Se: 0.67 Sp: 1 Acc: 0.82 | |||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Falcón-Cano, G.; Molina, C.; Cabrera-Pérez, M.Á. Reliable Prediction of Caco-2 Permeability by Supervised Recursive Machine Learning Approaches. Pharmaceutics 2022, 14, 1998. https://doi.org/10.3390/pharmaceutics14101998
Falcón-Cano G, Molina C, Cabrera-Pérez MÁ. Reliable Prediction of Caco-2 Permeability by Supervised Recursive Machine Learning Approaches. Pharmaceutics. 2022; 14(10):1998. https://doi.org/10.3390/pharmaceutics14101998
Chicago/Turabian StyleFalcón-Cano, Gabriela, Christophe Molina, and Miguel Ángel Cabrera-Pérez. 2022. "Reliable Prediction of Caco-2 Permeability by Supervised Recursive Machine Learning Approaches" Pharmaceutics 14, no. 10: 1998. https://doi.org/10.3390/pharmaceutics14101998
APA StyleFalcón-Cano, G., Molina, C., & Cabrera-Pérez, M. Á. (2022). Reliable Prediction of Caco-2 Permeability by Supervised Recursive Machine Learning Approaches. Pharmaceutics, 14(10), 1998. https://doi.org/10.3390/pharmaceutics14101998