
A General Small-Angle X-ray Scattering-Based Screening Protocol for Studying Physical Stability of Protein Formulations

 ,
, 
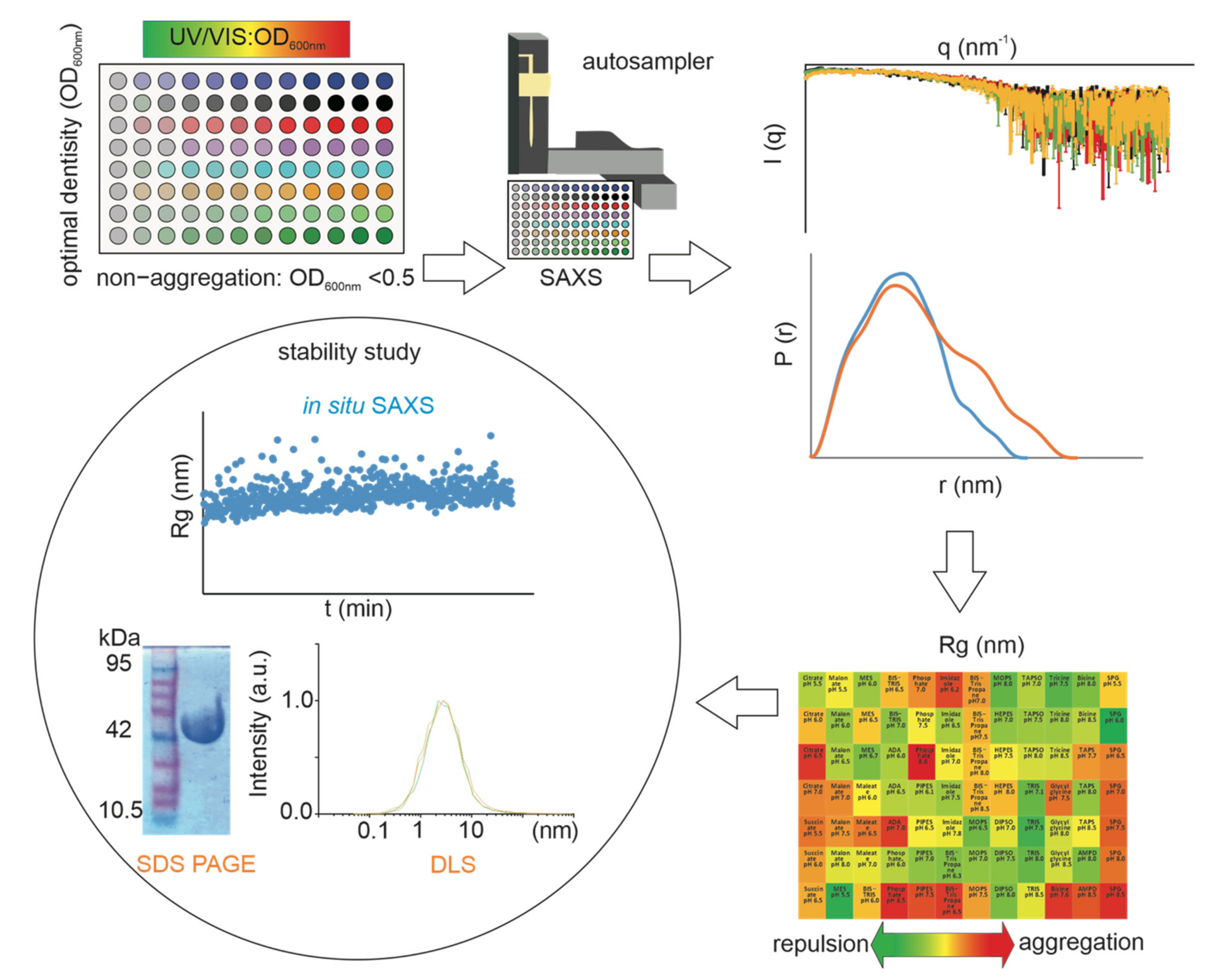
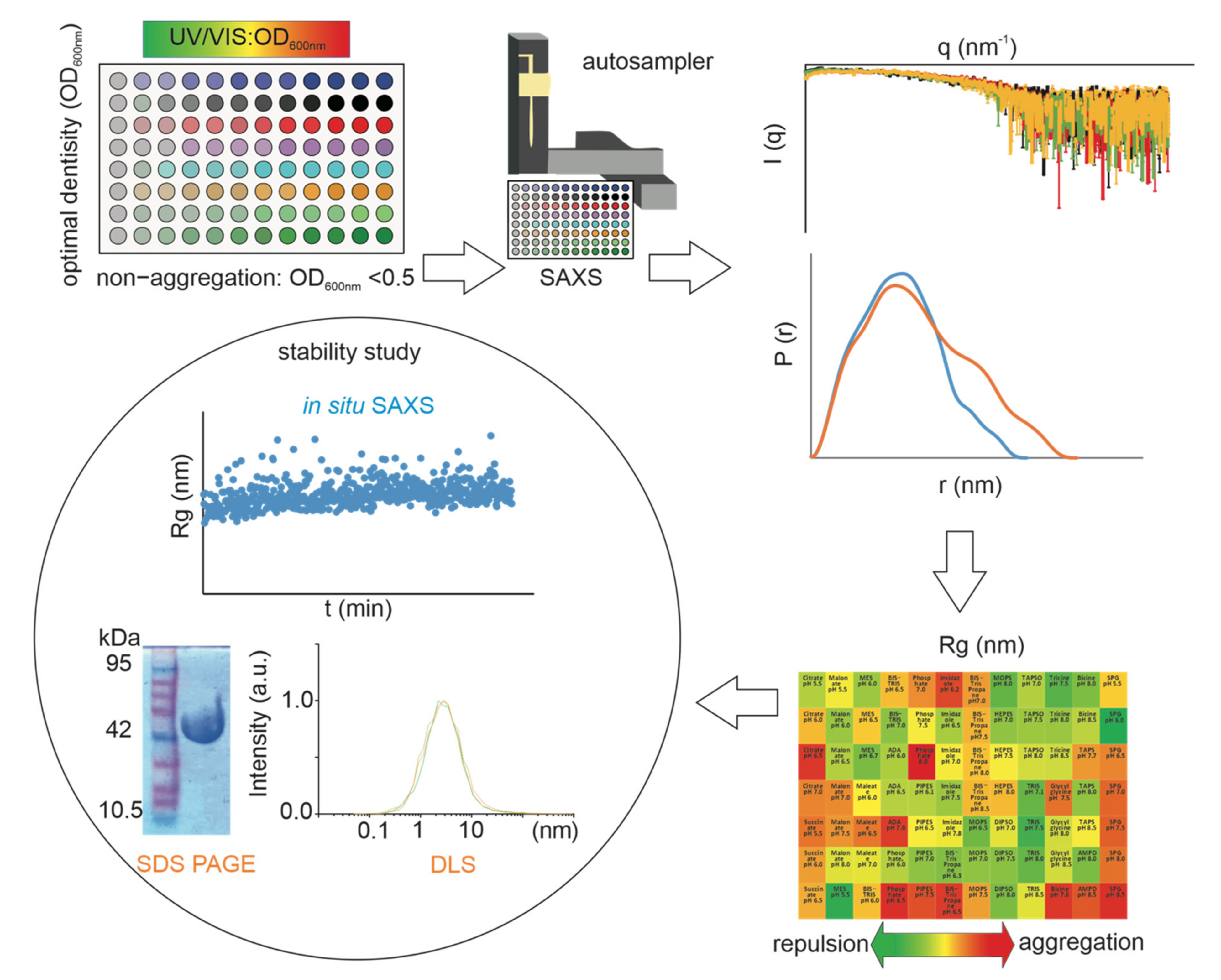{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Turbidity Assay
2.3. Small-Angle X-ray Scattering
2.4. Dynamic Light Scattering
2.5. SDS-PAGE
3. Results
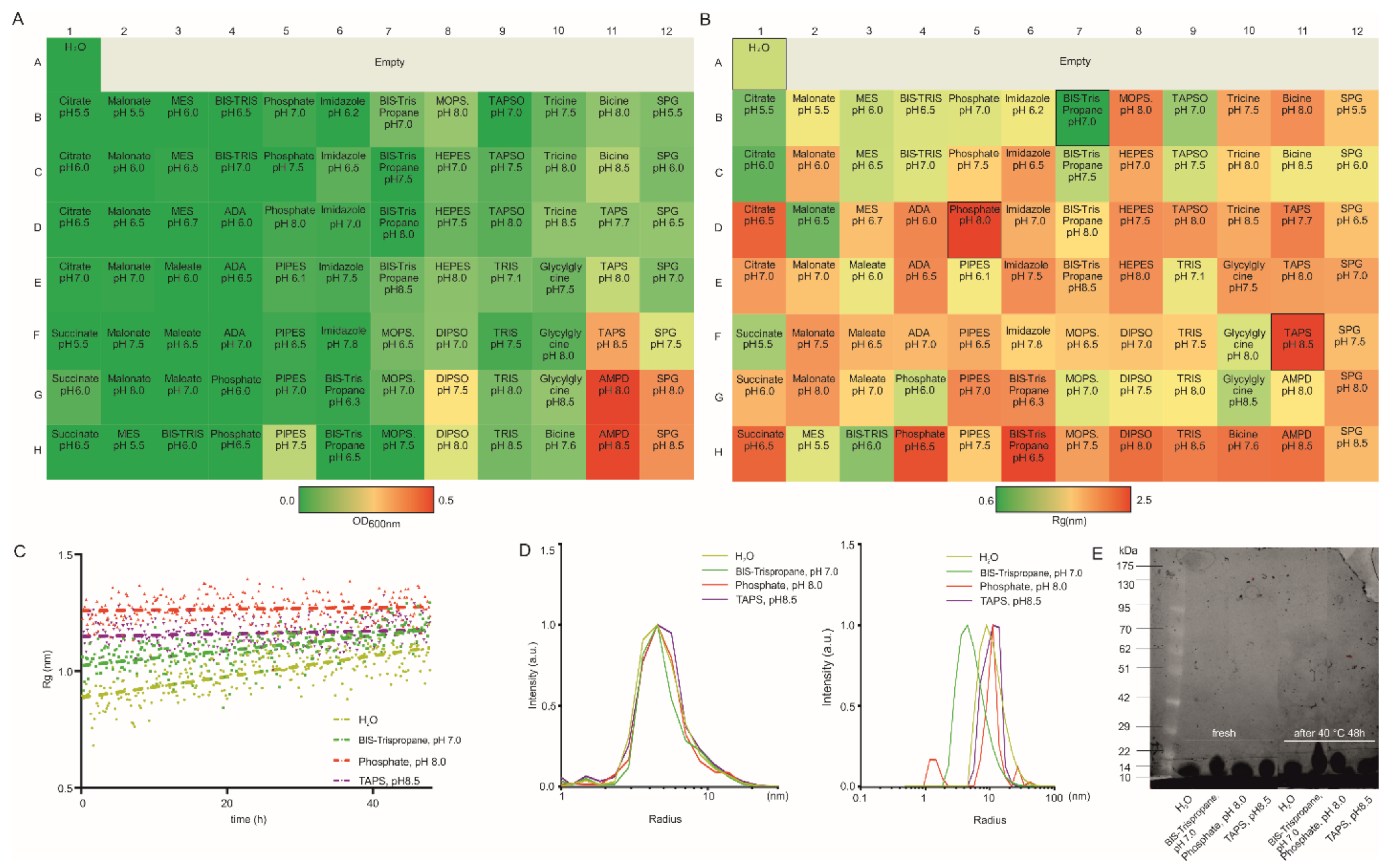
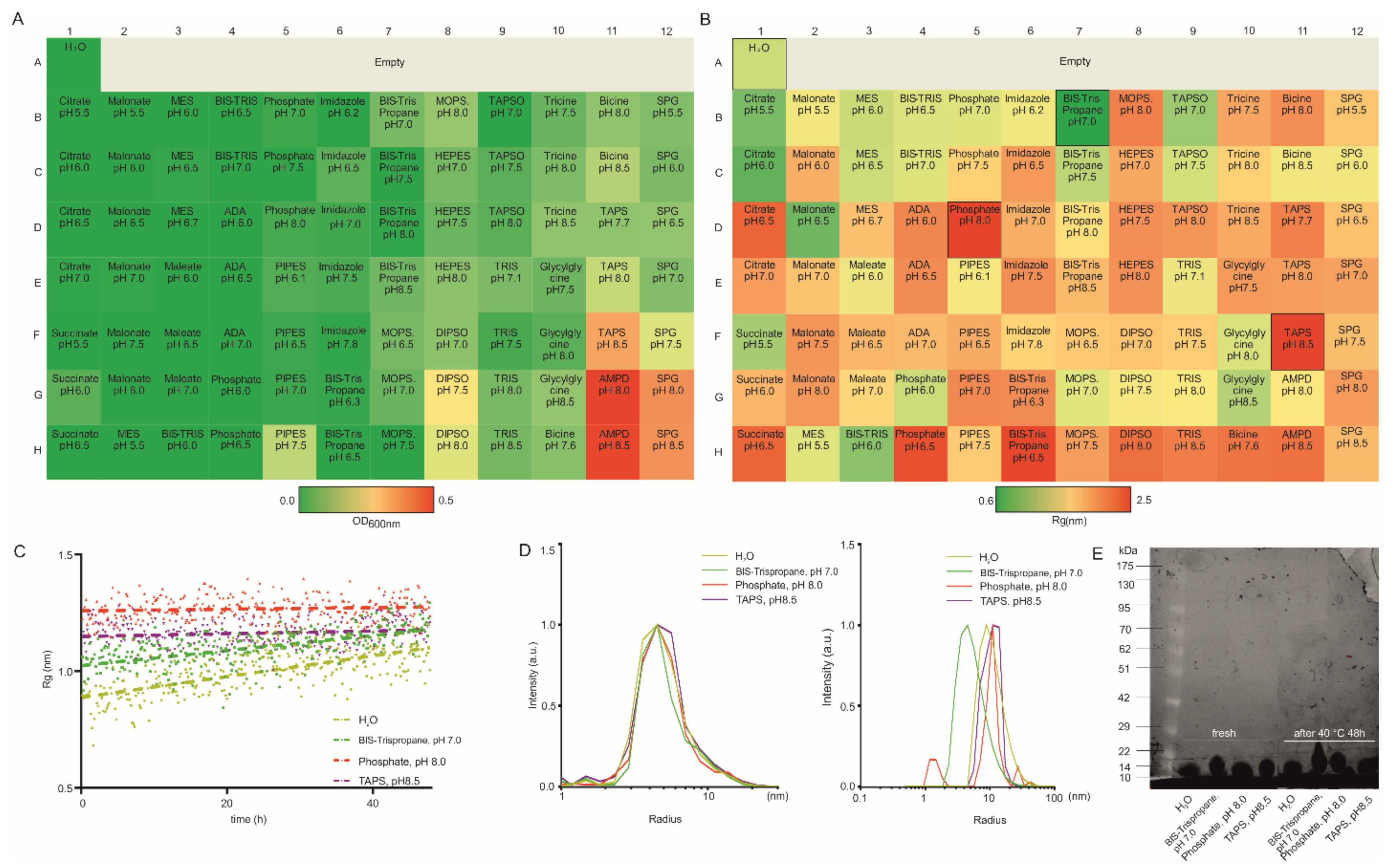
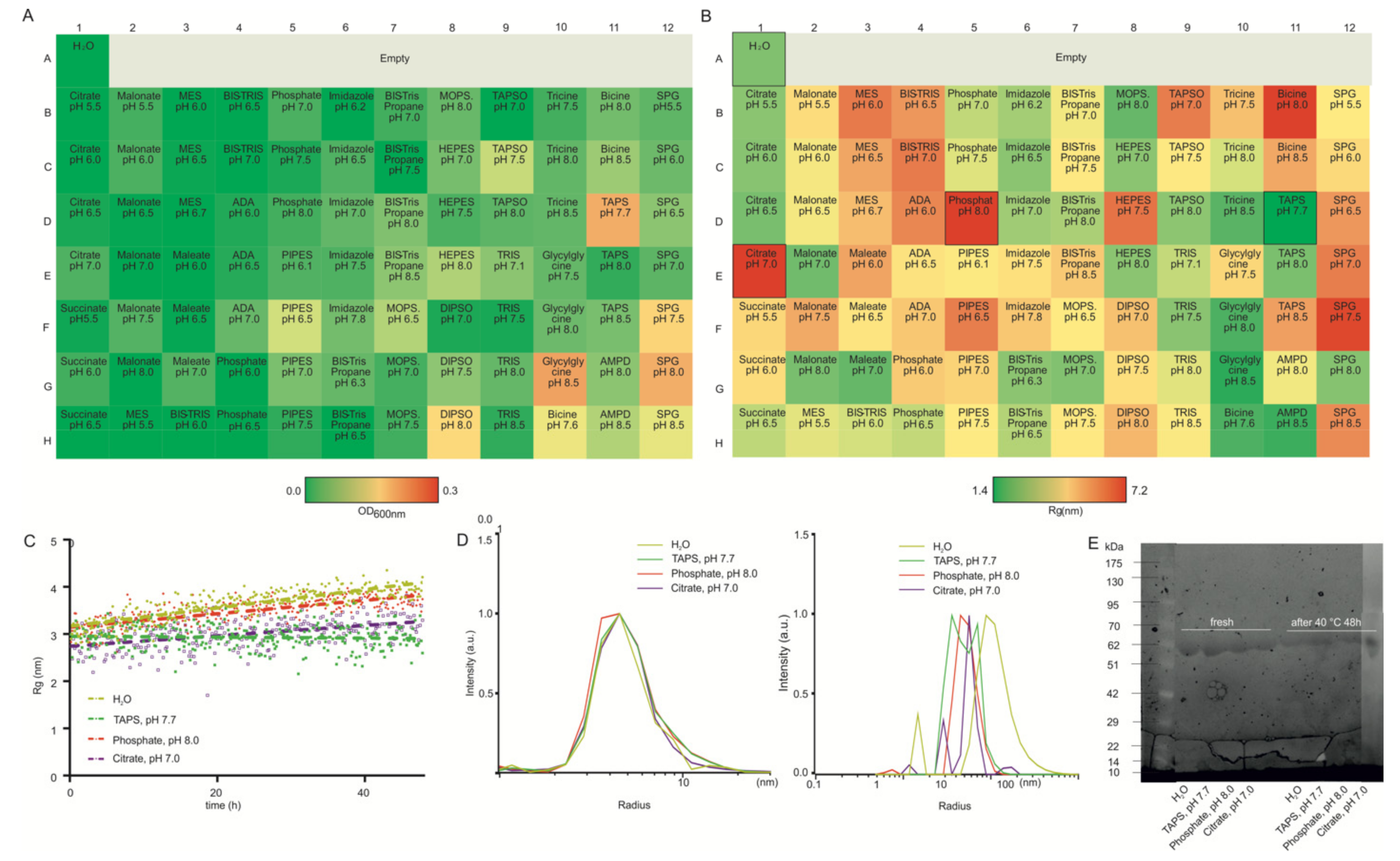
3.1. Lysozyme as a Model Protein
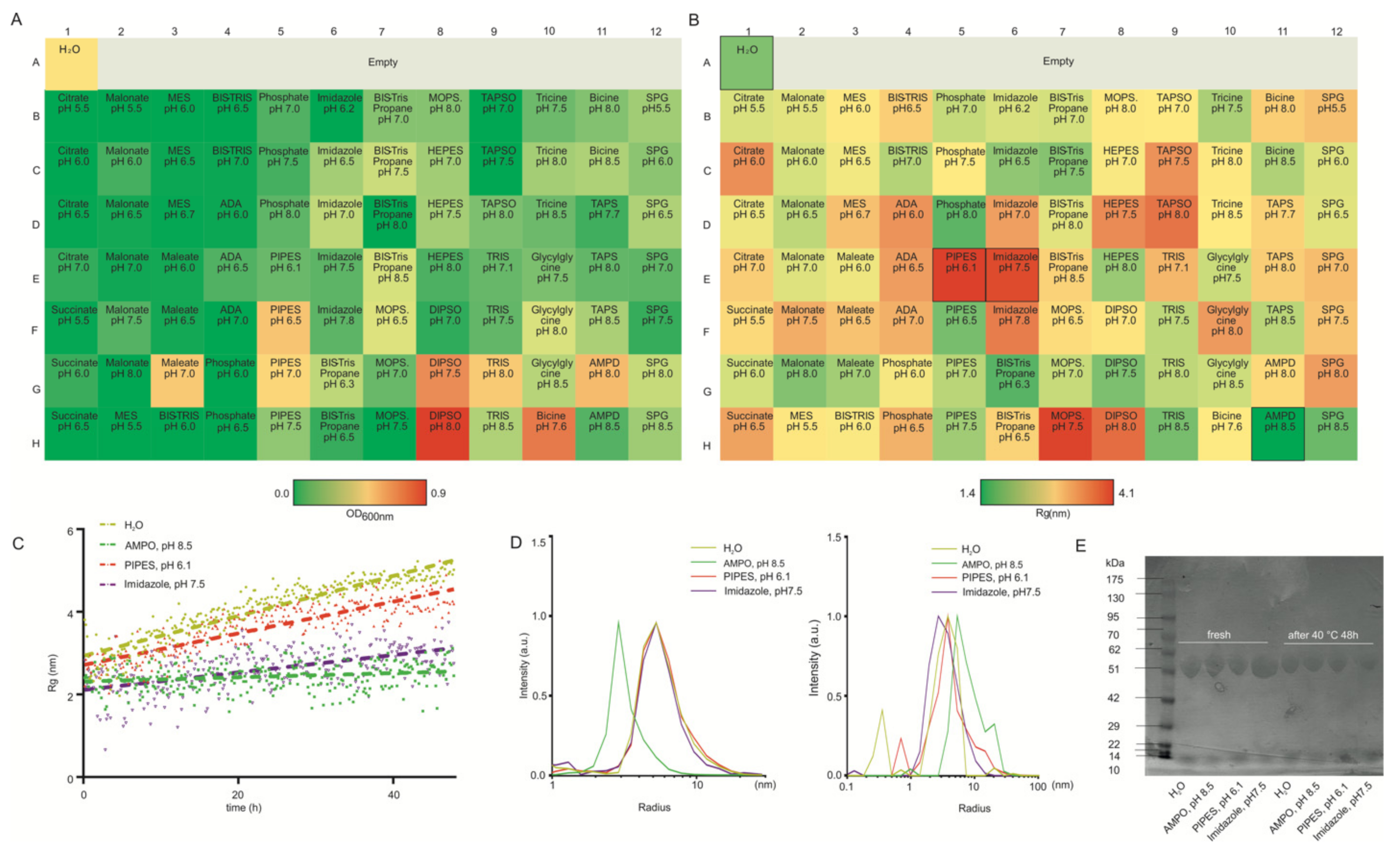
3.2. HSA as a Model Protein
3.3. Therapeutic Antibody Fragment as a Model Protein
4. Discussion
5. Limitation
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lagassé, H.D.; Alexaki, A.; Simhadri, V.L.; Katagiri, N.H.; Jankowski, W.; Sauna, Z.E.; Kimchi-Sarfaty, C. Recent advances in (therapeutic protein) drug development. F1000Research 2017, 6, 113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chennamsetty, N.; Voynov, V.; Kayser, V.; Helk, B.; Trout, B.L. Design of therapeutic proteins with enhanced stability. Proc. Natl. Acad. Sci. USA 2009, 106, 11937–11942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walsh, G. Biopharmaceutical benchmarks 2018. Nat. Biotechnol. 2018, 36, 1136–1145. [Google Scholar] [CrossRef] [PubMed]
- Kessler, M.; Goldsmith, D.; Schellekens, H. Immunogenicity of biopharmaceuticals. Nephrol. Dial. Transplant. 2006, 21, v9–v12. [Google Scholar] [CrossRef] [PubMed]
- Sauerborn, M.; Brinks, V.; Jiskoot, W.; Schellekens, H. Immunological mechanism underlying the immune response to recombinant human protein therapeutics. Trends Pharmacol. Sci. 2010, 31, 53–59. [Google Scholar] [CrossRef]
- Berkowitz, S.A.; Engen, J.R.; Mazzeo, J.R.; Jones, G.B. Analytical tools for characterizing biopharmaceuticals and the implications for biosimilars. Nat. Rev. Drug Discov. 2012, 11, 527–540. [Google Scholar] [CrossRef] [Green Version]
- Hermeling, S.; Crommelin, D.J.; Schellekens, H.; Jiskoot, W. Structure-immunogenicity relationships of therapeutic proteins. Pharm. Res. 2004, 21, 897–903. [Google Scholar] [CrossRef]
- Braun, A.; Kwee, L.; Labow, M.A.; Alsenz, J. Protein aggregates seem to play a key role among the parameters influencing the antigenicity of interferon alpha (IFN-α) in normal and transgenic mice. Pharm. Res. 1997, 14, 1472–1478. [Google Scholar] [CrossRef]
- Xu, A.Y.; Castellanos, M.M.; Mattison, K.; Krueger, S.; Curtis, J.E. Studying excipient modulated physical stability and viscosity of monoclonal antibody formulations using small-angle scattering. Mol. Pharm. 2019, 16, 4319–4338. [Google Scholar] [CrossRef]
- Zapadka, K.L.; Becher, F.J.; Gomes dos Santos, A.; Jackson, S.E. Factors affecting the physical stability (aggregation) of peptide therapeutics. Interface Focus 2017, 7, 20170030. [Google Scholar] [CrossRef] [Green Version]
- Chi, E.Y.; Krishnan, S.; Randolph, T.W.; Carpenter, J.F. Physical stability of proteins in aqueous solution: Mechanism and driving forces in nonnative protein aggregation. Pharm. Res. 2003, 20, 1325–1336. [Google Scholar] [CrossRef]
- Dumetz, A.C.; Snellinger-O’Brien, A.M.; Kaler, E.W.; Lenhoff, A.M. Patterns of protein–protein interactions in salt solutions and implications for protein crystallization. Protein Sci. 2007, 16, 1867–1877. [Google Scholar] [CrossRef]
- Garidel, P.; Hegyi, M.; Bassarab, S.; Weichel, M. A rapid, sensitive and economical assessment of monoclonal antibody conformational stability by intrinsic tryptophan fluorescence spectroscopy. Biotechnol. J. Healthc. Nutr. Technol. 2008, 3, 1201–1211. [Google Scholar] [CrossRef] [PubMed]
- Kamerzell, T.J.; Esfandiary, R.; Joshi, S.B.; Middaugh, C.R.; Volkin, D.B. Protein–excipient interactions: Mechanisms and biophysical characterization applied to protein formulation development. Adv. Drug Deliv. Rev. 2011, 63, 1118–1159. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, A.; Kim, Y.C.; Mittal, J. Protein–protein interactions in a crowded environment. Biophys. Rev. 2013, 5, 99–108. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.C.; Best, R.B.; Mittal, J. Macromolecular crowding effects on protein–protein binding affinity and specificity. J. Chem. Phys. 2010, 133, 11B608. [Google Scholar] [CrossRef] [PubMed]
- Tomaszewska, E.; Soliwoda, K.; Kadziola, K.; Tkacz-Szczesna, B.; Celichowski, G.; Cichomski, M.; Szmaja, W.; Grobelny, J. Detection limits of DLS and UV-Vis spectroscopy in characterization of polydisperse nanoparticles colloids. J. Nanomater. 2013, 2013, 60. [Google Scholar] [CrossRef] [Green Version]
- Corvari, V.; Narhi, L.O.; Spitznagel, T.M.; Afonina, N.; Cao, S.; Cash, P.; Cecchini, I.; DeFelippis, M.R.; Garidel, P.; Herre, A. Subvisible (2–100 μm) particle analysis during biotherapeutic drug product development: Part 2, experience with the application of subvisible particle analysis. Biologicals 2015, 43, 457–473. [Google Scholar] [CrossRef] [Green Version]
- Patel, A.R.; Lau, D.; Liu, J. Quantification and characterization of micrometer and submicrometer subvisible particles in protein therapeutics by use of a suspended microchannel resonator. Anal. Chem. 2012, 84, 6833–6840. [Google Scholar] [CrossRef] [PubMed]
- Ripple, D.C.; Dimitrova, M.N. Protein particles: What we know and what we do not know. J. Pharm. Sci. 2012, 101, 3568–3579. [Google Scholar] [CrossRef]
- Vasudev, R.; Mathew, S.; Afonina, N. Characterization of submicron (0.1–1 μm) particles in therapeutic proteins by nanoparticle tracking analysis. J. Pharm. Sci. 2015, 104, 1622–1631. [Google Scholar] [CrossRef] [PubMed]
- Gross-Rother, J.; Blech, M.; Preis, E.; Bakowsky, U.; Garidel, P. Particle Detection and Characterization for Biopharmaceutical Applications: Current Principles of Established and Alternative Techniques. Pharmaceutics 2020, 12, 1112. [Google Scholar] [CrossRef]
- Garcia-Cañas, V.; Lorbetskie, B.; Girard, M. Rapid and selective characterization of influenza virus constituents in monovalent and multivalent preparations using non-porous reversed-phase high performance liquid chromatography columns. J. Chromatogr. A 2006, 1123, 225–232. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Canas, V.; Lorbetskie, B.; Cyr, T.D.; Hefford, M.A.; Smith, S.; Girard, M. Approach to the profiling and characterization of influenza vaccine constituents by the combined use of size-exclusion chromatography, gel electrophoresis and mass spectrometry. Biologicals 2010, 38, 294–302. [Google Scholar] [CrossRef]
- Yang, Y.; Li, H.; Li, Z.; Zhang, Y.; Zhang, S.; Chen, Y.; Yu, M.; Ma, G.; Su, Z. Size-exclusion HPLC provides a simple, rapid, and versatile alternative method for quality control of vaccines by characterizing the assembly of antigens. Vaccine 2015, 33, 1143–1150. [Google Scholar] [CrossRef]
- Durowoju, I.B.; Bhandal, K.S.; Hu, J.; Carpick, B.; Kirkitadze, M. Differential scanning calorimetry—A method for assessing the thermal stability and conformation of protein antigen. JoVE J. Vis. Exp. 2017, 121, e55262. [Google Scholar] [CrossRef] [Green Version]
- Cueto, M.; Dorta, M.J.; Munguía, O.; Llabrés, M. New approach to stability assessment of protein solution formulations by differential scanning calorimetry. Int. J. Pharm. 2003, 252, 159–166. [Google Scholar] [CrossRef]
- Wen, J.; Arthur, K.; Chemmalil, L.; Muzammil, S.; Gabrielson, J.; Jiang, Y. Applications of differential scanning calorimetry for thermal stability analysis of proteins: Qualification of DSC. J. Pharm. Sci. 2012, 101, 955–964. [Google Scholar] [CrossRef]
- Hura, G.L.; Menon, A.L.; Hammel, M.; Rambo, R.P.; Poole Ii, F.L.; Tsutakawa, S.E.; Jenney, F.E., Jr.; Classen, S.; Frankel, K.A.; Hopkins, R.C. Robust, high-throughput solution structural analyses by small angle X-ray scattering (SAXS). Nat. Methods 2009, 6, 606–612. [Google Scholar] [CrossRef]
- Dixit, S.M.; Ruotolo, B.T. A Semi-Empirical Framework for Interpreting Traveling Wave Ion Mobility Arrival Time Distributions. J. Am. Soc. Mass Spectrom. 2019, 30, 956–966. [Google Scholar] [CrossRef] [PubMed]
- Chan-Yao-Chong, M.; Durand, D.; Ha-Duong, T. Molecular Dynamics Simulations Combined with Nuclear Magnetic Resonance and/or Small-Angle X-ray Scattering Data for Characterizing Intrinsically Disordered Protein Conformational Ensembles. J. Chem. Inf. Model. 2019, 59, 1743–1758. [Google Scholar] [CrossRef]
- Mertens, H.D.; Svergun, D.I. Structural characterization of proteins and complexes using small-angle X-ray solution scattering. J. Struct. Biol. 2010, 172, 128–141. [Google Scholar] [CrossRef]
- Rambo, R.P.; Tainer, J.A. Characterizing flexible and intrinsically unstructured biological macromolecules by SAS using the Porod-Debye law. Biopolymers 2011, 95, 559–571. [Google Scholar] [CrossRef] [Green Version]
- Zhang, F.; Skoda, M.W.; Jacobs, R.M.; Martin, R.A.; Martin, C.M.; Schreiber, F. Protein interactions studied by SAXS: Effect of ionic strength and protein concentration for BSA in aqueous solutions. J. Phys. Chem. B 2007, 111, 251–259. [Google Scholar] [CrossRef]
- Mosbæk, C.R.; Konarev, P.V.; Svergun, D.I.; Rischel, C.; Vestergaard, B. High concentration formulation studies of an IgG2 antibody using small angle X-ray scattering. Pharm. Res. 2012, 29, 2225–2235. [Google Scholar] [CrossRef] [PubMed]
- Jacques, D.A.; Trewhella, J. Small-angle scattering for structural biology—Expanding the frontier while avoiding the pitfalls. Protein Sci. 2010, 19, 642–657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silva, B.F. SAXS on a chip: From dynamics of phase transitions to alignment phenomena at interfaces studied with microfluidic devices. Phys. Chem. Chem. Phys. 2017, 19, 23690–23703. [Google Scholar] [CrossRef]
- Yaghmur, A.; Rappolt, M.; Jonassen, A.L.U.; Schmitt, M.; Larsen, S.W. In situ monitoring of the formation of lipidic non-lamellar liquid crystalline depot formulations in synovial fluid. J. Colloid Interface Sci. 2021, 582, 773–781. [Google Scholar] [CrossRef]
- Xu, J.; Wang, R.; Li, Y. A review of available technologies for seasonal thermal energy storage. Sol. Energy 2014, 103, 610–638. [Google Scholar] [CrossRef]
- Johnson, L.N. The structure and function of lysozyme. Sci. Progress 1933 1966, 54, 367–385. [Google Scholar]
- Jollès, P.; Jollès, J. What’s new in lysozyme research? Mol. Cell. Biochem. 1984, 63, 165–189. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.; Brinda, K.; Vishveshwara, S. Dynamics of lysozyme structure network: Probing the process of unfolding. Biophys. J. 2007, 92, 2523–2535. [Google Scholar] [CrossRef] [Green Version]
- Phan-Xuan, T.; Bogdanova, E.; Millqvist Fureby, A.; Fransson, J.; Terry, A.E.; Kocherbitov, V. Hydration-Induced Structural Changes in the Solid State of Protein: A SAXS/WAXS Study on Lysozyme. Mol. Pharm. 2020, 17, 3246–3258. [Google Scholar] [CrossRef] [PubMed]
- Schwenzfeier, A.; Lech, F.; Wierenga, P.A.; Eppink, M.H.; Gruppen, H. Foam properties of algae soluble protein isolate: Effect of pH and ionic strength. Food Hydrocoll. 2013, 33, 111–117. [Google Scholar] [CrossRef]
- Sugio, S.; Kashima, A.; Mochizuki, S.; Noda, M.; Kobayashi, K. Crystal structure of human serum albumin at 2.5 Å resolution. Protein Eng. Des. Sel. 1999, 12, 439–446. [Google Scholar] [CrossRef]
- Chubarov, A.; Spitsyna, A.; Krumkacheva, O.; Mitin, D.; Suvorov, D.; Tormyshev, V.; Fedin, M.; Bowman, M.K.; Bagryanskaya, E. Reversible dimerization of human serum albumin. Molecules 2021, 26, 108. [Google Scholar] [CrossRef]
- Sollenne, N.P.; Wu, H.-L.; Means, G.E. Disruption of the tryptophan binding site in the human serum albumin dimer. Arch. Biochem. Biophys. 1981, 207, 264–269. [Google Scholar] [CrossRef]
- Reščič, J.; Vlachy, V.; Jamnik, A.; Glatter, O. Osmotic pressure, small-angle X-ray, and dynamic light scattering studies of human serum albumin in aqueous solutions. J. Colloid Interface Sci. 2001, 239, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Zunszain, P.; Monie, T.; Konarev, P.; Svergun, D.; Curry, S. A Structural Analysis of Conformational Changes in Human Serum Albumin Associated with Ligand Binding and pH. 2003. Available online: http://hasyweb.desy.de/science/annual_reports/2003_report/part2/contrib/73/9952.pdf (accessed on 22 December 2021).
- Pandey, N.K.; Ghosh, S.; Tripathy, D.R.; Dasgupta, S. Effect of temperature and solvent on fibrillation of human serum albumin. Protein Pept. Lett. 2015, 22, 112–118. [Google Scholar] [CrossRef]
- Maciążek-Jurczyk, M.; Janas, K.; Pożycka, J.; Szkudlarek, A.; Rogóż, W.; Owczarzy, A.; Kulig, K. Human Serum Albumin Aggregation/Fibrillation and its Abilities to Drugs Binding. Molecules 2020, 25, 618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makurvet, F.D. Biologics vs. small molecules: Drug costs and patient access. Med. Drug Discov. 2021, 9, 100075. [Google Scholar] [CrossRef]
- Song, J.G.; Lee, S.H.; Han, H.-K. The stabilization of biopharmaceuticals: Current understanding and future perspectives. J. Pharm. Investig. 2017, 47, 475–496. [Google Scholar] [CrossRef]
- Kesik-Brodacka, M. Progress in biopharmaceutical development. Biotechnol. Appl. Biochem. 2018, 65, 306–322. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Nema, S.; Teagarden, D. Protein aggregation—Pathways and influencing factors. Int. J. Pharm. 2010, 390, 89–99. [Google Scholar] [CrossRef]
- Wang, W.; Roberts, C.J. Protein aggregation—Mechanisms, detection, and control. Int. J. Pharm. 2018, 550, 251–268. [Google Scholar] [CrossRef]
- Manning, M.C.; Liu, J.; Li, T.; Holcomb, R.E. Rational design of liquid formulations of proteins. Adv. Protein Chem. Struct. Biol. 2018, 112, 1–59. [Google Scholar]
- Mahler, H.-C.; Müller, R.; Frieβ, W.; Delille, A.; Matheus, S. Induction and analysis of aggregates in a liquid IgG1-antibody formulation. Eur. J. Pharm. Biopharm. 2005, 59, 407–417. [Google Scholar] [CrossRef] [PubMed]
- Arakawa, T.; Philo, J.S.; Ejima, D.; Tsumoto, K.; Arisaka, F. Aggregation analysis of therapeutic proteins, part 2. Bioprocess Int. 2007, 5, 36–47. [Google Scholar]
- Den Engelsman, J.; Garidel, P.; Smulders, R.; Koll, H.; Smith, B.; Bassarab, S.; Seidl, A.; Hainzl, O.; Jiskoot, W. Strategies for the assessment of protein aggregates in pharmaceutical biotech product development. Pharm. Res. 2011, 28, 920–933. [Google Scholar] [CrossRef] [Green Version]
- Fang, C.; Bhattarai, N.; Sun, C.; Zhang, M. Functionalized nanoparticles with long-term stability in biological media. Small 2009, 5, 1637–1641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewis, E.N.; Qi, W.; Kidder, L.H.; Amin, S.; Kenyon, S.M.; Blake, S. Combined dynamic light scattering and Raman spectroscopy approach for characterizing the aggregation of therapeutic proteins. Molecules 2014, 19, 20888–20905. [Google Scholar] [CrossRef] [Green Version]
- Bisht, M.; Kumar, A.; Venkatesu, P. Analysis of the driving force that rule the stability of lysozyme in alkylammonium-based ionic liquids. Int. J. Biol. Macromol. 2015, 81, 1074–1081. [Google Scholar] [CrossRef] [PubMed]
- Aminlari, L.; Mohammadi Hashemi, M.; Aminlari, M. Modified lysozymes as novel broad spectrum natural antimicrobial agents in foods. J. Food Sci. 2014, 79, R1077–R1090. [Google Scholar] [CrossRef]
- Wetzel, R.; Becker, M.; Behlke, J.; Billwitz, H.; Böhm, S.; Ebert, B.; Hamann, H.; Krumbiegel, J.; Lassmann, G. Temperature behaviour of human serum albumin. Eur. J. Biochem. 1980, 104, 469–478. [Google Scholar] [CrossRef]
- Pavani, P.; Kumar, K.; Rani, A.; Venkatesu, P.; Lee, M.-J. The influence of sodium phosphate buffer on the stability of various proteins: Insights into protein-buffer interactions. J. Mol. Liq. 2021, 331, 115753. [Google Scholar] [CrossRef]
- Vlasak, J.; Ionescu, R. Fragmentation of monoclonal antibodies. In Proceedings of the MAbs; Taylor & Francis: Abingdon, UK, 2011; pp. 253–263. [Google Scholar]
- Borzooeian, Z.; Taslim, M.; Borzooeian, G.; Ghasemi, O.; Aminlari, M. Activity and stability analysis of covalent conjugated lysozyme-single walled carbon nanotubes: Potential biomedical and industrial applications. RSC Adv. 2017, 7, 48692–48701. [Google Scholar] [CrossRef] [Green Version]
- Goda, D.A.; Bassiouny, A.R.; Abdel Monem, N.M.; Soliman, N.A.; Abdel-Fattah, Y.R. Feather protein lysate optimization and feather meal formation using YNDH protease with keratinolytic activity afterward enzyme partial purification and characterization. Sci. Rep. 2021, 11, 14543. [Google Scholar] [CrossRef] [PubMed]
- Sadeghi, A.; Nikkhah, A.; Shawrang, P.; Shahrebabak, M. Protein degradation kinetics of untreated and treated soybean meal using SDS-PAGE. Anim. Feed Sci. Technol. 2006, 126, 121–133. [Google Scholar] [CrossRef]
- Fukuda, M.; Moriyama, C.; Yamazaki, T.; Imaeda, Y.; Koga, A. Quantitative correlation between viscosity of concentrated MAb solutions and particle size parameters obtained from small-angle X-ray scattering. Pharm. Res. 2015, 32, 3803–3812. [Google Scholar] [CrossRef]
- Schmid, P.W.; Lim, N.C.; Peters, C.; Back, K.C.; Bourgeois, B.; Pirolt, F.; Richter, B.; Peschek, J.; Puk, O.; Amarie, O.V. Imbalances in the eye lens proteome are linked to cataract formation. Nat. Struct. Mol. Biol. 2021, 28, 143–151. [Google Scholar] [CrossRef]
- Schilcher, I.; Ledinski, G.; Radulović, S.; Hallström, S.; Eichmann, T.; Madl, T.; Zhang, F.; Leitinger, G.; Kolb-Lenz, D.; Darnhofer, B. Endothelial lipase increases antioxidative capacity of high-density lipoprotein. Biochim. Biophys. Acta BBA Mol. Cell Biol. Lipids 2019, 1864, 1363–1374. [Google Scholar] [CrossRef] [PubMed]
- Janowski, R.; Scanu, S.; Niessing, D.; Madl, T. Crystal and solution structural studies of mouse phospholipid hydroperoxide glutathione peroxidase 4. Acta Crystallogr. Sect. F Struct. Biol. Commun. 2016, 72, 743–749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sonntag, M.; Jagtap, P.K.A.; Simon, B.; Appavou, M.S.; Geerlof, A.; Stehle, R.; Gabel, F.; Hennig, J.; Sattler, M. Segmental, Domain-Selective Perdeuteration and Small-Angle Neutron Scattering for Structural Analysis of Multi-Domain Proteins. Angew. Chem. 2017, 129, 9450–9453. [Google Scholar] [CrossRef]
- Kooshapur, H.; Choudhury, N.R.; Simon, B.; Mühlbauer, M.; Jussupow, A.; Fernandez, N.; Jones, A.N.; Dallmann, A.; Gabel, F.; Camilloni, C. Structural basis for terminal loop recognition and stimulation of pri-miRNA-18a processing by hnRNP A1. Nat. Commun. 2018, 9, 2479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trewhella, J.; Duff, A.P.; Durand, D.; Gabel, F.; Guss, J.M.; Hendrickson, W.A.; Hura, G.L.; Jacques, D.A.; Kirby, N.M.; Kwan, A.H. 2017 publication guidelines for structural modelling of small-angle scattering data from biomolecules in solution: An update. Acta Crystallogr. Sect. D Struct. Biol. 2017, 73, 710–728. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.-L.; Kwan, A.H.; Yammine, A.; Zhou, X.; Trewhella, J.; Hugrass, B.M.; Collins, D.A.; Horne, J.; Ye, P.; Harty, D. Structural properties of a haemophore facilitate targeted elimination of the pathogen Porphyromonas gingivalis. Nat. Commun. 2018, 9, 4097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Damoiseaux, R.; George, S.; Li, M.; Pokhrel, S.; Ji, Z.; France, B.; Xia, T.; Suarez, E.; Rallo, R.; Mädler, L. No time to lose—High throughput screening to assess nanomaterial safety. Nanoscale 2011, 3, 1345–1360. [Google Scholar] [CrossRef] [PubMed]
- David, G.; Pérez, J. Combined sampler robot and high-performance liquid chromatography: A fully automated system for biological small-angle X-ray scattering experiments at the Synchrotron SOLEIL SWING beamline. J. Appl. Crystallogr. 2009, 42, 892–900. [Google Scholar] [CrossRef]
- Blanchet, C.E.; Svergun, D.I. Small-angle X-ray scattering on biological macromolecules and nanocomposites in solution. Annu. Rev. Phys. Chem. 2013, 64, 37–54. [Google Scholar] [CrossRef]
- Lilyestrom, W.G.; Shire, S.J.; Scherer, T.M. Influence of the cosolute environment on IgG solution structure analyzed by small-angle X-ray scattering. J. Phys. Chem. B 2012, 116, 9611–9618. [Google Scholar] [CrossRef] [PubMed]
- Blanchet, C.E.; Zozulya, A.V.; Kikhney, A.G.; Franke, D.; Konarev, P.V.; Shang, W.; Klaering, R.; Robrahn, B.; Hermes, C.; Cipriani, F. Instrumental setup for high-throughput small-and wide-angle solution scattering at the X33 beamline of EMBL Hamburg. J. Appl. Crystallogr. 2012, 45, 489–495. [Google Scholar] [CrossRef]
- Frewein, M.P.; Doktorova, M.; Heberle, F.A.; Scott, H.L.; Semeraro, E.F.; Porcar, L.; Pabst, G. Structure and Interdigitation of Chain-Asymmetric Phosphatidylcholines and Milk Sphingomyelin in the Fluid Phase. Symmetry 2021, 13, 1441. [Google Scholar] [CrossRef]
- Kaltenegger, M.; Kremser, J.; Frewein, M.P.; Bonthuis, D.J.; Ziherl, P.; Pabst, G. Intrinsic lipid curvatures of mammalian plasma membrane outer leaflet lipids and ceramides. bioRxiv 2021. preprint. [Google Scholar] [CrossRef]
- Scherdel, C.; Miller, E.; Reichenauer, G.; Schmitt, J. Advances in the Development of Sol-Gel Materials Combining Small-Angle X-ray Scattering (SAXS) and Machine Learning (ML). Processes 2021, 9, 672. [Google Scholar] [CrossRef]
- Franke, D.; Jeffries, C.M.; Svergun, D.I. Machine learning methods for X-ray scattering data analysis from biomacromolecular solutions. Biophys. J. 2018, 114, 2485–2492. [Google Scholar] [CrossRef] [Green Version]
- Do, C.; Chen, W.-R.; Lee, S. Small angle scattering data analysis assisted by machine learning methods. MRS Adv. 2020, 5, 1577–1584. [Google Scholar] [CrossRef]
- Demerdash, O.; Shrestha, U.R.; Petridis, L.; Smith, J.C.; Mitchell, J.C.; Ramanathan, A. Using small-angle scattering data and parametric machine learning to optimize force field parameters for intrinsically disordered proteins. Front. Mol. Biosci. 2019, 6, 64. [Google Scholar] [CrossRef] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, F.; Richter, G.; Bourgeois, B.; Spreitzer, E.; Moser, A.; Keilbach, A.; Kotnik, P.; Madl, T. A General Small-Angle X-ray Scattering-Based Screening Protocol for Studying Physical Stability of Protein Formulations. Pharmaceutics 2022, 14, 69. https://doi.org/10.3390/pharmaceutics14010069
Zhang F, Richter G, Bourgeois B, Spreitzer E, Moser A, Keilbach A, Kotnik P, Madl T. A General Small-Angle X-ray Scattering-Based Screening Protocol for Studying Physical Stability of Protein Formulations. Pharmaceutics. 2022; 14(1):69. https://doi.org/10.3390/pharmaceutics14010069
Chicago/Turabian StyleZhang, Fangrong, Gesa Richter, Benjamin Bourgeois, Emil Spreitzer, Armin Moser, Andreas Keilbach, Petra Kotnik, and Tobias Madl. 2022. "A General Small-Angle X-ray Scattering-Based Screening Protocol for Studying Physical Stability of Protein Formulations" Pharmaceutics 14, no. 1: 69. https://doi.org/10.3390/pharmaceutics14010069
APA StyleZhang, F., Richter, G., Bourgeois, B., Spreitzer, E., Moser, A., Keilbach, A., Kotnik, P., & Madl, T. (2022). A General Small-Angle X-ray Scattering-Based Screening Protocol for Studying Physical Stability of Protein Formulations. Pharmaceutics, 14(1), 69. https://doi.org/10.3390/pharmaceutics14010069