
Targeting Histone Deacetylases: Opportunities for Cancer Treatment and Chemoprevention

 ,
,  , ,
, , 


Abstract
:1. Introduction
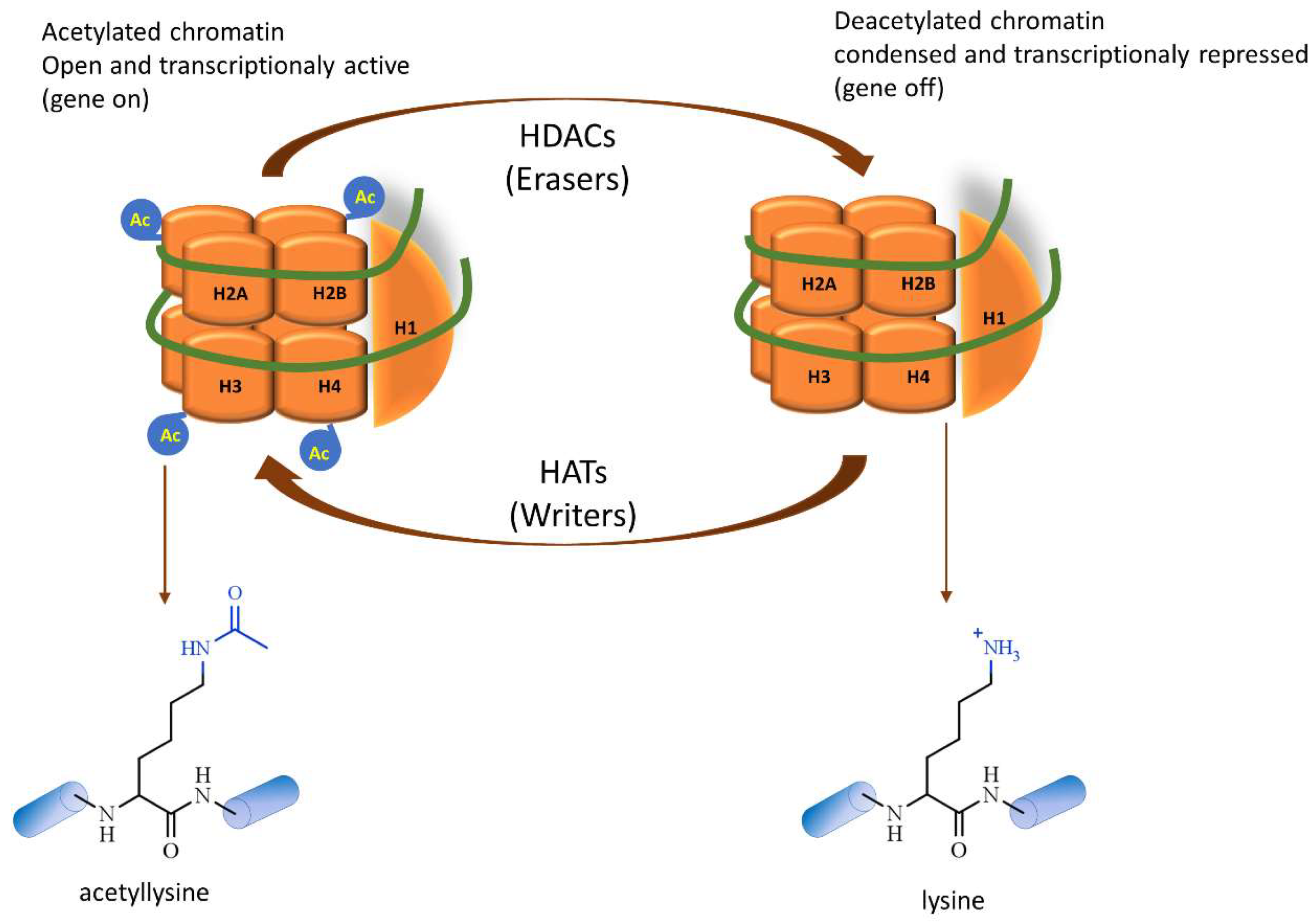
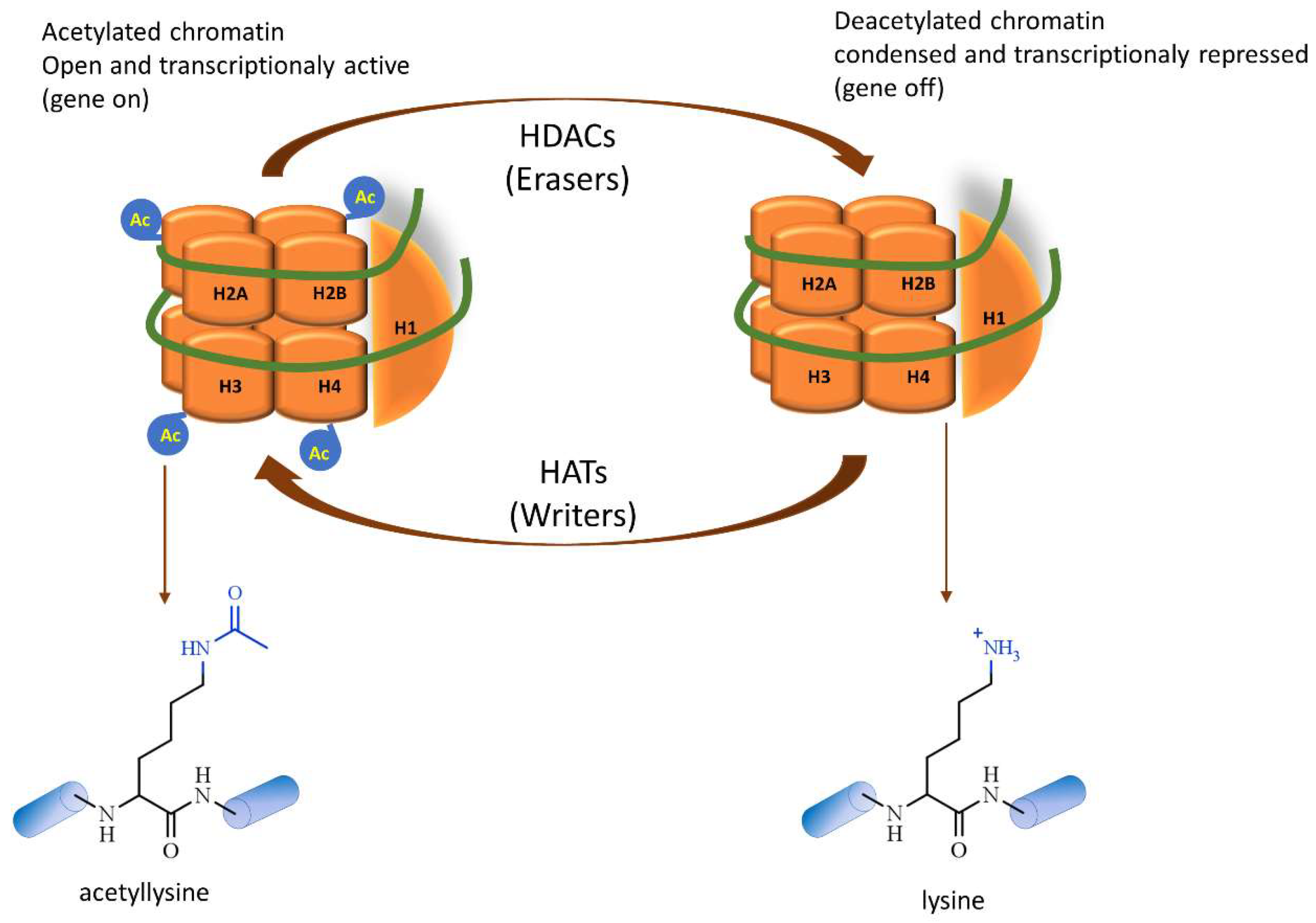
2. Histone Deacetylases Overview
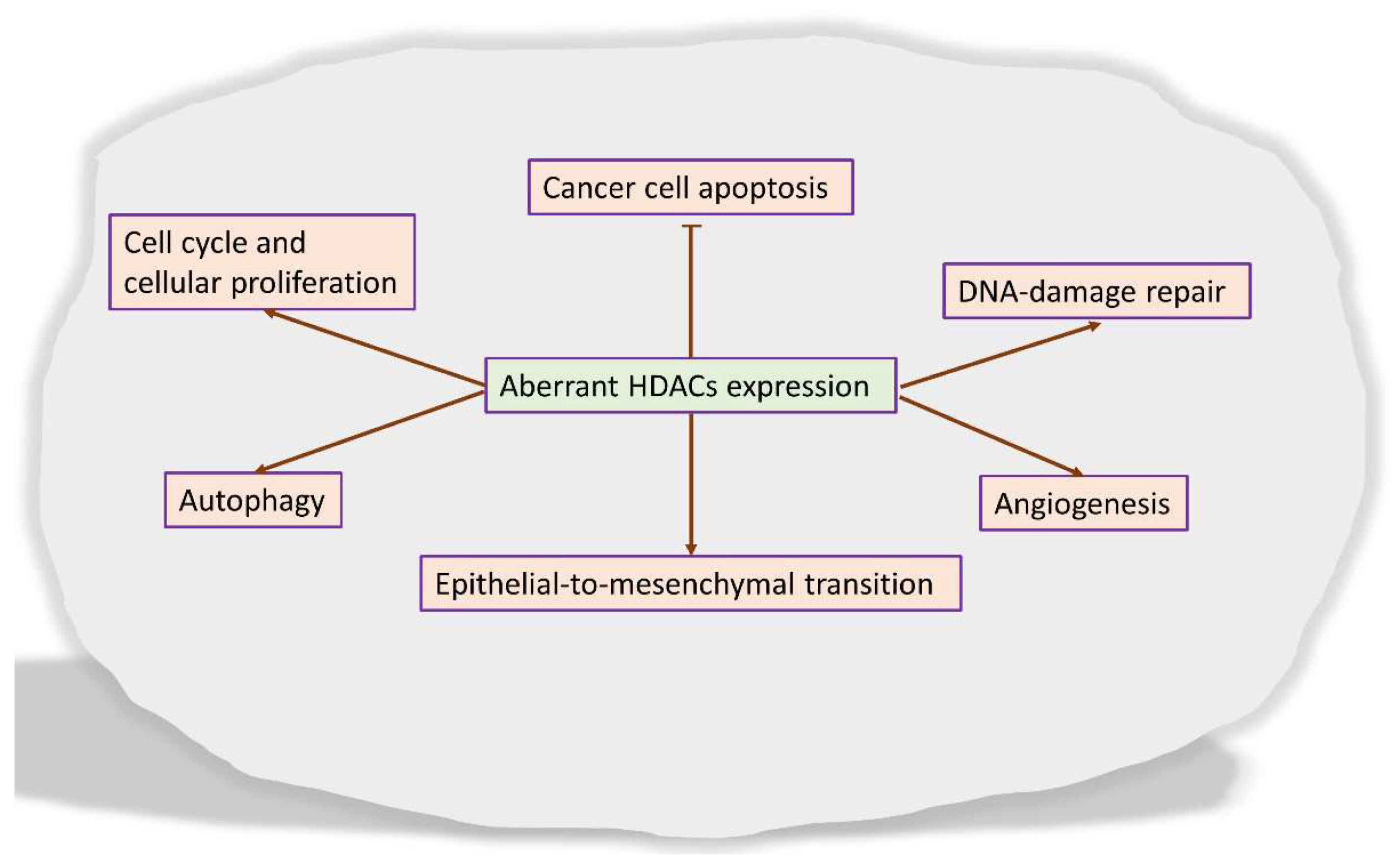
3. Histone Deacetylases Inhibitors in the Treatment of Cancer
3.1. Histone Deacetylase Inhibitors
3.1.1. Aliphatic Acids
3.1.2. Hydroxamic Acids
3.1.3. Benzamides
3.1.4. Cyclic Peptides
3.2. Natural Compounds in Targeting Histone Deacetylases for Cancer Chemoprevention
4. Perspectives in the Development of New HDAC Inhibitors
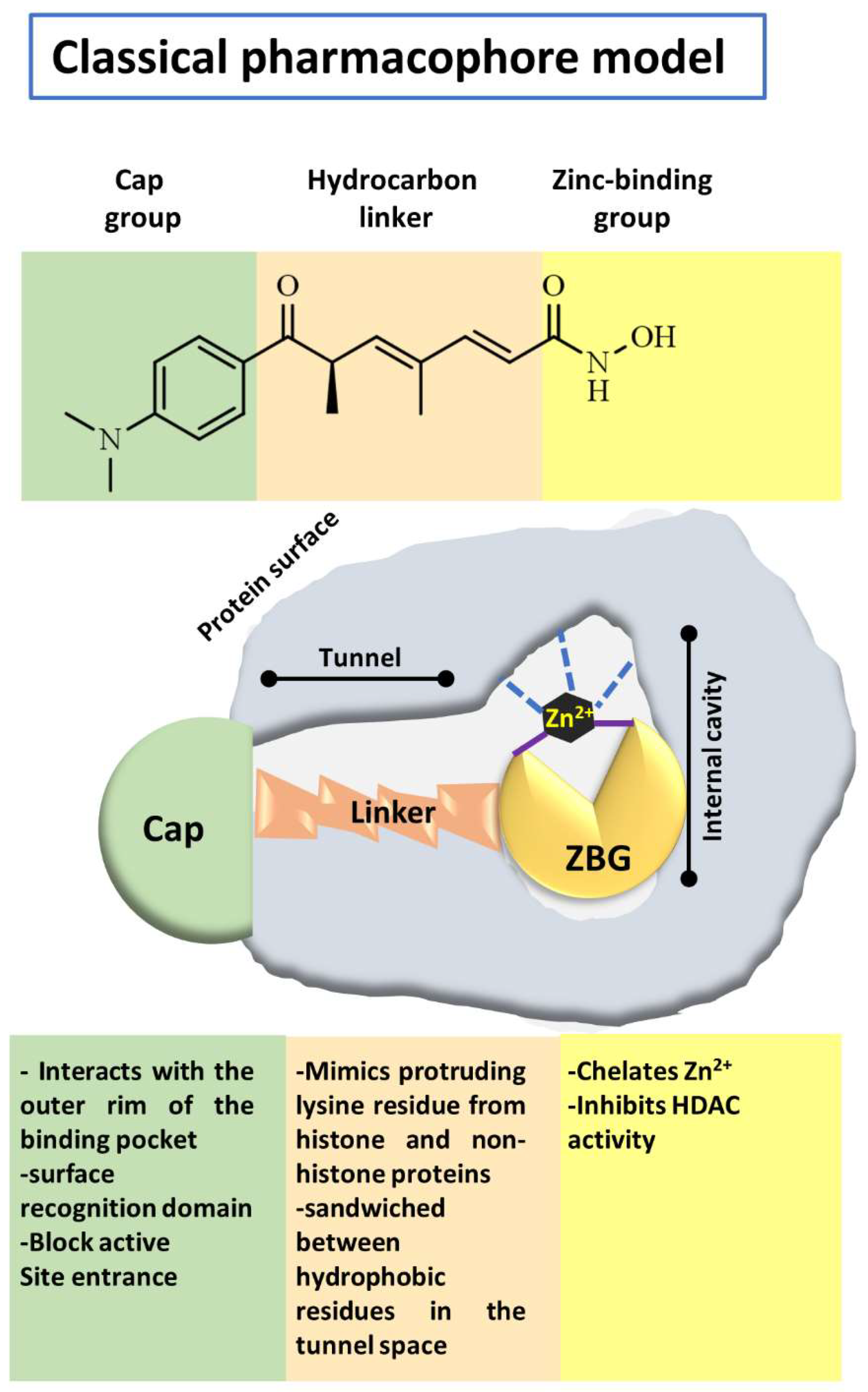
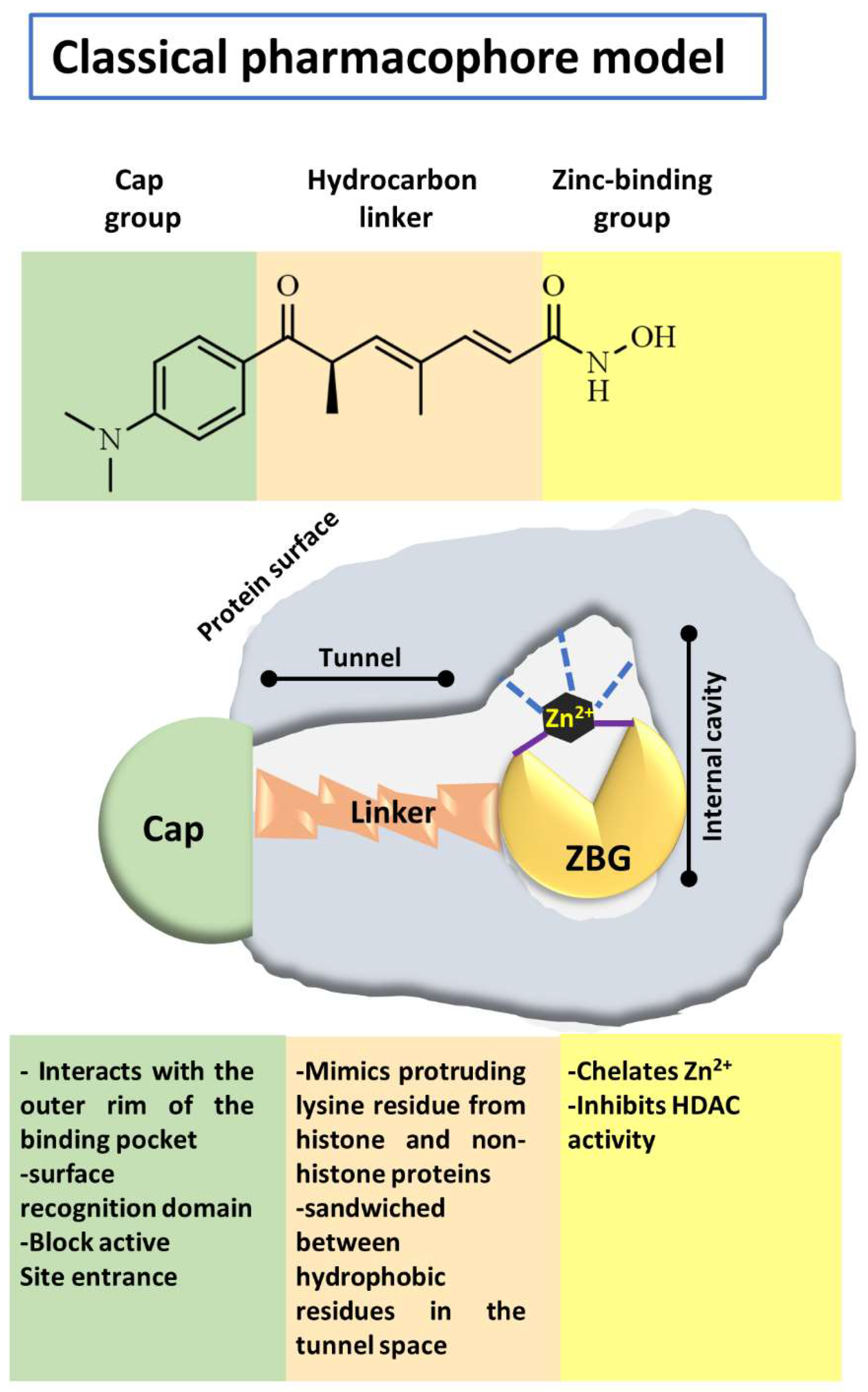
4.1. Structural Considerations to Design Histone Deacetylases Inhibitors
4.2. Bifunctional Histone Deacetylase Inhibitors
4.3. Proteolysis Targeting Chimeras
5. Combined Clinical Strategies with Histone Deacetylases Inhibitors
6. Conclusions and Concluding Remarks
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Feinberg, A.P.; Ohlsson, R.; Henikoff, S. The epigenetic progenitor origin of human cancer. Nat. Rev. Genet. 2006, 7, 21–33. [Google Scholar] [CrossRef]
- Feinberg, A.P.; Vogelstein, B. Hypomethylation distinguishes genes of some human cancers from their normal counterparts. Nature 1983, 301, 89–92. [Google Scholar] [CrossRef]
- Herman, J.G.; Baylin, S.B. Gene silencing in cancer in association with promoter hypermethylation. N. Engl. J. Med. 2003, 349, 2042–2054. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Lin, G.-L.; Yan, P.; Huang, J.; Wang, Y.-L. The expression of H3K9Ac, H3K14Ac, and H4K20TriMe in epithelial ovarian tumors and the clinical significance. Int. J. Gynecol. Cancer 2010, 20, 82–86. [Google Scholar] [CrossRef]
- Fraga, M.F.; Ballestar, E.; Villar-Garea, A.; Boix-Chornet, M.; Espada, J.; Schotta, G.; Bonaldi, T.; Haydon, C.; Ropero, S.; Petrie, K.; et al. Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nat. Genet. 2005, 37, 391–400. [Google Scholar] [CrossRef]
- Talbert, P.B.; Henikoff, S. Histone variants at a glance. J. Cell Sci. 2021, 134, jcs244749. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Carey, M.; Workman, J.L. The role of chromatin during transcription. Cell 2007, 128, 707–719. [Google Scholar] [CrossRef] [Green Version]
- Willhoft, O.; Costa, A. A structural framework for DNA replication and transcription through chromatin. Curr. Opin. Struct. Biol. 2021, 71, 51–58. [Google Scholar] [CrossRef]
- Rothbart, S.B.; Strahl, B.D. Interpreting the language of histone and DNA modifications. Biochim. Biophys. Acta 2014, 1839, 627–643. [Google Scholar] [CrossRef] [Green Version]
- Nitsch, S.; Zorro Shahidian, L.; Schneider, R. Histone acylations and chromatin dynamics: Concepts, challenges, and links to metabolism. EMBO Rep. 2021, 22, e52774. [Google Scholar] [CrossRef]
- Seto, E.; Yoshida, M. Erasers of histone acetylation: The histone deacetylase enzymes. Cold Spring Harb. Perspect. Biol. 2014, 6, a018713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Losson, H.; Schnekenburger, M.; Dicato, M.; Diederich, M. Natural Compound Histone Deacetylase Inhibitors (HDACi): Synergy with Inflammatory Signaling Pathway Modulators and Clinical Applications in Cancer. Molecules 2016, 21, 1608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Archer, S.Y.; Hodin, R.A. Histone acetylation and cancer. Curr. Opin. Genet. Dev. 1999, 9, 171–174. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.L.; Luo, M.; Huang, C.; Chen, H.N.; Zhou, Z.G. Epigenetic Regulation of Epithelial to Mesenchymal Transition in the Cancer Metastatic Cascade: Implications for Cancer Therapy. Front. Oncol. 2021, 11, 657546. [Google Scholar] [CrossRef]
- Mirzadeh Azad, F.; Atlasi, Y. Deregulation of Transcriptional Enhancers in Cancer. Cancers 2021, 13, 3532. [Google Scholar] [CrossRef] [PubMed]
- You, J.S.; Jones, P.A. Cancer genetics and epigenetics: Two sides of the same coin? Cancer Cell 2012, 22, 9–20. [Google Scholar] [CrossRef] [Green Version]
- Kanwal, R.; Gupta, S. Epigenetic modifications in cancer. Clin. Genet. 2012, 81, 303–311. [Google Scholar] [CrossRef] [Green Version]
- Peng, L.; Seto, E. Deacetylation of nonhistone proteins by HDACs and the implications in cancer. Handb. Exp. Pharmacol. 2011, 206, 39–56. [Google Scholar] [CrossRef]
- Darwiche, N. Epigenetic mechanisms and the hallmarks of cancer: An intimate affair. Am. J. Cancer Res. 2020, 10, 1954–1978. [Google Scholar]
- Pant, K.; Peixoto, E.; Richard, S.; Gradilone, S.A. Role of Histone Deacetylases in Carcinogenesis: Potential Role in Cholangiocarcinoma. Cells 2020, 9, 780. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Seto, E. HDACs and HDAC Inhibitors in Cancer Development and Therapy. Cold Spring Harb. Perspect. Med. 2016, 6, a026831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.B.; Ki, S.W.; Yoshida, M.; Horinouchi, S. Mechanism of cell cycle arrest caused by histone deacetylase inhibitors in human carcinoma cells. J. Antibiot. 2000, 53, 1191–1200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chun, P. Histone deacetylase inhibitors in hematological malignancies and solid tumors. Arch. Pharmacal Res. 2015, 38, 933–949. [Google Scholar] [CrossRef] [PubMed]
- Gong, P.; Wang, Y.; Jing, Y. Apoptosis Induction byHistone Deacetylase Inhibitors in Cancer Cells: Role of Ku70. Int. J. Mol. Sci. 2019, 20, 1601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Zhong, Q. Histone deacetylase inhibitors and cell death. Cell. Mol. Life Sci. 2014, 71, 3885–3901. [Google Scholar] [CrossRef]
- Roos, W.P.; Krumm, A. The multifaceted influence of histone deacetylases on DNA damage signalling and DNA repair. Nucleic Acids Res. 2016, 44, 10017–10030. [Google Scholar] [CrossRef] [Green Version]
- Koeneke, E.; Witt, O.; Oehme, I. HDAC Family Members Intertwined in the Regulation of Autophagy: A Druggable Vulnerability in Aggressive Tumor Entities. Cells 2015, 4, 135–168. [Google Scholar] [CrossRef]
- Rikiishi, H. Autophagic and apoptotic effects of HDAC inhibitors on cancer cells. J. Biomed. Biotechnol. 2011, 2011, 830260. [Google Scholar] [CrossRef]
- Mrakovcic, M.; Kleinheinz, J.; Fröhlich, L.F. Histone Deacetylase Inhibitor-Induced Autophagy in Tumor Cells: Implications for p53. Int. J. Mol. Sci. 2017, 18, 1883. [Google Scholar] [CrossRef] [Green Version]
- Sykes, S.M.; Mellert, H.S.; Holbert, M.A.; Li, K.; Marmorstein, R.; Lane, W.S.; McMahon, S.B. Acetylation of the p53 DNA-binding domain regulates apoptosis induction. Mol. Cell 2006, 24, 841–851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.L.; Yang, P.M.; Shun, C.T.; Wu, M.S.; Weng, J.R.; Chen, C.C. Autophagy potentiates the anti-cancer effects of the histone deacetylase inhibitors in hepatocellular carcinoma. Autophagy 2010, 6, 1057–1065. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, M.; Adachi, S.; Matsubara, H.; Imai, T.; Yui, Y.; Mizushima, Y.; Hiraumi, Y.; Watanabe, K.; Kamitsuji, Y.; Toyokuni, S.Y.; et al. Induction of autophagy in malignant rhabdoid tumor cells by the histone deacetylase inhibitor FK228 through AIF translocation. Int. J. Cancer 2009, 124, 55–67. [Google Scholar] [CrossRef] [Green Version]
- von Burstin, J.; Eser, S.; Paul, M.C.; Seidler, B.; Brandl, M.; Messer, M.; von Werder, A.; Schmidt, A.; Mages, J.; Pagel, P.; et al. E-cadherin regulates metastasis of pancreatic cancer in vivo and is suppressed by a SNAIL/HDAC1/HDAC2 repressor complex. Gastroenterology 2009, 137, 361–371.e3715. [Google Scholar] [CrossRef] [PubMed]
- Aghdassi, A.; Sendler, M.; Guenther, A.; Mayerle, J.; Behn, C.O.; Heidecke, C.D.; Friess, H.; Büchler, M.; Evert, M.; Lerch, M.M.; et al. Recruitment of histone deacetylases HDAC1 and HDAC2 by the transcriptional repressor ZEB1 downregulates E-cadherin expression in pancreatic cancer. Gut 2012, 61, 439–448. [Google Scholar] [CrossRef]
- Rajabi, M.; Mousa, S.A. The Role of Angiogenesis in Cancer Treatment. Biomedicines 2017, 5, 34. [Google Scholar] [CrossRef] [Green Version]
- Geng, H.; Harvey, C.T.; Pittsenbarger, J.; Liu, Q.; Beer, T.M.; Xue, C.; Qian, D.Z. HDAC4 protein regulates HIF1α protein lysine acetylation and cancer cell response to hypoxia. J. Biol. Chem. 2011, 286, 38095–38102. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Yin, C.; Lao, T.; Liang, D.; He, D.; Wang, C.; Sang, N. AMPK-HDAC5 pathway facilitates nuclear accumulation of HIF-1α and functional activation of HIF-1 by deacetylating Hsp70 in the cytosol. Cell Cycle 2015, 14, 2520–2536. [Google Scholar] [CrossRef] [Green Version]
- Allfrey, V.G.; Faulkner, R.; Mirsky, A.E. Acetylation and Methylation of histones and their possible role in the regulation of RNA synthesis. Proc. Natl. Acad. Sci. USA 1964, 51, 786–794. [Google Scholar] [CrossRef] [Green Version]
- Inoue, A.; Fujimoto, D. Enzymatic deacetylation of histone. Biochem. Biophys. Res. Commun. 1969, 36, 146–150. [Google Scholar] [CrossRef]
- Taunton, J.; Hassig, C.A.; Schreiber, S.L. A mammalian histone deacetylase related to the yeast transcriptional regulator Rpd3p. Science 1996, 272, 408–411. [Google Scholar] [CrossRef]
- Ho, T.; Chan, A.; Ganesan, A. Thirty Years of HDAC Inhibitors: 2020 Insight and Hindsight. J. Med. Chem. 2020, 63, 12460–12484. [Google Scholar] [CrossRef]
- Leipe, D.D.; Landsman, D. Histone deacetylases, acetoin utilization proteins and acetylpolyamine amidohydrolases are members of an ancient protein superfamily. Nucleic Acids Res. 1997, 25, 3693–3697. [Google Scholar] [CrossRef] [Green Version]
- Gao, L.; Cueto, M.A.; Asselbergs, F.; Atadja, P. Cloning and functional characterization of HDAC11, a novel member of the human histone deacetylase family. J. Biol. Chem. 2002, 277, 25748–25755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Ruijter, A.J.; van Gennip, A.H.; Caron, H.N.; Kemp, S.; van Kuilenburg, A.B. Histone deacetylases (HDACs): Characterization of the classical HDAC family. Biochem. J. 2003, 370 Pt 3, 737–749. [Google Scholar] [CrossRef] [PubMed]
- Grozinger, C.M.; Hassig, C.A.; Schreiber, S.L. Three proteins define a class of human histone deacetylases related to yeast Hda1p. Proc. Natl. Acad. Sci. USA 1999, 96, 4868–4873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, X.J.; Seto, E. The Rpd3/Hda1 family of lysine deacetylases: From bacteria and yeast to mice and men. Nat. Rev. Mol. Cell Biol. 2008, 9, 206–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, B.; Hong, J. An overview of naturally occurring histone deacetylase inhibitors. Curr. Top. Med. Chem. 2015, 14, 2759–2782. [Google Scholar] [CrossRef] [PubMed]
- Adimoolam, S.; Sirisawad, M.; Chen, J.; Thiemann, P.; Ford, J.M.; Buggy, J.J. HDAC inhibitor PCI-24781 decreases RAD51 expression and inhibits homologous recombination. Proc. Natl. Acad. Sci. USA 2007, 104, 19482–19487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patra, S.; Panigrahi, D.P.; Praharaj, P.P.; Bhol, C.S.; Mahapatra, K.K.; Mishra, S.R.; Behera, B.P.; Jena, M.; Bhutia, S.K. Dysregulation of histone deacetylases in carcinogenesis and tumor progression: A possible link to apoptosis and autophagy. Cell. Mol. Life Sci. 2019, 76, 3263–3282. [Google Scholar] [CrossRef] [PubMed]
- Wawruszak, A.; Kalafut, J.; Okon, E.; Czapinski, J.; Halasa, M.; Przybyszewska, A.; Miziak, P.; Okla, K.; Rivero-Muller, A.; Stepulak, A. Histone Deacetylase Inhibitors and Phenotypical Transformation of Cancer Cells. Cancers 2019, 11, 148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bassett, S.A.; Barnett, M.P. The role of dietary histone deacetylases (HDACs) inhibitors in health and disease. Nutrients 2014, 6, 4273–4301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.P.; Zhao, Y.T.; Zhao, T.C. Histone deacetylases and mechanisms of regulation of gene expression. Crit. Rev. Oncog. 2015, 20, 35–47. [Google Scholar] [CrossRef]
- Galluzzi, L.; Buqué, A.; Kepp, O.; Zitvogel, L.; Kroemer, G. Immunological Effects of Conventional Chemotherapy and Targeted Anticancer Agents. Cancer Cell 2015, 28, 690–714. [Google Scholar] [CrossRef] [Green Version]
- Eckschlager, T.; Plch, J.; Stiborova, M.; Hrabeta, J. Histone Deacetylase Inhibitors as Anticancer Drugs. Int. J. Mol. Sci. 2017, 18, 1414. [Google Scholar] [CrossRef]
- Nussinov, R.; Zhang, M.; Maloney, R.; Tsai, C.J.; Yavuz, B.R.; Tuncbag, N.; Jang, H. Mechanism of activation and the rewired network: New drug design concepts. Med. Res. Rev. 2021. [Google Scholar] [CrossRef]
- de Melo, F.; Oliveira, J.S.; Sartorelli, V.; Montor, W.R. Cancer Chemoprevention: Classic and Epigenetic Mechanisms Inhibiting Tumorigenesis. What Have We Learned So Far? Front. Oncol. 2018, 8, 644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perri, F.; Longo, F.; Giuliano, M.; Sabbatino, F.; Favia, G.; Ionna, F.; Addeo, R.; Della Vittoria Scarpati, G.; Di Lorenzo, G.; Pisconti, S. Epigenetic control of gene expression: Potential implications for cancer treatment. Crit. Rev. Oncol. Hematol. 2017, 111, 166–172. [Google Scholar] [CrossRef]
- Dawson, M.A.; Kouzarides, T. Cancer epigenetics: From mechanism to therapy. Cell 2012, 150, 12–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mair, B.; Kubicek, S.; Nijman, S.M. Exploiting epigenetic vulnerabilities for cancer therapeutics. Trends Pharmacol. Sci. 2014, 35, 136–145. [Google Scholar] [CrossRef]
- Koschmann, C.; Nunez, F.J.; Mendez, F.; Brosnan-Cashman, J.A.; Meeker, A.K.; Lowenstein, P.R.; Castro, M.G. Mutated Chromatin Regulatory Factors as Tumor Drivers in Cancer. Cancer Res. 2017, 77, 227–233. [Google Scholar] [CrossRef] [Green Version]
- Park, S.Y.; Kim, J.S. A short guide to histone deacetylases including recent progress on class II enzymes. Exp. Mol. Med. 2020, 52, 204–212. [Google Scholar] [CrossRef]
- Lombardi, P.M.; Cole, K.E.; Dowling, D.P.; Christianson, D.W. Structure, mechanism, and inhibition of histone deacetylases and related metalloenzymes. Curr. Opin. Struct. Biol. 2011, 21, 735–743. [Google Scholar] [CrossRef] [Green Version]
- Roche, J.; Bertrand, P. Inside HDACs with more selective HDAC inhibitors. Eur. J. Med. Chem. 2016, 121, 451–483. [Google Scholar] [CrossRef] [PubMed]
- Smalley, J.P.; Cowley, S.M.; Hodgkinson, J.T. Bifunctional HDAC Therapeutics: One Drug to Rule Them All? Molecules 2020, 25, 4394. [Google Scholar] [CrossRef] [PubMed]
- Jung, M.; Brosch, G.; Kölle, D.; Scherf, H.; Gerhäuser, C.; Loidl, P. Amide analogues of trichostatin A as inhibitors of histone deacetylase and inducers of terminal cell differentiation. J. Med. Chem. 1999, 42, 4669–4679. [Google Scholar] [CrossRef]
- Melesina, J.; Praetorius, L.; Simoben, C.V.; Robaa, D.; Sippl, W. Design of selective histone deacetylase inhibitors: Rethinking classical pharmacophore. Future Med. Chem. 2018, 10, 1537–1540. [Google Scholar] [CrossRef]
- Beshore, D.C.; Adam, G.C.; Barnard, R.; Burlein, C.; Gallicchio, S.N.; Holloway, M.K.; Krosky, D.; Lemaire, W.; Myers, R.W.; Patel, S.; et al. Redefining the Histone Deacetylase Inhibitor Pharmacophore: High Potency with No Zinc Cofactor Interaction. ACS Med. Chem. Lett. 2021, 12, 540–547. [Google Scholar] [CrossRef]
- Dokmanovic, M.; Marks, P.A. Prospects: Histone deacetylase inhibitors. J. Cell. Biochem. 2005, 96, 293–304. [Google Scholar] [CrossRef]
- Fass, D.M.; Shah, R.; Ghosh, B.; Hennig, K.; Norton, S.; Zhao, W.N.; Reis, S.A.; Klein, P.S.; Mazitschek, R.; Maglathlin, R.L.; et al. Effect of Inhibiting Histone Deacetylase with Short-Chain Carboxylic Acids and Their Hydroxamic Acid Analogs on Vertebrate Development and Neuronal Chromatin. ACS Med. Chem. Lett. 2010, 2, 39–42. [Google Scholar] [CrossRef] [PubMed]
- Kruh, J. Effects of sodium butyrate, a new pharmacological agent, on cells in culture. Mol. Cell. Biochem. 1982, 42, 65–82. [Google Scholar] [CrossRef]
- Sekhavat, A.; Sun, J.M.; Davie, J.R. Competitive inhibition of histone deacetylase activity by trichostatin A and butyrate. Biochem. Cell Biol. 2007, 85, 751–758. [Google Scholar] [CrossRef] [PubMed]
- Sowa, Y.; Sakai, T. Butyrate as a model for “gene-regulating chemoprevention and chemotherapy”. BioFactors 2000, 12, 283–287. [Google Scholar] [CrossRef] [PubMed]
- Scharlau, D.; Borowicki, A.; Habermann, N.; Hofmann, T.; Klenow, S.; Miene, C.; Munjal, U.; Stein, K.; Glei, M. Mechanisms of primary cancer prevention by butyrate and other products formed during gut flora-mediated fermentation of dietary fibre. Mutat. Res. 2009, 682, 39–53. [Google Scholar] [CrossRef] [PubMed]
- Spira, A.I.; Carducci, M.A. Differentiation therapy. Curr. Opin. Pharmacol. 2003, 3, 338–343. [Google Scholar] [CrossRef]
- Home-ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ (accessed on 15 July 2021).
- Blaheta, R.A.; Cinatl, J., Jr. Anti-tumor mechanisms of valproate: A novel role for an old drug. Med. Res. Rev. 2002, 22, 492–511. [Google Scholar] [CrossRef] [PubMed]
- Göttlicher, M.; Minucci, S.; Zhu, P.; Krämer, O.H.; Schimpf, A.; Giavara, S.; Sleeman, J.P.; Lo Coco, F.; Nervi, C.; Pelicci, P.G.; et al. Valproic acid defines a novel class of HDAC inhibitors inducing differentiation of transformed cells. EMBO J. 2001, 20, 6969–6978. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, M.; Kijima, M.; Akita, M.; Beppu, T. Potent and specific inhibition of mammalian histone deacetylase both in vivo and in vitro by trichostatin A. J. Biol. Chem. 1990, 265, 17174–17179. [Google Scholar] [CrossRef]
- Charrier, C.; Bertrand, P.; Gesson, J.P.; Roche, J. Synthesis of rigid trichostatin A analogs as HDAC inhibitors. Bioorg. Med. Chem. Lett. 2006, 16, 5339–5344. [Google Scholar] [CrossRef] [PubMed]
- Kudo, K.; Ozaki, T.; Shin-Ya, K.; Nishiyama, M.; Kuzuyama, T. Biosynthetic Origin of the Hydroxamic Acid Moiety of Trichostatin A: Identification of Unprecedented Enzymatic Machinery Involved in Hydroxylamine Transfer. J. Am. Chem. Soc. 2017, 139, 6799–6802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wetzel, M.; Premkumar, D.R.; Arnold, B.; Pollack, I.F. Effect of trichostatin A, a histone deacetylase inhibitor, on glioma proliferation in vitro by inducing cell cycle arrest and apoptosis. J. Neurosurg. 2005, 103 (Suppl. 6), 549–556. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, A.; Trivedi, C.M.; Damera, G.; Jiang, M.; Jester, W.; Hoshi, T.; Epstein, J.A.; Panettieri, R.A., Jr. Trichostatin A abrogates airway constriction, but not inflammation, in murine and human asthma models. Am. J. Respir. Cell Mol. Biol. 2012, 46, 132–138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chuang, D.M.; Leng, Y.; Marinova, Z.; Kim, H.J.; Chiu, C.T. Multiple roles of HDAC inhibition in neurodegenerative conditions. Trends Neurosci. 2009, 32, 591–601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finnin, M.S.; Donigian, J.R.; Cohen, A.; Richon, V.M.; Rifkind, R.A.; Marks, P.A.; Breslow, R.; Pavletich, N.P. Structures of a histone deacetylase homologue bound to the TSA and SAHA inhibitors. Nature 1999, 401, 188–193. [Google Scholar] [CrossRef]
- Yoon, S.; Eom, G.H. HDAC and HDAC Inhibitor: From Cancer to Cardiovascular Diseases. Chonnam Med. J. 2016, 52, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tran, K.; Risingsong, R.; Royce, D.B.; Williams, C.R.; Sporn, M.B.; Pioli, P.A.; Gediya, L.K.; Njar, V.C.; Liby, K.T. The combination of the histone deacetylase inhibitor vorinostat and synthetic triterpenoids reduces tumorigenesis in mouse models of cancer. Carcinogenesis 2013, 34, 199–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gowda, R.; Madhunapantula, S.V.; Desai, D.; Amin, S.; Robertson, G.P. Selenium-containing histone deacetylase inhibitors for melanoma management. Cancer Biol. Ther. 2012, 13, 756–765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nowotarski, S.L.; Woster, P.M.; Casero, R.A., Jr. Polyamines and cancer: Implications for chemotherapy and chemoprevention. Expert Rev. Mol. Med. 2013, 15, e3. [Google Scholar] [CrossRef] [Green Version]
- Varghese, S.; Senanayake, T.; Murray-Stewart, T.; Doering, K.; Fraser, A.; Casero, R.A., Jr.; Woster, P.M. Polyaminohydroxamic acids and polyaminobenzamides as isoform selective histone deacetylase inhibitors. J. Med. Chem. 2008, 51, 2447–2456. [Google Scholar] [CrossRef] [Green Version]
- Plumb, J.A.; Finn, P.W.; Williams, R.J.; Bandara, M.J.; Romero, M.R.; Watkins, C.J.; La Thangue, N.B.; Brown, R. Pharmacodynamic response and inhibition of growth of human tumor xenografts by the novel histone deacetylase inhibitor PXD101. Mol. Cancer Ther. 2003, 2, 721–728. [Google Scholar]
- Foss, F.; Advani, R.; Duvic, M.; Hymes, K.B.; Intragumtornchai, T.; Lekhakula, A.; Shpilberg, O.; Lerner, A.; Belt, R.J.; Jacobsen, E.D.; et al. A Phase II trial of Belinostat (PXD101) in patients with relapsed or refractory peripheral or cutaneous T-cell lymphoma. Br. J. Haematol. 2015, 168, 811–819. [Google Scholar] [CrossRef]
- Sawas, A.; Radeski, D.; O’Connor, O.A. Belinostat in patients with refractory or relapsed peripheral T-cell lymphoma: A perspective review. Ther. Adv. Hematol. 2015, 6, 202–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prince, H.M.; Bishton, M.J.; Johnstone, R.W. Panobinostat (LBH589): A potent pan-deacetylase inhibitor with promising activity against hematologic and solid tumors. Future Oncol. 2009, 5, 601–612. [Google Scholar] [CrossRef]
- Atadja, P. Development of the pan-DAC inhibitor panobinostat (LBH589): Successes and challenges. Cancer Lett. 2009, 280, 233–241. [Google Scholar] [CrossRef]
- Cohen, L.A.; Amin, S.; Marks, P.A.; Rifkind, R.A.; Desai, D.; Richon, V.M. Chemoprevention of carcinogen-induced mammary tumorigenesis by the hybrid polar cytodifferentiation agent, suberanilohydroxamic acid (SAHA). Anticancer Res. 1999, 19, 4999–5005. [Google Scholar]
- Desai, D.; Das, A.; Cohen, L.; el-Bayoumy, K.; Amin, S. Chemopreventive efficacy of suberoylanilide hydroxamic acid (SAHA) against 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK)-induced lung tumorigenesis in female A/J mice. Anticancer Res. 2003, 23, 499–503. [Google Scholar]
- Singh, A.; Patel, V.K.; Jain, D.K.; Patel, P.; Rajak, H. Panobinostat as Pan-deacetylase Inhibitor for the Treatment of Pancreatic Cancer: Recent Progress and Future Prospects. Oncol. Ther. 2016, 4, 73–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Helland, Ø.; Popa, M.; Bischof, K.; Gjertsen, B.T.; McCormack, E.; Bjørge, L. The HDACi Panobinostat Shows Growth Inhibition Both In Vitro and in a Bioluminescent Orthotopic Surgical Xenograft Model of Ovarian Cancer. PLoS ONE 2016, 11, e0158208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bots, M.; Verbrugge, I.; Martin, B.P.; Salmon, J.M.; Ghisi, M.; Baker, A.; Stanley, K.; Shortt, J.; Ossenkoppele, G.J.; Zuber, J.; et al. Differentiation therapy for the treatment of t(8;21) acute myeloid leukemia using histone deacetylase inhibitors. Blood 2014, 123, 1341–1352. [Google Scholar] [CrossRef] [PubMed]
- Scuto, A.; Kirschbaum, M.; Buettner, R.; Kujawski, M.; Cermak, J.M.; Atadja, P.; Jove, R. SIRT1 activation enhances HDAC inhibition-mediated upregulation of GADD45G by repressing the binding of NF-κB/STAT3 complex to its promoter in malignant lymphoid cells. Cell Death Dis. 2013, 4, e635. [Google Scholar] [CrossRef] [PubMed]
- Merarchi, M.; Sethi, G.; Shanmugam, M.K.; Fan, L.; Arfuso, F.; Ahn, K.S. Role of Natural Products in Modulating Histone Deacetylases in Cancer. Molecules 2019, 24, 1047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porter, N.J.; Mahendran, A.; Breslow, R.; Christianson, D.W. Unusual zinc-binding mode of HDAC6-selective hydroxamate inhibitors. Proc. Natl. Acad. Sci. USA 2017, 114, 13459–13464. [Google Scholar] [CrossRef] [Green Version]
- Santo, L.; Hideshima, T.; Kung, A.L.; Tseng, J.C.; Tamang, D.; Yang, M.; Jarpe, M.; van Duzer, J.H.; Mazitschek, R.; Ogier, W.C.; et al. Preclinical activity, pharmacodynamic, and pharmacokinetic properties of a selective HDAC6 inhibitor, ACY-1215, in combination with bortezomib in multiple myeloma. Blood 2012, 119, 2579–2589. [Google Scholar] [CrossRef] [PubMed]
- Yee, A.J.; Bensinger, W.I.; Supko, J.G.; Voorhees, P.M.; Berdeja, J.G.; Richardson, P.G.; Libby, E.N.; Wallace, E.E.; Birrer, N.E.; Burke, J.N.; et al. Ricolinostat plus lenalidomide, and dexamethasone in relapsed or refractory multiple myeloma: A multicentre phase 1b trial. Lancet Oncol. 2016, 17, 1569–1578. [Google Scholar] [CrossRef]
- Amengual, J.E.; Lue, J.K.; Ma, H.; Lichtenstein, R.; Shah, B.; Cremers, S.; Jones, S.; Sawas, A. First-in-Class Selective HDAC6 Inhibitor (ACY-1215) Has a Highly Favorable Safety Profile in Patients with Relapsed and Refractory Lymphoma. Oncologist 2021, 26, e184–e366. [Google Scholar] [CrossRef]
- Wang, F.; Zhong, B.W.; Zhao, Z.R. ACY 1215, a histone deacetylase 6 inhibitor, inhibits cancer cell growth in melanoma. J. Biol. Regul. Homeost. Agents 2018, 32, 851–858. [Google Scholar]
- Lee, D.H.; Won, H.R.; Ryu, H.W.; Han, J.M.; Kwon, S.H. The HDAC6 inhibitor ACY-1215 enhances the anticancer activity of oxaliplatin in colorectal cancer cells. Int. J. Oncol. 2018, 53, 844–854. [Google Scholar] [CrossRef] [Green Version]
- Asthana, J.; Kapoor, S.; Mohan, R.; Panda, D. Inhibition of HDAC6 deacetylase activity increases its binding with microtubules and suppresses microtubule dynamic instability in MCF-7 cells. J. Biol. Chem. 2013, 288, 22516–22526. [Google Scholar] [CrossRef] [Green Version]
- Huang, P.; Almeciga-Pinto, I.; Jarpe, M.; van Duzer, J.H.; Mazitschek, R.; Yang, M.; Jones, S.S.; Quayle, S.N. Selective HDAC inhibition by ACY-241 enhances the activity of paclitaxel in solid tumor models. Oncotarget 2017, 8, 2694–2707. [Google Scholar] [CrossRef] [Green Version]
- Connolly, R.M.; Rudek, M.A.; Piekarz, R. Entinostat: A promising treatment option for patients with advanced breast cancer. Future Oncol. 2017, 13, 1137–1148. [Google Scholar] [CrossRef] [PubMed]
- Lim, B.; Murthy, R.K.; Lee, J.; Jackson, S.A.; Iwase, T.; Davis, D.W.; Willey, J.S.; Wu, J.; Shen, Y.; Tripathy, D.; et al. A phase Ib study of entinostat plus lapatinib with or without trastuzumab in patients with HER2-positive metastatic breast cancer that progressed during trastuzumab treatment. Br. J. Cancer 2019, 120, 1105–1112. [Google Scholar] [CrossRef]
- Yeruva, S.; Zhao, F.; Miller, K.D.; Tevaarwerk, A.J.; Wagner, L.I.; Gray, R.J.; Sparano, J.A.; Connolly, R.M. E2112: Randomized phase iii trial of endocrine therapy plus entinostat/placebo in patients with hormone receptor-positive advanced breast cancer. NPJ Breast Cancer 2018, 4, 1. [Google Scholar] [CrossRef] [Green Version]
- Trapani, D.; Esposito, A.; Criscitiello, C.; Mazzarella, L.; Locatelli, M.; Minchella, I.; Minucci, S.; Curigliano, G. Entinostat for the treatment of breast cancer. Expert Opin. Investig. Drugs 2017, 26, 965–971. [Google Scholar] [CrossRef] [PubMed]
- Ning, Z.Q.; Li, Z.B.; Newman, M.J.; Shan, S.; Wang, X.H.; Pan, D.S.; Zhang, J.; Dong, M.; Du, X.; Lu, X.P. Chidamide (CS055/HBI-8000): A new histone deacetylase inhibitor of the benzamide class with antitumor activity and the ability to enhance immune cell-mediated tumor cell cytotoxicity. Cancer Chemother. Pharmacol. 2012, 69, 901–909. [Google Scholar] [CrossRef]
- Shi, Y.; Jia, B.; Xu, W.; Li, W.; Liu, T.; Liu, P.; Zhao, W.; Zhang, H.; Sun, X.; Yang, H.; et al. Chidamide in relapsed or refractory peripheral T cell lymphoma: A multicenter real-world study in China. J. Hematol. Oncol. 2017, 10, 69. [Google Scholar] [CrossRef] [Green Version]
- Zhao, S.; Guo, J.; Zhao, Y.; Fei, C.; Zheng, Q.; Li, X.; Chang, C. Chidamide, a novel histone deacetylase inhibitor, inhibits the viability of MDS and AML cells by suppressing JAK2/STAT3 signaling. Am. J. Transl. Res. 2016, 8, 3169–3178. [Google Scholar]
- Chang, T.Y.; Nepali, K.; Chen, Y.Y.; Yang, Y.; Hsu, K.C.; Yen, Y.; Pan, S.L.; Liou, J.P.; Lee, S.B. A novel histone deacetylase inhibitor MPT0L184 dysregulates cell-cycle checkpoints and initiates unscheduled mitotic signaling. Biomed. Pharmacother. 2021, 138, 111485. [Google Scholar] [CrossRef] [PubMed]
- Boumber, Y.; Younes, A.; Garcia-Manero, G. Mocetinostat (MGCD0103): A review of an isotype-specific histone deacetylase inhibitor. Expert Opin. Investig. Drugs 2011, 20, 823–829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Tourneau, C.; Siu, L.L. Promising antitumor activity with MGCD0103, a novel isotype-selective histone deacetylase inhibitor. Expert Opin. Investig. Drugs 2008, 17, 1247–1254. [Google Scholar] [CrossRef]
- Choy, E.; Ballman, K.; Chen, J.; Dickson, M.A.; Chugh, R.; George, S.; Okuno, S.; Pollock, R.; Patel, R.M.; Hoering, A.; et al. SARC018_SPORE02: Phase II Study of Mocetinostat Administered with Gemcitabine for Patients with Metastatic Leiomyosarcoma with Progression or Relapse following Prior Treatment with Gemcitabine-Containing Therapy. Sarcoma 2018, 2018, 2068517. [Google Scholar] [CrossRef] [Green Version]
- Batlevi, C.L.; Crump, M.; Andreadis, C.; Rizzieri, D.; Assouline, S.E.; Fox, S.; van der Jagt, R.; Copeland, A.; Potvin, D.; Chao, R.; et al. A phase 2 study of mocetinostat, a histone deacetylase inhibitor, in relapsed or refractory lymphoma. Br. J. Haematol. 2017, 178, 434–441. [Google Scholar] [CrossRef]
- VanderMolen, K.M.; McCulloch, W.; Pearce, C.J.; Oberlies, N.H. Romidepsin (Istodax, NSC 630176, FR901228, FK228, depsipeptide): A natural product recently approved for cutaneous T-cell lymphoma. J. Antibiot. 2011, 64, 525–531. [Google Scholar] [CrossRef]
- Barbarotta, L.; Hurley, K. Romidepsin for the Treatment of Peripheral T-Cell Lymphoma. J. Adv. Pract. Oncol. 2015, 6, 22–36. [Google Scholar]
- Ueda, H.; Nakajima, H.; Hori, Y.; Fujita, T.; Nishimura, M.; Goto, T.; Okuhara, M. FR901228, a novel antitumor bicyclic depsipeptide produced by Chromobacterium violaceum No. 968. I. Taxonomy, fermentation, isolation, physico-chemical and biological properties, and antitumor activity. J. Antibiot. 1994, 47, 301–310. [Google Scholar] [CrossRef] [Green Version]
- Shigematsu, N.; Ueda, H.; Takase, S.; Tanaka, H.; Yamamoto, K.; Tada, T. FR901228, a novel antitumor bicyclic depsipeptide produced by Chromobacterium violaceum No. 968. II. Structure determination. J. Antibiot. 1994, 47, 311–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furumai, R.; Matsuyama, A.; Kobashi, N.; Lee, K.H.; Nishiyama, M.; Nakajima, H.; Tanaka, A.; Komatsu, Y.; Nishino, N.; Yoshida, M.; et al. FK228 (depsipeptide) as a natural prodrug that inhibits class I histone deacetylases. Cancer Res. 2002, 62, 4916–4921. [Google Scholar]
- Rosato, R.R.; Almenara, J.A.; Grant, S. The histone deacetylase inhibitor MS-275 promotes differentiation or apoptosis in human leukemia cells through a process regulated by generation of reactive oxygen species and induction of p21CIP1/WAF1 1. Cancer Res. 2003, 63, 3637–3645. [Google Scholar] [PubMed]
- Seidel, C.; Schnekenburger, M.; Dicato, M.; Diederich, M. Histone deacetylase modulators provided by Mother Nature. Genes Nutr. 2012, 7, 357–367. [Google Scholar] [CrossRef] [Green Version]
- Nakajima, H.; Kim, Y.B.; Terano, H.; Yoshida, M.; Horinouchi, S. FR901228, a potent antitumor antibiotic, is a novel histone deacetylase inhibitor. Exp. Cell Res. 1998, 241, 126–133. [Google Scholar] [CrossRef] [PubMed]
- Wagner, J.M.; Hackanson, B.; Lübbert, M.; Jung, M. Histone deacetylase (HDAC) inhibitors in recent clinical trials for cancer therapy. Clin. Epigenet. 2010, 1, 117–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Issa, J.P. Cancer prevention: Epigenetics steps up to the plate. Cancer Prev. Res. 2008, 1, 219–222. [Google Scholar] [CrossRef] [Green Version]
- Jenke, R.; Reßing, N.; Hansen, F.K.; Aigner, A.; Büch, T. Anticancer Therapy with HDAC Inhibitors: Mechanism-Based Combination Strategies and Future Perspectives. Cancers 2021, 13, 634. [Google Scholar] [CrossRef] [PubMed]
- DiMarco-Crook, C.; Xiao, H. Diet-based strategies for cancer chemoprevention: The role of combination regimens using dietary bioactive components. Annu. Rev. Food Sci. Technol. 2015, 6, 505–526. [Google Scholar] [CrossRef]
- Landis-Piwowar, K.R.; Iyer, N.R. Cancer chemoprevention: Current state of the art. Cancer Growth Metastasis 2014, 7, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Koklesova, L.; Liskova, A.; Samec, M.; Qaradakhi, T.; Zulli, A.; Smejkal, K.; Kajo, K.; Jakubikova, J.; Behzadi, P.; Pec, M.; et al. Genoprotective activities of plant natural substances in cancer and chemopreventive strategies in the context of 3P medicine. EPMA J. 2020, 11, 261–287. [Google Scholar] [CrossRef]
- Steward, W.P.; Brown, K. Cancer chemoprevention: A rapidly evolving field. Br. J. Cancer 2013, 109, 1–7. [Google Scholar] [CrossRef]
- Gan, F.F.; Ling, H.; Ang, X.; Reddy, S.A.; Lee, S.S.; Yang, H.; Tan, S.H.; Hayes, J.D.; Chui, W.K.; Chew, E.H. A novel shogaol analog suppresses cancer cell invasion and inflammation, and displays cytoprotective effects through modulation of NF-κB and Nrf2-Keap1 signaling pathways. Toxicol. Appl. Pharmacol. 2013, 272, 852–862. [Google Scholar] [CrossRef] [PubMed]
- Din, F.V.; Valanciute, A.; Houde, V.P.; Zibrova, D.; Green, K.A.; Sakamoto, K.; Alessi, D.R.; Dunlop, M.G. Aspirin inhibits mTOR signaling, activates AMP-activated protein kinase, and induces autophagy in colorectal cancer cells. Gastroenterology 2012, 142, 1504–1515.e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs over the last 25 years. J. Nat. Prod. 2007, 70, 461–477. [Google Scholar] [CrossRef] [Green Version]
- Newman, D.J.; Cragg, G.M. Natural Products as Sources of New Drugs over the Nearly Four Decades from 01/1981 to 09/2019. J. Nat. Prod. 2020, 83, 770–803. [Google Scholar] [CrossRef]
- Carlson, E.E. Natural products as chemical probes. ACS Chem. Biol. 2010, 5, 639–653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atanasov, A.G.; Zotchev, S.B.; Dirsch, V.M.; International Natural Product Sciences Taskforce; Supuran, C.T. Natural products in drug discovery: Advances and opportunities. Nat. Rev. Drug Discov. 2021, 20, 200–216. [Google Scholar] [CrossRef] [PubMed]
- Schnekenburger, M.; Dicato, M.; Diederich, M. Plant-derived epigenetic modulators for cancer treatment and prevention. Biotechnol. Adv. 2014, 32, 1123–1132. [Google Scholar] [CrossRef]
- Mijatović, S.; Bramanti, A.; Nicoletti, F.; Fagone, P.; Kaluđerović, G.N.; Maksimović-Ivanić, D. Naturally occurring compounds in differentiation based therapy of cancer. Biotechnol. Adv. 2018, 36, 1622–1632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tabolacci, C.; Forni, C.; Jadeja, R.N.; Facchiano, F. Natural Compounds against Cancer, Inflammation, and Oxidative Stress. BioMed Res. Int. 2019, 2019, 9495628. [Google Scholar] [CrossRef]
- Damiani, E.; Duran, M.N.; Mohan, N.; Rajendran, P.; Dashwood, R.H. Targeting Epigenetic ‘Readers’ with Natural Compounds for Cancer Interception. J. Cancer Prev. 2020, 25, 189–203. [Google Scholar] [CrossRef]
- Kim, E.; Bisson, W.H.; Löhr, C.V.; Williams, D.E.; Ho, E.; Dashwood, R.H.; Rajendran, P. Histone and Non-Histone Targets of Dietary Deacetylase Inhibitors. Curr. Top. Med. Chem. 2016, 16, 714–731. [Google Scholar] [CrossRef]
- Mazzone, R.; Zwergel, C.; Artico, M.; Taurone, S.; Ralli, M.; Greco, A.; Mai, A. The emerging role of epigenetics in human autoimmune disorders. Clin. Epigenet. 2019, 11, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kraan, C.M.; Godler, D.E.; Amor, D.J. Epigenetics of fragile X syndrome and fragile X-related disorders. Dev. Med. Child Neurol. 2019, 61, 121–127. [Google Scholar] [CrossRef] [Green Version]
- Riggs, M.G.; Whittaker, R.G.; Neumann, J.R.; Ingram, V.M. n-Butyrate causes histone modification in HeLa and Friend erythroleukaemia cells. Nature 1977, 268, 462–464. [Google Scholar] [CrossRef] [PubMed]
- Hassig, C.A.; Tong, J.K.; Schreiber, S.L. Fiber-derived butyrate and the prevention of colon cancer. Chem. Biol. 1997, 4, 783–789. [Google Scholar] [CrossRef] [Green Version]
- Lea, M.A.; Randolph, V.M.; Lee, J.E.; desBordes, C. Induction of histone acetylation in mouse erythroleukemia cells by some organosulfur compounds including allyl isothiocyanate. Int. J. Cancer 2001, 92, 784–789. [Google Scholar] [CrossRef] [PubMed]
- Nian, H.; Delage, B.; Pinto, J.T.; Dashwood, R.H. Allyl mercaptan, a garlic-derived organosulfur compound, inhibits histone deacetylase and enhances Sp3 binding on the P21WAF1 promoter. Carcinogenesis 2008, 29, 1816–1824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wisnieski, F.; Calcagno, D.Q.; Leal, M.F.; Santos, L.C.; Gigek, C.O.; Chen, E.S.; Demachki, S.; Artigiani, R.; Assumpção, P.P.; Lourenço, L.G.; et al. CDKN1A histone acetylation and gene expression relationship in gastric adenocarcinomas. Clin. Exp. Med. 2017, 17, 121–129. [Google Scholar] [CrossRef]
- Ocker, M.; Bitar, S.A.; Monteiro, A.C.; Gali-Muhtasib, H.; Schneider-Stock, R. Epigenetic Regulation of p21cip1/waf1 in Human Cancer. Cancers 2019, 11, 1343. [Google Scholar] [CrossRef] [Green Version]
- Druesne, N.; Pagniez, A.; Mayeur, C.; Thomas, M.; Cherbuy, C.; Duée, P.H.; Martel, P.; Chaumontet, C. Diallyl disulfide (DADS) increases histone acetylation and p21(waf1/cip1) expression in human colon tumor cell lines. Carcinogenesis 2004, 25, 1227–1236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stan, S.D.; Singh, S.V.; Whitcomb, D.C.; Brand, R.E. Phenethyl isothiocyanate inhibits proliferation and induces apoptosis in pancreatic cancer cells in vitro and in a MIAPaca2 xenograft animal model. Nutr. Cancer 2014, 66, 747–755. [Google Scholar] [CrossRef] [Green Version]
- Mitsiogianni, M.; Mantso, T.; Trafalis, D.T.; Vasantha Rupasinghe, H.P.; Zoumpourlis, V.; Franco, R.; Botaitis, S.; Pappa, A.; Panayiotidis, M.I. Allyl isothiocyanate regulates lysine acetylation and methylation marks in an experimental model of malignant melanoma. Eur. J. Nutr. 2020, 59, 557–569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Batra, S.; Sahu, R.P.; Kandala, P.K.; Srivastava, S.K. Benzyl isothiocyanate-mediated inhibition of histone deacetylase leads to NF-kappaB turnoff in human pancreatic carcinoma cells. Mol. Cancer Ther. 2010, 9, 1596–1608. [Google Scholar] [CrossRef] [Green Version]
- Myzak, M.C.; Karplus, P.A.; Chung, F.L.; Dashwood, R.H. A novel mechanism of chemoprotection by sulforaphane: Inhibition of histone deacetylase. Cancer Res. 2004, 64, 5767–5774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Myzak, M.C.; Dashwood, W.M.; Orner, G.A.; Ho, E.; Dashwood, R.H. Sulforaphane inhibits histone deacetylase in vivo and suppresses tumorigenesis in Apc-minus mice. FASEB J. 2006, 20, 506–508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.; Su, Z.Y.; Khor, T.O.; Shu, L.; Kong, A.N. Sulforaphane enhances Nrf2 expression in prostate cancer TRAMP C1 cells through epigenetic regulation. Biochem. Pharmacol. 2013, 85, 1398–1404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.G.; Liu, X.M.; Fang, Y.; Dai, W.; Chiao, F.B.; Puccio, G.M.; Feng, J.; Liu, D.; Chiao, J.W. De-repression of the p21 promoter in prostate cancer cells by an isothiocyanate via inhibition of HDACs and c-Myc. Int. J. Oncol. 2008, 33, 375–380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cang, S.; Ma, Y.; Chiao, J.W.; Liu, D. Phenethyl isothiocyanate and paclitaxel synergistically enhanced apoptosis and alpha-tubulin hyperacetylation in breast cancer cells. Exp. Hematol. Oncol. 2014, 3, 5. [Google Scholar] [CrossRef] [Green Version]
- Chou, Y.C.; Chang, M.Y.; Lee, H.T.; Shen, C.C.; Harnod, T.; Liang, Y.J.; Wu, R.S.; Lai, K.C.; Hsu, F.T.; Chung, J.G. Phenethyl Isothiocyanate Inhibits In Vivo Growth of Xenograft Tumors of Human Glioblastoma Cells. Molecules 2018, 23, 2305. [Google Scholar] [CrossRef] [Green Version]
- Park, J.E.; Sun, Y.; Lim, S.K.; Tam, J.P.; Dekker, M.; Chen, H.; Sze, S.K. Dietary phytochemical PEITC restricts tumor development via modulation of epigenetic writers and erasers. Sci. Rep. 2017, 7, 40569. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, A.; Sakr, W.A.; Rahman, K.M. Anticancer properties of indole compounds: Mechanism of apoptosis induction and role in chemotherapy. Curr. Drug Targets 2010, 11, 652–666. [Google Scholar] [CrossRef]
- Bhatnagar, N.; Li, X.; Chen, Y.; Zhou, X.; Garrett, S.H.; Guo, B. 3,3’-diindolylmethane enhances the efficacy of butyrate in colon cancer prevention through down-regulation of survivin. Cancer Prev. Res. 2009, 2, 581–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Li, X.; Guo, B. Chemopreventive agent 3,3’-diindolylmethane selectively induces proteasomal degradation of class I histone deacetylases. Cancer Res. 2010, 70, 646–654. [Google Scholar] [CrossRef] [Green Version]
- Liskova, A.; Samec, M.; Koklesova, L.; Brockmueller, A.; Zhai, K.; Abdellatif, B.; Siddiqui, M.; Biringer, K.; Kudela, E.; Pec, M.; et al. Flavonoids as an effective sensitizer for anti-cancer therapy: Insights into multi-faceted mechanisms and applicability towards individualized patient profiles. EPMA J. 2021, 12, 155–176. [Google Scholar] [CrossRef]
- Abotaleb, M.; Samuel, S.M.; Varghese, E.; Varghese, S.; Kubatka, P.; Liskova, A.; Büsselberg, D. Flavonoids in Cancer and Apoptosis. Cancers 2018, 11, 28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ijaz, S.; Akhtar, N.; Khan, M.S.; Hameed, A.; Irfan, M.; Arshad, M.A.; Ali, S.; Asrar, M. Plant derived anticancer agents: A green approach towards skin cancers. Biomed. Pharmacother. 2018, 103, 1643–1651. [Google Scholar] [CrossRef]
- Biswas, S.; Reddy, N.D.; Jayashree, B.S.; Rao, C.M. Evaluation of Novel 3-Hydroxyflavone Analogues as HDAC Inhibitors against Colorectal Cancer. Adv. Pharmacol. Sci. 2018, 2018, 4751806. [Google Scholar] [CrossRef] [PubMed]
- Priyadarsini, R.V.; Vinothini, G.; Murugan, R.S.; Manikandan, P.; Nagini, S. The flavonoid quercetin modulates the hallmark capabilities of hamster buccal pouch tumors. Nutr. Cancer 2011, 63, 218–226. [Google Scholar] [CrossRef] [PubMed]
- Salehi, B.; Venditti, A.; Sharifi-Rad, M.; Kręgiel, D.; Sharifi-Rad, J.; Durazzo, A.; Lucarini, M.; Santini, A.; Souto, E.B.; Novellino, E.; et al. The Therapeutic Potential of Apigenin. Int. J. Mol. Sci. 2019, 20, 1305. [Google Scholar] [CrossRef] [Green Version]
- Shukla, S.; Gupta, S. Apigenin-induced cell cycle arrest is mediated by modulation of MAPK, PI3K-Akt, and loss of cyclin D1 associated retinoblastoma dephosphorylation in human prostate cancer cells. Cell Cycle 2007, 6, 1102–1114. [Google Scholar] [CrossRef] [PubMed]
- Pandey, M.; Kaur, P.; Shukla, S.; Abbas, A.; Fu, P.; Gupta, S. Plant flavone apigenin inhibits HDAC and remodels chromatin to induce growth arrest and apoptosis in human prostate cancer cells: In Vitro and In Vivo study. Mol. Carcinog. 2012, 51, 952–962. [Google Scholar] [CrossRef] [Green Version]
- Ganai, S.A.; Sheikh, F.A.; Baba, Z.A. Plant flavone Chrysin as an emerging histone deacetylase inhibitor for prosperous epigenetic-based anticancer therapy. Phytother. Res. 2021, 35, 823–834. [Google Scholar] [CrossRef]
- Middleton, E., Jr.; Kandaswami, C.; Theoharides, T.C. The effects of plant flavonoids on mammalian cells: Implications for inflammation, heart disease, and cancer. Pharmacol. Rev. 2000, 52, 673–751. [Google Scholar]
- Pal-Bhadra, M.; Ramaiah, M.J.; Reddy, T.L.; Krishnan, A.; Pushpavalli, S.N.; Babu, K.S.; Tiwari, A.K.; Rao, J.M.; Yadav, J.S.; Bhadra, U. Plant HDAC inhibitor chrysin arrest cell growth and induce p21WAF1 by altering chromatin of STAT response element in A375 cells. BMC Cancer 2012, 12, 180. [Google Scholar] [CrossRef] [Green Version]
- Jin, T.R. Curcumin and dietary polyphenol research: Beyond drug discovery. Acta Pharmacol. Sin. 2018, 39, 779–786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schnekenburger, M.; Dicato, M.; Diederich, M.F. Anticancer potential of naturally occurring immunoepigenetic modulators: A promising avenue? Cancer 2019, 125, 1612–1628. [Google Scholar] [CrossRef] [PubMed]
- Teiten, M.H.; Dicato, M.; Diederich, M. Curcumin as a regulator of epigenetic events. Mol. Nutr. Food Res. 2013, 57, 1619–1629. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.; Krauthauser, C.; Maduskuie, V.; Fawcett, P.T.; Olson, J.M.; Rajasekaran, S.A. Curcumin-induced HDAC inhibition and attenuation of medulloblastoma growth in vitro and in vivo. BMC Cancer 2011, 11, 144. [Google Scholar] [CrossRef] [Green Version]
- Sarkar, R.; Mukherjee, A.; Mukherjee, S.; Biswas, R.; Biswas, J.; Roy, M. Curcumin augments the efficacy of antitumor drugs used in leukemia by modulation of heat shock proteins via HDAC6. J. Environ. Pathol. Toxicol. Oncol. 2014, 33, 247–263. [Google Scholar] [CrossRef]
- Chen, J.; Wang, G.; Wang, L.; Kang, J.; Wang, J. Curcumin p38-dependently enhances the anticancer activity of valproic acid in human leukemia cells. Eur. J. Pharm. Sci. 2010, 41, 210–218. [Google Scholar] [CrossRef]
- Lecumberri, E.; Dupertuis, Y.M.; Miralbell, R.; Pichard, C. Green tea polyphenol epigallocatechin-3-gallate (EGCG) as adjuvant in cancer therapy. Clin. Nutr. 2013, 32, 894–903. [Google Scholar] [CrossRef] [Green Version]
- Singh, B.N.; Shankar, S.; Srivastava, R.K. Green tea catechin, epigallocatechin-3-gallate (EGCG): Mechanisms, perspectives and clinical applications. Biochem. Pharmacol. 2011, 82, 1807–1821. [Google Scholar] [CrossRef] [Green Version]
- Pandey, M.; Shukla, S.; Gupta, S. Promoter demethylation and chromatin remodeling by green tea polyphenols leads to re-expression of GSTP1 in human prostate cancer cells. Int. J. Cancer 2010, 126, 2520–2533. [Google Scholar] [CrossRef] [Green Version]
- Nandakumar, V.; Vaid, M.; Katiyar, S.K. (-)-Epigallocatechin-3-gallate reactivates silenced tumor suppressor genes, Cip1/p21 and p16INK4a, by reducing DNA methylation and increasing histones acetylation in human skin cancer cells. Carcinogenesis 2011, 32, 537–544. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Yuan, Y.Y.; Meeran, S.M.; Tollefsbol, T.O. Synergistic epigenetic reactivation of estrogen receptor-α (ERα) by combined green tea polyphenol and histone deacetylase inhibitor in ERα-negative breast cancer cells. Mol. Cancer 2010, 9, 274. [Google Scholar] [CrossRef] [Green Version]
- Vanden Berghe, W. Epigenetic impact of dietary polyphenols in cancer chemoprevention: Lifelong remodeling of our epigenomes. Pharmacol. Res. 2012, 65, 565–576. [Google Scholar] [CrossRef] [PubMed]
- Varoni, E.M.; Lo Faro, A.F.; Sharifi-Rad, J.; Iriti, M. Anticancer Molecular Mechanisms of Resveratrol. Front. Nutr. 2016, 3, 8. [Google Scholar] [CrossRef] [Green Version]
- Venturelli, S.; Berger, A.; Böcker, A.; Busch, C.; Weiland, T.; Noor, S.; Leischner, C.; Schleicher, S.; Mayer, M.; Weiss, T.S.; et al. Resveratrol as a pan-HDAC inhibitor alters the acetylation status of histone proteins in human-derived hepatoblastoma cells. PLoS ONE 2013, 8, e73097. [Google Scholar] [CrossRef]
- Farhan, M.; Ullah, M.F.; Faisal, M.; Farooqi, A.A.; Sabitaliyevich, U.Y.; Biersack, B.; Ahmad, A. Differential Methylation and Acetylation as the Epigenetic Basis of Resveratrol’s Anticancer Activity. Medicines 2019, 6, 24. [Google Scholar] [CrossRef] [Green Version]
- Spagnuolo, C.; Russo, G.L.; Orhan, I.E.; Habtemariam, S.; Daglia, M.; Sureda, A.; Nabavi, S.F.; Devi, K.P.; Loizzo, M.R.; Tundis, R.; et al. Genistein and cancer: Current status, challenges, and future directions. Adv. Nutr. 2015, 6, 408–419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tuli, H.S.; Tuorkey, M.J.; Thakral, F.; Sak, K.; Kumar, M.; Sharma, A.K.; Sharma, U.; Jain, A.; Aggarwal, V.; Bishayee, A. Molecular Mechanisms of Action of Genistein in Cancer: Recent Advances. Front. Pharmacol. 2019, 10, 1336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Chen, H.; Hardy, T.M.; Tollefsbol, T.O. Epigenetic regulation of multiple tumor-related genes leads to suppression of breast tumorigenesis by dietary genistein. PLoS ONE 2013, 8, e54369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Meeran, S.M.; Patel, S.N.; Chen, H.; Hardy, T.M.; Tollefsbol, T.O. Epigenetic reactivation of estrogen receptor-α (ERα) by genistein enhances hormonal therapy sensitivity in ERα-negative breast cancer. Mol. Cancer 2013, 12, 9. [Google Scholar] [CrossRef] [Green Version]
- Groh, I.A.; Chen, C.; Lüske, C.; Cartus, A.T.; Esselen, M. Plant polyphenols and oxidative metabolites of the herbal alkenylbenzene methyleugenol suppress histone deacetylase activity in human colon carcinoma cells. J. Nutr. Metab. 2013, 2013, 821082. [Google Scholar] [CrossRef]
- Phillip, C.J.; Giardina, C.K.; Bilir, B.; Cutler, D.J.; Lai, Y.H.; Kucuk, O.; Moreno, C.S. Genistein cooperates with the histone deacetylase inhibitor vorinostat to induce cell death in prostate cancer cells. BMC Cancer 2012, 12, 145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sundaram, M.K.; Ansari, M.Z.; Al Mutery, A.; Ashraf, M.; Nasab, R.; Rai, S.; Rais, N.; Hussain, A. Genistein Induces Alterations of Epigenetic Modulatory Signatures in Human Cervical Cancer Cells. Anti-Cancer Agents Med. Chem. 2018, 18, 412–421. [Google Scholar] [CrossRef] [PubMed]
- Sundaram, M.K.; Unni, S.; Somvanshi, P.; Bhardwaj, T.; Mandal, R.K.; Hussain, A.; Haque, S. Genistein Modulates Signaling Pathways and Targets Several Epigenetic Markers in HeLa Cells. Genes 2019, 10, 955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mottamal, M.; Zheng, S.; Huang, T.L.; Wang, G. Histone deacetylase inhibitors in clinical studies as templates for new anticancer agents. Molecules 2015, 20, 3898–3941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, P.; Wang, Z.; Liu, J. Role of HDACs in normal and malignant hematopoiesis. Mol. Cancer 2020, 19, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rana, Z.; Diermeier, S.; Hanif, M.; Rosengren, R.J. Understanding Failure and Improving Treatment Using HDAC Inhibitors for Prostate Cancer. Biomedicines 2020, 8, 22. [Google Scholar] [CrossRef] [Green Version]
- Shah, R.R. Safety and Tolerability of Histone Deacetylase (HDAC) Inhibitors in Oncology. Drug Saf. 2019, 42, 235–245. [Google Scholar] [CrossRef]
- Melesina, J.; Simoben, C.V.; Praetorius, L.; Bülbül, E.F.; Robaa, D.; Sippl, W. Strategies To Design Selective Histone Deacetylase Inhibitors. ChemMedChem 2021, 16, 1336–1359. [Google Scholar] [CrossRef]
- Micelli, C.; Rastelli, G. Histone deacetylases: Structural determinants of inhibitor selectivity. Drug Discov. Today 2015, 20, 718–735. [Google Scholar] [CrossRef]
- Simoben, C.V.; Robaa, D.; Chakrabarti, A.; Schmidtkunz, K.; Marek, M.; Lancelot, J.; Kannan, S.; Melesina, J.; Shaik, T.B.; Pierce, R.J.; et al. A Novel Class of Schistosoma mansoni Histone Deacetylase 8 (HDAC8) Inhibitors Identified by Structure-Based Virtual Screening and In Vitro Testing. Molecules 2018, 23, 566. [Google Scholar] [CrossRef] [Green Version]
- Stoddard, V.S.; Dodson, K.; Adams, K.; Watkins, D.L. In silico Design of Novel Histone Deacetylase 4 Inhibitors: Design Guidelines for Improved Binding Affinity. Int. J. Mol. Sci. 2019, 21, 219. [Google Scholar] [CrossRef] [Green Version]
- Mwakwari, S.C.; Patil, V.; Guerrant, W.; Oyelere, A.K. Macrocyclic histone deacetylase inhibitors. Curr. Top. Med. Chem. 2010, 10, 1423–1440. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhang, J.; Jiang, Q.; Zhang, L.; Song, W. Zinc binding groups for histone deacetylase inhibitors. J. Enzym. Inhib. Med. Chem. 2018, 33, 714–721. [Google Scholar] [CrossRef]
- Wagner, F.F.; Weїwer, M.; Lewis, M.C.; Holson, E.B. Small molecule inhibitors of zinc-dependent histone deacetylases. Neurotherapeutics 2013, 10, 589–604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lauffer, B.E.; Mintzer, R.; Fong, R.; Mukund, S.; Tam, C.; Zilberleyb, I.; Flicke, B.; Ritscher, A.; Fedorowicz, G.; Vallero, R.; et al. Histone deacetylase (HDAC) inhibitor kinetic rate constants correlate with cellular histone acetylation but not transcription and cell viability. J. Biol. Chem. 2013, 288, 26926–26943. [Google Scholar] [CrossRef] [Green Version]
- Madsen, A.S.; Kristensen, H.M.; Lanz, G.; Olsen, C.A. The effect of various zinc binding groups on inhibition of histone deacetylases 1-11. ChemMedChem 2014, 9, 614–626. [Google Scholar] [CrossRef] [PubMed]
- Lobera, M.; Madauss, K.P.; Pohlhaus, D.T.; Wright, Q.G.; Trocha, M.; Schmidt, D.R.; Baloglu, E.; Trump, R.P.; Head, M.S.; Hofmann, G.A.; et al. Selective class IIa histone deacetylase inhibition via a nonchelating zinc-binding group. Nat. Chem. Biol. 2013, 9, 319–325. [Google Scholar] [CrossRef]
- Wagner, F.F.; Olson, D.E.; Gale, J.P.; Kaya, T.; Weïwer, M.; Aidoud, N.; Thomas, M.; Davoine, E.L.; Lemercier, B.C.; Zhang, Y.L.; et al. Potent and selective inhibition of histone deacetylase 6 (HDAC6) does not require a surface-binding motif. J. Med. Chem. 2013, 56, 1772–1776. [Google Scholar] [CrossRef]
- Haggarty, S.J.; Koeller, K.M.; Wong, J.C.; Grozinger, C.M.; Schreiber, S.L. Domain-selective small-molecule inhibitor of histone deacetylase 6 (HDAC6)-mediated tubulin deacetylation. Proc. Natl. Acad. Sci. USA 2003, 100, 4389–4394. [Google Scholar] [CrossRef] [Green Version]
- Yu, W.; Liu, J.; Yu, Y.; Zhang, V.; Clausen, D.; Kelly, J.; Wolkenberg, S.; Beshore, D.; Duffy, J.L.; Chung, C.C.; et al. Discovery of ethyl ketone-based HDACs 1, 2, and 3 selective inhibitors for HIV latency reactivation. Bioorg. Med. Chem. Lett. 2020, 30, 127197. [Google Scholar] [CrossRef]
- Liu, J.; Kelly, J.; Yu, W.; Clausen, D.; Yu, Y.; Kim, H.; Duffy, J.L.; Chung, C.C.; Myers, R.W.; Carroll, S.; et al. Selective Class I HDAC Inhibitors Based on Aryl Ketone Zinc Binding Induce HIV-1 Protein for Clearance. ACS Med. Chem. Lett. 2020, 11, 1476–1483. [Google Scholar] [CrossRef] [PubMed]
- Maolanon, A.R.; Kristensen, H.M.; Leman, L.J.; Ghadiri, M.R.; Olsen, C.A. Natural and Synthetic Macrocyclic Inhibitors of the Histone Deacetylase Enzymes. ChemBioChem 2017, 18, 5–49. [Google Scholar] [CrossRef] [PubMed]
- Krennhrubec, K.; Marshall, B.L.; Hedglin, M.; Verdin, E.; Ulrich, S.M. Design and evaluation of ‘Linkerless’ hydroxamic acids as selective HDAC8 inhibitors. Bioorg. Med. Chem. Lett. 2007, 17, 2874–2878. [Google Scholar] [CrossRef]
- Balasubramanian, S.; Ramos, J.; Luo, W.; Sirisawad, M.; Verner, E.; Buggy, J.J. A novel histone deacetylase 8 (HDAC8)-specific inhibitor PCI-34051 induces apoptosis in T-cell lymphomas. Leukemia 2008, 22, 1026–1034. [Google Scholar] [CrossRef] [PubMed]
- Hontecillas-Prieto, L.; Flores-Campos, R.; Silver, A.; de Álava, E.; Hajji, N.; García-Domínguez, D.J. Synergistic Enhancement of Cancer Therapy Using HDAC Inhibitors: Opportunity for Clinical Trials. Front. Genet. 2020, 11, 578011. [Google Scholar] [CrossRef] [PubMed]
- Thurn, K.T.; Thomas, S.; Moore, A.; Munster, P.N. Rational therapeutic combinations with histone deacetylase inhibitors for the treatment of cancer. Future Oncol. 2011, 7, 263–283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Attwood, M.M.; Fabbro, D.; Sokolov, A.V.; Knapp, S.; Schiöth, H.B. Trends in kinase drug discovery: Targets, indications and inhibitor design. Nat. Rev. Drug Discov. 2021, 20, 839–861. [Google Scholar] [CrossRef]
- Musso, L.; Dallavalle, S.; Zunino, F. Perspectives in the development of hybrid bifunctional antitumour agents. Biochem. Pharmacol. 2015, 96, 297–305. [Google Scholar] [CrossRef] [PubMed]
- Pisa, R.; Kapoor, T.M. Chemical strategies to overcome resistance against targeted anticancer therapeutics. Nat. Chem. Biol. 2020, 16, 817–825. [Google Scholar] [CrossRef]
- Anighoro, A.; Bajorath, J.; Rastelli, G. Polypharmacology: Challenges and opportunities in drug discovery. J. Med. Chem. 2014, 57, 7874–7887. [Google Scholar] [CrossRef]
- Proschak, E.; Stark, H.; Merk, D. Polypharmacology by Design: A Medicinal Chemist’s Perspective on Multitargeting Compounds. J. Med. Chem. 2019, 62, 420–444. [Google Scholar] [CrossRef] [PubMed]
- Lai, C.J.; Bao, R.; Tao, X.; Wang, J.; Atoyan, R.; Qu, H.; Wang, D.G.; Yin, L.; Samson, M.; Forrester, J.; et al. CUDC-101, a multitargeted inhibitor of histone deacetylase, epidermal growth factor receptor, and human epidermal growth factor receptor 2, exerts potent anticancer activity. Cancer Res. 2010, 70, 3647–3656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Zhang, Y.; Mehta, A.; Boufraqech, M.; Davis, S.; Wang, J.; Tian, Z.; Yu, Z.; Boxer, M.B.; Kiefer, J.A.; et al. Dual inhibition of HDAC and EGFR signaling with CUDC-101 induces potent suppression of tumor growth and metastasis in anaplastic thyroid cancer. Oncotarget 2015, 6, 9073–9085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moertl, S.; Payer, S.; Kell, R.; Winkler, K.; Anastasov, N.; Atkinson, M.J. Comparison of Radiosensitization by HDAC Inhibitors CUDC-101 and SAHA in Pancreatic Cancer Cells. Int. J. Mol. Sci. 2019, 20, 3259. [Google Scholar] [CrossRef] [Green Version]
- Liffers, K.; Kolbe, K.; Westphal, M.; Lamszus, K.; Schulte, A. Histone Deacetylase Inhibitors Resensitize EGFR/EGFRvIII-Overexpressing, Erlotinib-Resistant Glioblastoma Cells to Tyrosine Kinase Inhibition. Target. Oncol. 2016, 11, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Kong, Y.; Zhang, J.; Su, M.; Zhou, Y.; Zang, Y.; Li, J.; Chen, Y.; Fang, Y.; Zhang, X.; et al. Design, synthesis and biological evaluation of colchicine derivatives as novel tubulin and histone deacetylase dual inhibitors. Eur. J. Med. Chem. 2015, 95, 127–135. [Google Scholar] [CrossRef] [PubMed]
- Yang, E.G.; Mustafa, N.; Tan, E.C.; Poulsen, A.; Ramanujulu, P.M.; Chng, W.J.; Yen, J.J.; Dymock, B.W. Design and Synthesis of Janus Kinase 2 (JAK2) and Histone Deacetlyase (HDAC) Bispecific Inhibitors Based on Pacritinib and Evidence of Dual Pathway Inhibition in Hematological Cell Lines. J. Med. Chem. 2016, 59, 8233–8262. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.G.; Wynder, C.; Bochar, D.A.; Hakimi, M.A.; Cooch, N.; Shiekhattar, R. Functional interplay between histone demethylase and deacetylase enzymes. Mol. Cell. Biol. 2006, 26, 6395–6402. [Google Scholar] [CrossRef] [Green Version]
- Duan, Y.C.; Ma, Y.C.; Qin, W.P.; Ding, L.N.; Zheng, Y.C.; Zhu, Y.L.; Zhai, X.Y.; Yang, J.; Ma, C.Y.; Guan, Y.Y. Design and synthesis of tranylcypromine derivatives as novel LSD1/HDACs dual inhibitors for cancer treatment. Eur. J. Med. Chem. 2017, 140, 392–402. [Google Scholar] [CrossRef]
- He, M.; Lv, W.; Rao, Y. Opportunities and Challenges of Small Molecule Induced Targeted Protein Degradation. Front. Cell Dev. Biol. 2021, 9, 685106. [Google Scholar] [CrossRef]
- Utsugi, Y.; Miyamae, Y. Strategies for Post-Translational Control of Protein Expression and Their Applications. Appl. Sci. 2021, 11, 8300. [Google Scholar] [CrossRef]
- Burslem, G.M.; Crews, C.M. Proteolysis-Targeting Chimeras as Therapeutics and Tools for Biological Discovery. Cell 2020, 181, 102–114. [Google Scholar] [CrossRef] [PubMed]
- Amm, I.; Sommer, T.; Wolf, D.H. Protein quality control and elimination of protein waste: The role of the ubiquitin-proteasome system. Biochim. Biophys. Acta 2014, 1843, 182–196. [Google Scholar] [CrossRef] [Green Version]
- Burslem, G.M.; Smith, B.E.; Lai, A.C.; Jaime-Figueroa, S.; McQuaid, D.C.; Bondeson, D.P.; Toure, M.; Dong, H.; Qian, Y.; Wang, J.; et al. The Advantages of Targeted Protein Degradation Over Inhibition: An RTK Case Study. Cell Chem. Biol. 2018, 25, 67–77.e3. [Google Scholar] [CrossRef] [PubMed]
- Nalawansha, D.A.; Crews, C.M. PROTACs: An Emerging Therapeutic Modality in Precision Medicine. Cell Chem. Biol. 2020, 27, 998–1014. [Google Scholar] [CrossRef]
- Fischer, F.; Alves Avelar, L.A.; Murray, L.; Kurz, T. Designing HDAC-PROTACs: Lessons learned so far. Future Med. Chem. 2021. [Google Scholar] [CrossRef]
- Yang, K.; Song, Y.; Xie, H.; Wu, H.; Wu, Y.T.; Leisten, E.D.; Tang, W. Development of the first small molecule histone deacetylase 6 (HDAC6) degraders. Bioorg. Med. Chem. Lett. 2018, 28, 2493–2497. [Google Scholar] [CrossRef]
- An, Z.; Lv, W.; Su, S.; Wu, W.; Rao, Y. Developing potent PROTACs tools for selective degradation of HDAC6 protein. Protein Cell 2019, 10, 606–609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.; Lv, W.; He, M.; Deng, H.; Li, H.; Wu, W.; Rao, Y. Plasticity in designing PROTACs for selective and potent degradation of HDAC6. Chem. Commun. 2019, 55, 14848–14851. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Wu, H.; Zhang, Z.; Leisten, E.D.; Nie, X.; Liu, B.; Wen, Z.; Zhang, J.; Cunningham, M.D.; Tang, W. Development of Selective Histone Deacetylase 6 (HDAC6) Degraders Recruiting Von Hippel-Lindau (VHL) E3 Ubiquitin Ligase. ACS Med. Chem. Lett. 2020, 11, 575–581. [Google Scholar] [CrossRef]
- Xiao, Y.; Wang, J.; Zhao, L.Y.; Chen, X.; Zheng, G.; Zhang, X.; Liao, D. Discovery of histone deacetylase 3 (HDAC3)-specific PROTACs. Chem. Commun. 2020, 56, 9866–9869. [Google Scholar] [CrossRef]
- Smalley, J.P.; Adams, G.E.; Millard, C.J.; Song, Y.; Norris, J.; Schwabe, J.; Cowley, S.M.; Hodgkinson, J.T. PROTAC-mediated degradation of class I histone deacetylase enzymes in corepressor complexes. Chem. Commun. 2020, 56, 4476–4479. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Zhao, W.; Zhao, C.; Liu, Q.; Li, S.; Zhang, G.; Chou, C.J.; Zhang, Y. Development of a Bestatin-SAHA Hybrid with Dual Inhibitory Activity against APN and HDAC. Molecules 2020, 25, 4991. [Google Scholar] [CrossRef]
- Bertrand, M.J.; Milutinovic, S.; Dickson, K.M.; Ho, W.C.; Boudreault, A.; Durkin, J.; Gillard, J.W.; Jaquith, J.B.; Morris, S.J.; Barker, P.A. cIAP1 and cIAP2 facilitate cancer cell survival by functioning as E3 ligases that promote RIP1 ubiquitination. Mol. Cell 2008, 30, 689–700. [Google Scholar] [CrossRef]
- Lee, J.H.; Choy, M.L.; Marks, P.A. Mechanisms of resistance to histone deacetylase inhibitors. Adv. Cancer Res. 2012, 116, 39–86. [Google Scholar] [CrossRef] [PubMed]
- Halsall, J.A.; Turner, B.M. Histone deacetylase inhibitors for cancer therapy: An evolutionarily ancient resistance response may explain their limited success. BioEssays 2016, 38, 1102–1110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suraweera, A.; O’Byrne, K.J.; Richard, D.J. Combination Therapy With Histone Deacetylase Inhibitors (HDACi) for the Treatment of Cancer: Achieving the Full Therapeutic Potential of HDACi. Front. Oncol. 2018, 8, 92. [Google Scholar] [CrossRef] [Green Version]
- Huang, L.; Jiang, S.; Shi, Y. Tyrosine kinase inhibitors for solid tumors in the past 20 years (2001–2020). J. Hematol. Oncol. 2020, 13, 143. [Google Scholar] [CrossRef]
- Kavarthapu, R.; Anbazhagan, R.; Dufau, M.L. Crosstalk between PRLR and EGFR/HER2 Signaling Pathways in Breast Cancer. Cancers 2021, 13, 4685. [Google Scholar] [CrossRef]
- Jin, J.S.; Tsao, T.Y.; Sun, P.C.; Yu, C.P.; Tzao, C. SAHA inhibits the growth of colon tumors by decreasing histone deacetylase and the expression of cyclin D1 and survivin. Pathol. Oncol. Res. 2021, 18, 713–720. [Google Scholar] [CrossRef]
- Han, J.Y.; Lee, S.H.; Lee, G.K.; Yun, T.; Lee, Y.J.; Hwang, K.H.; Kim, J.Y.; Kim, H.T. Phase I/II study of gefitinib (Iressa(®)) and vorinostat (IVORI) in previously treated patients with advanced non-small cell lung cancer. Cancer Chemother. Pharmacol. 2015, 75, 475–483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Apte, R.S.; Chen, D.S.; Ferrara, N. VEGF in Signaling and Disease: Beyond Discovery and Development. Cell 2019, 176, 1248–1264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pili, R.; Liu, G.; Chintala, S.; Verheul, H.; Rehman, S.; Attwood, K.; Lodge, M.A.; Wahl, R.; Martin, J.I.; Miles, K.M.; et al. Combination of the histone deacetylase inhibitor vorinostat with bevacizumab in patients with clear-cell renal cell carcinoma: A multicentre, single-arm phase I/II clinical trial. Br. J. Cancer 2017, 116, 874–883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, W.J.; Zhao, G.; Zhang, C.Y.; Yang, C.Q.; Zeng, X.B.; Li, J.; Zhu, K.; Zhao, S.Q.; Lu, H.M.; Yin, D.C.; et al. Comparison of the roles of estrogens and androgens in breast cancer and prostate cancer. J. Cell. Biochem. 2020, 121, 2756–2769. [Google Scholar] [CrossRef]
- Jiang, Z.; Li, W.; Hu, X.; Zhang, Q.; Sun, T.; Cui, S.; Wang, S.; Ouyang, Q.; Yin, Y.; Geng, C.; et al. Tucidinostat plus exemestane for postmenopausal patients with advanced, hormone receptor-positive breast cancer (ACE): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2019, 20, 806–815. [Google Scholar] [CrossRef]
- Lin, J.; Elkon, J.; Ricart, B.; Palmer, E.; Zevallos-Delgado, C.; Noonepalle, S.; Burgess, B.; Siegel, R.; Ma, Y.; Villagra, A. Phase I Study of Entinostat in Combination with Enzalutamide for Treatment of Patients with Metastatic Castration-Resistant Prostate Cancer. Oncologist 2021, 26, e2136–e2142. [Google Scholar] [CrossRef]
- Yardley, D.A.; Ismail-Khan, R.R.; Melichar, B.; Lichinitser, M.; Munster, P.N.; Klein, P.M.; Cruickshank, S.; Miller, K.D.; Lee, M.J.; Trepel, J.B. Randomized phase II, double-blind, placebo-controlled study of exemestane with or without entinostat in postmenopausal women with locally recurrent or metastatic estrogen receptor-positive breast cancer progressing on treatment with a nonsteroidal aromatase inhibitor. J. Clin. Oncol. 2013, 31, 2128–2135. [Google Scholar] [CrossRef] [Green Version]
- Connolly, R.M.; Zhao, F.; Miller, K.D.; Lee, M.J.; Piekarz, R.L.; Smith, K.L.; Brown-Glaberman, U.A.; Winn, J.S.; Faller, B.A.; Onitilo, A.A.; et al. E2112: Randomized Phase III Trial of Endocrine Therapy Plus Entinostat or Placebo in Hormone Receptor-Positive Advanced Breast Cancer. A Trial of the ECOG-ACRIN Cancer Research Group. J. Clin. Oncol. 2021, 39, 3171–3181. [Google Scholar] [CrossRef]
- Malone, C.F.; Emerson, C.; Ingraham, R.; Barbosa, W.; Guerra, S.; Yoon, H.; Liu, L.L.; Michor, F.; Haigis, M.; Macleod, K.F.; et al. mTOR and HDAC Inhibitors Converge on the TXNIP/Thioredoxin Pathway to Cause Catastrophic Oxidative Stress and Regression of RAS-Driven Tumors. Cancer Discov. 2017, 7, 1450–1463. [Google Scholar] [CrossRef] [Green Version]
- Wood, A.; George, S.; Adra, N.; Chintala, S.; Damayanti, N.; Pili, R. Phase I study of the mTOR inhibitor everolimus in combination with the histone deacetylase inhibitor panobinostat in patients with advanced clear cell renal cell carcinoma. Investig. New Drugs 2020, 38, 1108–1116. [Google Scholar] [CrossRef]
- Pan, S.T.; Li, Z.L.; He, Z.X.; Qiu, J.X.; Zhou, S.F. Molecular mechanisms for tumour resistance to chemotherapy. Clin. Exp. Pharmacol. Physiol. 2016, 43, 723–737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karagiannis, D.; Rampias, T. HDAC Inhibitors: Dissecting Mechanisms of Action to Counter Tumor Heterogeneity. Cancers 2021, 13, 3575. [Google Scholar] [CrossRef]
- Meng, Y.; Jin, J.; Gong, C.; Miao, H.; Tao, Z.; Li, T.; Cao, J.; Wang, L.; Wang, B.; Zhang, J.; et al. Phase II study of chidamide in combination with cisplatin in patients with metastatic triple-negative breast cancer. Ann. Palliat. Med. 2021, 10, 11255–11264. [Google Scholar] [CrossRef]
- Vu, K.; Wu, C.H.; Yang, C.Y.; Zhan, A.; Cavallone, E.; Berry, W.; Heeter, P.; Pincus, L.; Wieduwilt, M.J.; William, B.M.; et al. Romidepsin Plus Liposomal Doxorubicin Is Safe and Effective in Patients with Relapsed or Refractory T-Cell Lymphoma: Results of a Phase I Dose-Escalation Study. Clin. Cancer Res. 2020, 26, 1000–1008. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.S.; Park, S.B.; Kim, S.A.; Kwon, S.K.; Cha, H.; Lee, D.Y.; Ro, S.; Cho, J.M.; Song, S.Y. A novel HDAC inhibitor, CG200745, inhibits pancreatic cancer cell growth and overcomes gemcitabine resistance. Sci. Rep. 2017, 7, 41615. [Google Scholar] [CrossRef] [Green Version]
- Chan, E.; Chiorean, E.G.; O’Dwyer, P.J.; Gabrail, N.Y.; Alcindor, T.; Potvin, D.; Chao, R.; Hurwitz, H. Phase I/II study of mocetinostat in combination with gemcitabine for patients with advanced pancreatic cancer and other advanced solid tumors. Cancer Chemother. Pharmacol. 2018, 81, 355–364. [Google Scholar] [CrossRef]
- Blomberg, O.S.; Spagnuolo, L.; de Visser, K.E. Immune regulation of metastasis: Mechanistic insights and therapeutic opportunities. Dis. Models Mech. 2018, 11, dmm036236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Su, X.; Liu, R.; Pan, Y.; Fang, J.; Cao, L.; Feng, C.; Shang, Q.; Chen, Y.; Shao, C.; et al. HDAC inhibition potentiates anti-tumor activity of macrophages and enhances anti-PD-L1-mediated tumor suppression. Oncogene 2021, 40, 1836–1850. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, C.P.; Wu, Q.V.; Voutsinas, J.; Fromm, J.R.; Jiang, X.; Pillarisetty, V.G.; Lee, S.M.; Santana-Davila, R.; Goulart, B.; Baik, C.S.; et al. A Phase II Trial of Pembrolizumab and Vorinostat in Recurrent Metastatic Head and Neck Squamous Cell Carcinomas and Salivary Gland Cancer. Clin. Cancer Res. 2020, 26, 837–845. [Google Scholar] [CrossRef] [PubMed]
- Ny, L.; Jespersen, H.; Karlsson, J.; Alsén, S.; Filges, S.; All-Eriksson, C.; Andersson, B.; Carneiro, A.; Helgadottir, H.; Levin, M.; et al. The PEMDAC phase 2 study of pembrolizumab and entinostat in patients with metastatic uveal melanoma. Nat. Commun. 2021, 12, 5155. [Google Scholar] [CrossRef]
- Awad, M.M.; Le Bruchec, Y.; Lu, B.; Ye, J.; Miller, J.; Lizotte, P.H.; Cavanaugh, M.E.; Rode, A.J.; Dumitru, C.D.; Spira, A. Selective Histone Deacetylase Inhibitor ACY-241 (Citarinostat) Plus Nivolumab in Advanced Non-Small Cell Lung Cancer: Results From a Phase Ib Study. Front. Oncol. 2021, 11, 696512. [Google Scholar] [CrossRef] [PubMed]
- Mehta-Shah, N.; Lunning, M.A.; Moskowitz, A.J.; Boruchov, A.M.; Ruan, J.; Lynch, P.; Hamlin, P.A.; Leonard, J.; Matasar, M.J.; Myskowski, P.L.; et al. Romidepsin and lenalidomide-based regimens have efficacy in relapsed/refractory lymphoma: Combined analysis of two phase I studies with expansion cohorts. Am. J. Hematol. 2021, 96, 1211–1222. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Tian, Y.; Zhu, W.G. The Roles of Histone Deacetylases and Their Inhibitors in Cancer Therapy. Front. Cell Dev. Biol. 2020, 8, 576946. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Histone Deacetylases | |||||
|---|---|---|---|---|---|
| Class | Member | Cellular Localization | Chromosome Position | Aminoacids No (Molecular Weight, kD) | Basic Structure |
| I | HDAC1 | Nucleus | 1p35-p35.1 | 483 (51) |  |
| HDAC2 | 6q21 | 488 (55) |  | ||
| HDAC3 | 5q31.3 | 428 (49) |  | ||
| HDAC8 | Xq13.1 | 377 (42) |  | ||
| IIa | HDAC4 | Nucleus/ cytoplasm | 2q37.3 | 1084 (119) |  |
| HDAC5 | 17q21.31 | 1122 (122) |  | ||
| HDAC7 | 12q13.11 | 912 (103) |  | ||
| HDAC9 | 7p21 | 1069 (118) |  | ||
| IIb | HDAC6 | Cytoplasm | Xp11.23 | 1215 (131) |  |
| HDAC10 | 2q13.33 | 669 (71) |  | ||
| IV | HDAC11 | Nucleus | 3p25.1 | 343 (39) |  |
 N and C terminal regions,
N and C terminal regions,  Catalytic domains,
Catalytic domains,  Nuclear localization sequence,
Nuclear localization sequence,  Nuclear export sequence,
Nuclear export sequence,  cytoplasmic anchoring motif,
cytoplasmic anchoring motif,  Zinc finger motif.
Zinc finger motif.| Histone Deacetylases Inhibitors | ||||
|---|---|---|---|---|
| Clasification | Name | HDACs (IC50) | Structure | Ref. |
| Aliphatic carboxylic acids | Sodium butyrate | HDAC1 (16 mM), HDAC2 (12 μM), HDAC3 (9 μM), HDAC8 (15 μM) | 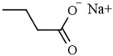 | [72] |
| Valproic acid | HDAC1 (38 mM), HDAC2 (62 mM), HDAC3 (161 μM), HDAC8 (103 μM) |  | [76,77,78] | |
| Hydroxamic acids | Vorinostat | HDAC1 (30 nM), HDAC2 (144 nM), HDAC3 (6 nM), HDAC6, (10 nM) HDAC8 (38 nM), HDAC10 (21 nM), HDAC11 (28 nM) | 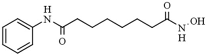 | [85,86] |
| Belinostat | HDAC1 (41 nM), HDAC2 (125 nM), HDAC3 (30 nM), HDAC4 (115 nM) HDAC6 (82 nM), HDAC7 (67 nM), HDAC8 (216 nM) |  | [91,92,93] | |
| Panobinostat | HDAC1 (3 nM), HDAC2 (3 nM), HDC3 (4 nM), HDAC4 (23 nM), HDAC6 (3 nM), HDAC7 (18 nM), HDAC8 (248 nM), |  | [94,95] | |
| Ricolinostat | HDAC1 (58 nM), HDAC2 (48 nM), HDAC3 (51 nM), HDAC6 (5 nM), HDAC8 (100 nM) | 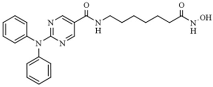 | [104] | |
| Citarinostat | HDAC1 (35 nM), HDAC2 (45 nM), HDAC3 (46 nM), HDAC6 (3 nM), HDAC8 (137 nM) |  | [110] | |
| Benzamides | Entinostat | HDAC1 (190 nM), HDAC2 (650 nM), HDC3 (600 nM) |  | [115,116,117] |
| Chidamide | HDAC1 (95 nM), HDAC2 (169 nM), HDAC3 (67 nM), HDAC10 (78 nM) | 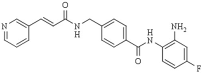 | [118,119,120] | |
| MPT0L184 | HDAC1 (90 nM), HDAC2 (400 nM), HDAC3 (2,3 μM) |  | [119] | |
| Mocetinostat | HDAC1 (9 nM), HDAC2 (34 nM), HDAC3 (265 nM) |  | [122] | |
| Cyclic peptides | Romidepsin | HDAC1, 3, -8 (<1 nM), HDAC4 (20 nM), HDAC6 (9 nM) | 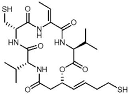 | [48,127,128,129,130] |
| Natural HDAC Inhibitors | ||||
|---|---|---|---|---|
| Compound Name and Structure | Source | HDAC Isoforms | Ref. | |
| Organosulfurs | allyl mercaptan | organosulfur compounds from garlic | HDAC 8 | [154] |
diallyl disulfide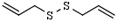 | ||||
| Isothiocyanates | Benzyl isothiocyanate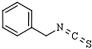 | brassica or cruciferous vegetables | HDAC1 and 3 | [160] |
sulforaphane  | -HDAC1, -4, -6 and -7 | [161,162] | ||
| Flavonoids | Quercetin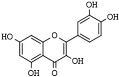 | plants and fruits | HDAC1, HDAC8 | [173,174,175] |
Apigenin | Asteraceae family | HDAC1 and -3 | [177,178] | |
Chrysin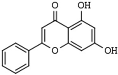 | mushrooms, olive oil, tea, red wine, and passion fruit flowers, as well as Thai propolis and honey | HDAC-2, 3 and 8 | [181] | |
| Polyphenols | Curcumin | Curcuma longa | HDAC1, -3, -4, -6 and -8 | [185,186,187] |
(−)Epigallocatechin-3-gallate (EGCG)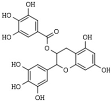 | curry spices, grapes, soy, and berries | HDAC1, -2, and -3 | [189,190,191,192] | |
Resveratrol | grapes and wine | pan-inhibitor | [195,196] | |
| Isoflavone | Genistein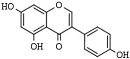 | soybeans | HDAC1, -5 and -6 | [199,200,201,202,203] |
| Bifunctional HDAC Inhibitors | |||
|---|---|---|---|
| Name | Targets (IC50) | Structure | Ref. |
| (1) CUDC-101 | HDAC (4.2 nM) EGFR (2.4 nM) HER2 (15.7 nM) | 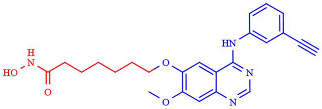 | [233] |
| (2) 6a | HDAC1 (1.5 μM) HDAC2 (0.19 μM) HDAC3 (1.49 μM | 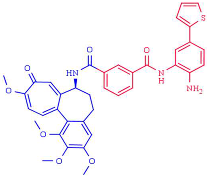 | [237] |
| (3) JAK/HDAC6 dual inhibitor | HDAC6 (2.1 nM) JAK2 (1.4 nM) HDAC3 (2.17 μM) |  | [238] |
| (4) LSD1/HDAC dual inhibitor | HDAC1 (15 nM) HDAC2 (23 nM) LSD1 (1.2 μM) |  | [240] |
| Proteolysis Targeting Chimeras | |||
|---|---|---|---|
| Name | Targets (DC50, Dmax) | Structure | Ref. |
| 9c—HDAC6 degrader | HDAC6 (DC50 = 34 nM, Dmax = 70.5%) | 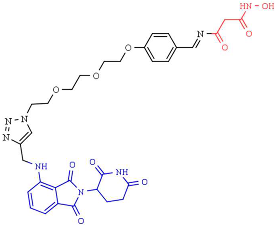 | [248] |
| NP8 | HDAC6 (DC50 = 3.8nM, Dmax = ND) | 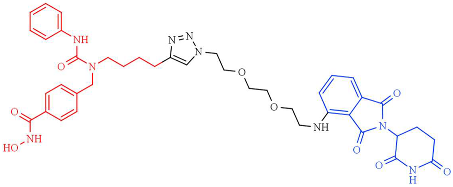 | [249] |
| NH2 | HDAC6 (DC50 = 3.2 nM, Dmax = ND) | [250] | |
| VHL-Next-A degrader | HDAC6 (DC50 = 7.1 nM, Dmax = 90%) |  | [251] |
| XZ9002 degrader | HDAC3 (DC50 = 42 nM, Dmax = 70%) |  | [252] |
| P1 | HDAC1, HDAC6, and HDAC8 |  | [254] |
| Combined Clinical Trails Strategies | |||
|---|---|---|---|
| HDACi | Combined Targeting | Cancer Type | Ref. |
| Vorinostat | Phase I/II, gefitinib- EGFR-TKi | Non-small-cell lung cancer | [262] |
| Vorinostat | Phase II, bevacizumab- angiogenic VEGF blocker | Metastatic clear-cell renal cell carcinoma | [264] |
| Tucidinostat | Phase III, exemestane-steroidal aromatase inhibitor, hormonal therapies | Hormone receptor-positive (HR+) and HER2 negative breast cancer | [266] |
| Entinostat | Phase I, testosterone antagonist therapy-enzalutamide, hormonal therapy | Castration-resistant prostate cancer | [267] |
| Entinostat | Placebo-controlled phase III study, exemestane-steroidal aromatase inhibitor, hormonal therapy | Hormone receptor-positive (HR+) and HER2-negative breast cancer | [269] |
| Panobinostat | Phase I dose-finding trial, -mTOR inhibitor-everolimus, autophagy | Advanced clear-cell renal cell carcinoma | [271] |
| Tucidinostat | Phase II, cisplatin, chemotherapy | Triple-negative breast cancer | [274] |
| Romidepsin | Phase I dose-escalation study, liposomal doxorubicin chemotherapy | Cutaneous T-cell lymphoma | [275] |
| Mocetinostat | Non-randomized phase I/II, gemcitabine chemotherapy | Various solid tumors, including advanced pancreatic cancer | [277] |
| Vorinostat | Phase II, immune checkpoint inhibitor anti–PD-1-pembrolizumab, immunotherapy | Recurrent/metastatic squamous cell carcinomas of the head and neck and salivary gland cancer | [280] |
| Entinostat | Phase II, immune checkpoint inhibitor anti–PD-1-pembrolizumab, immunotherapy | Metastatic uveal melanoma | [281] |
| Citarinostat | Phase Ib, immune checkpoint inhibitor anti–PD-1-nivolumab, immunotherapy | Non-small-cell lung cancer | [282] |
| Romidepsin | Phase I, immunomodulatory drug lenalidomide and the proteasome inhibitor carfilzomib | T-cell lymphoma and B-cell lymphoma | [283] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ruzic, D.; Djoković, N.; Srdić-Rajić, T.; Echeverria, C.; Nikolic, K.; Santibanez, J.F. Targeting Histone Deacetylases: Opportunities for Cancer Treatment and Chemoprevention. Pharmaceutics 2022, 14, 209. https://doi.org/10.3390/pharmaceutics14010209
Ruzic D, Djoković N, Srdić-Rajić T, Echeverria C, Nikolic K, Santibanez JF. Targeting Histone Deacetylases: Opportunities for Cancer Treatment and Chemoprevention. Pharmaceutics. 2022; 14(1):209. https://doi.org/10.3390/pharmaceutics14010209
Chicago/Turabian StyleRuzic, Dusan, Nemanja Djoković, Tatjana Srdić-Rajić, Cesar Echeverria, Katarina Nikolic, and Juan F. Santibanez. 2022. "Targeting Histone Deacetylases: Opportunities for Cancer Treatment and Chemoprevention" Pharmaceutics 14, no. 1: 209. https://doi.org/10.3390/pharmaceutics14010209
APA StyleRuzic, D., Djoković, N., Srdić-Rajić, T., Echeverria, C., Nikolic, K., & Santibanez, J. F. (2022). Targeting Histone Deacetylases: Opportunities for Cancer Treatment and Chemoprevention. Pharmaceutics, 14(1), 209. https://doi.org/10.3390/pharmaceutics14010209