
In Vitro and In Vivo Bioequivalence Study of 3D-Printed Instant-Dissolving Levetiracetam Tablets and Subsequent Personalized Dosing for Chinese Children Based on Physiological Pharmacokinetic Modeling

 ,
,
Abstract
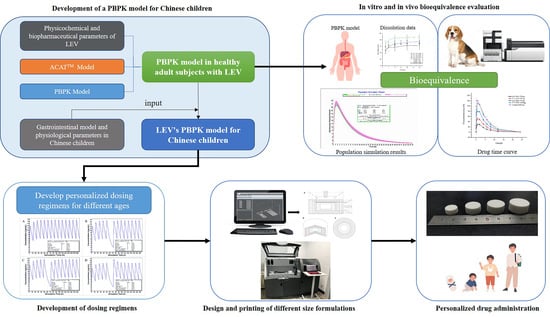
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Animals
2.3. In Vitro Dissolution Experiments
2.4. In Vivo Dissolution Experiments
2.4.1. Experimental Protocol and Sample Collection
2.4.2. LC-MS/MS Conditions
2.4.3. Sample Preparation
2.5. Pharmacokinetic Data Analysis
2.5.1. Drug–Time Curve Plotting and Calculation of Pharmacokinetic Parameters
2.5.2. Bioequivalence Evaluation in Dogs
2.6. PBPK Modeling of LEV in Chinese Adults
2.7. Prediction of Bioequivalence
2.8. PBPK Modeling of LEV in Chinese Children
2.9. The Application of the PBPK Model for Children in Personalized Medication and Dose Adjustment
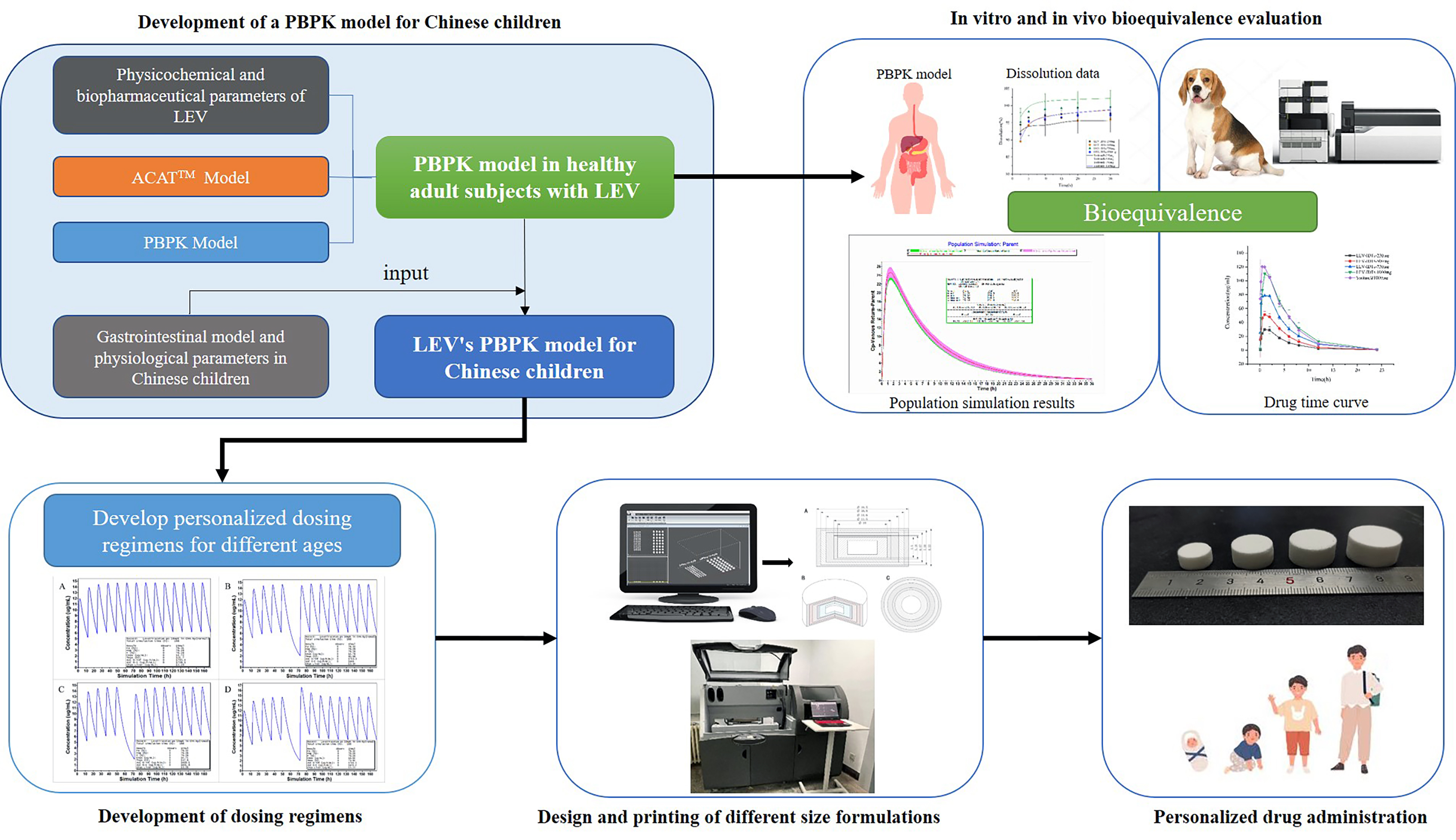
3. Results
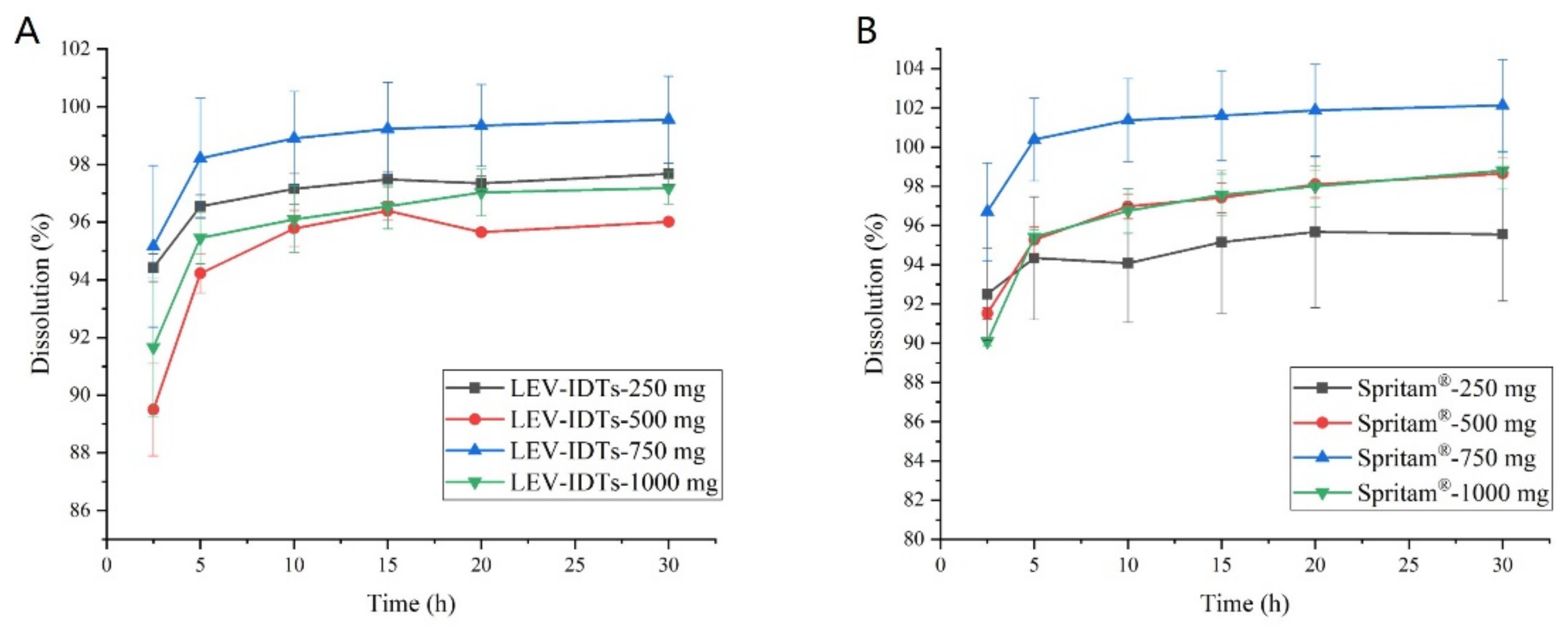
3.1. In Vitro Dissolution Experiment Results
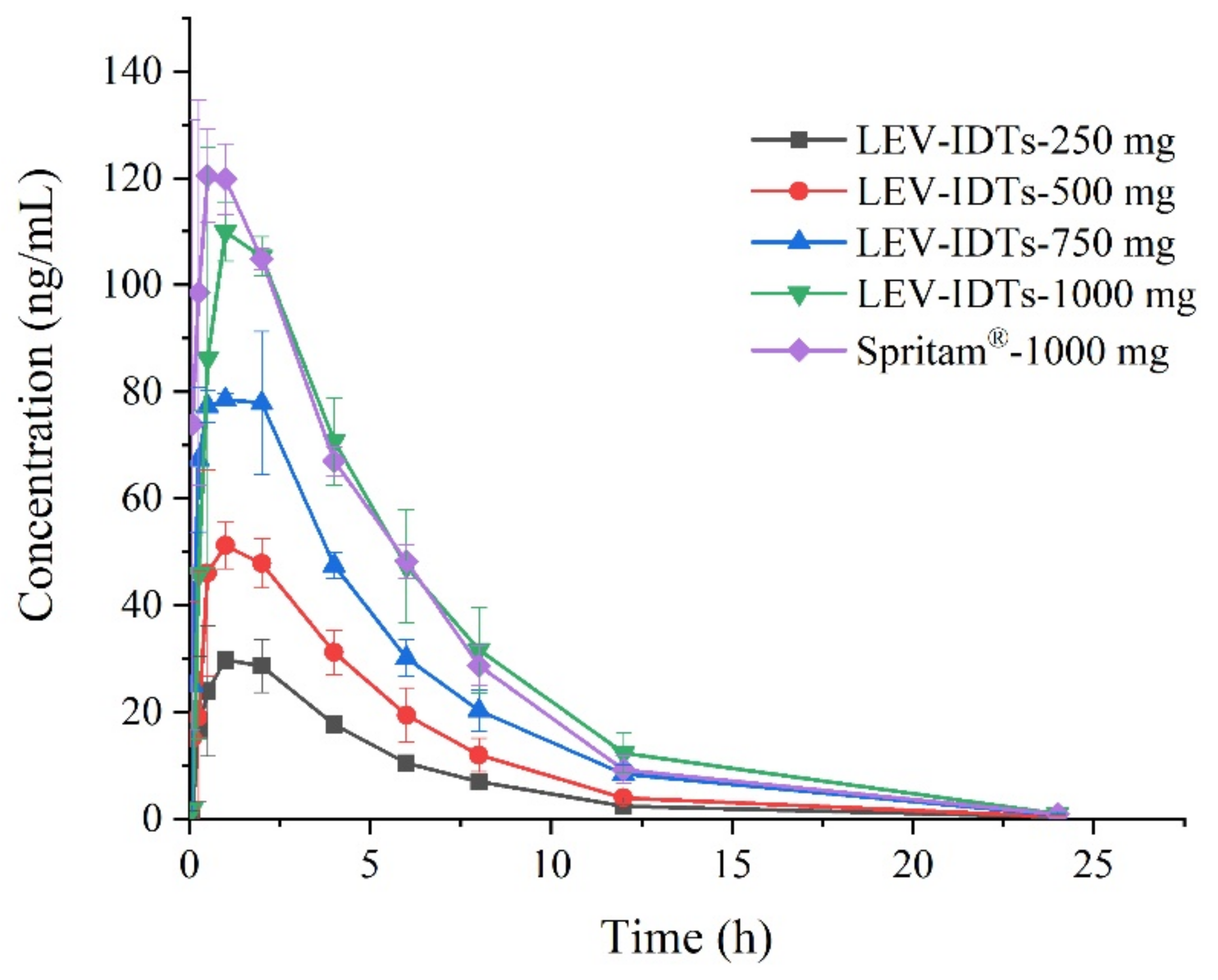
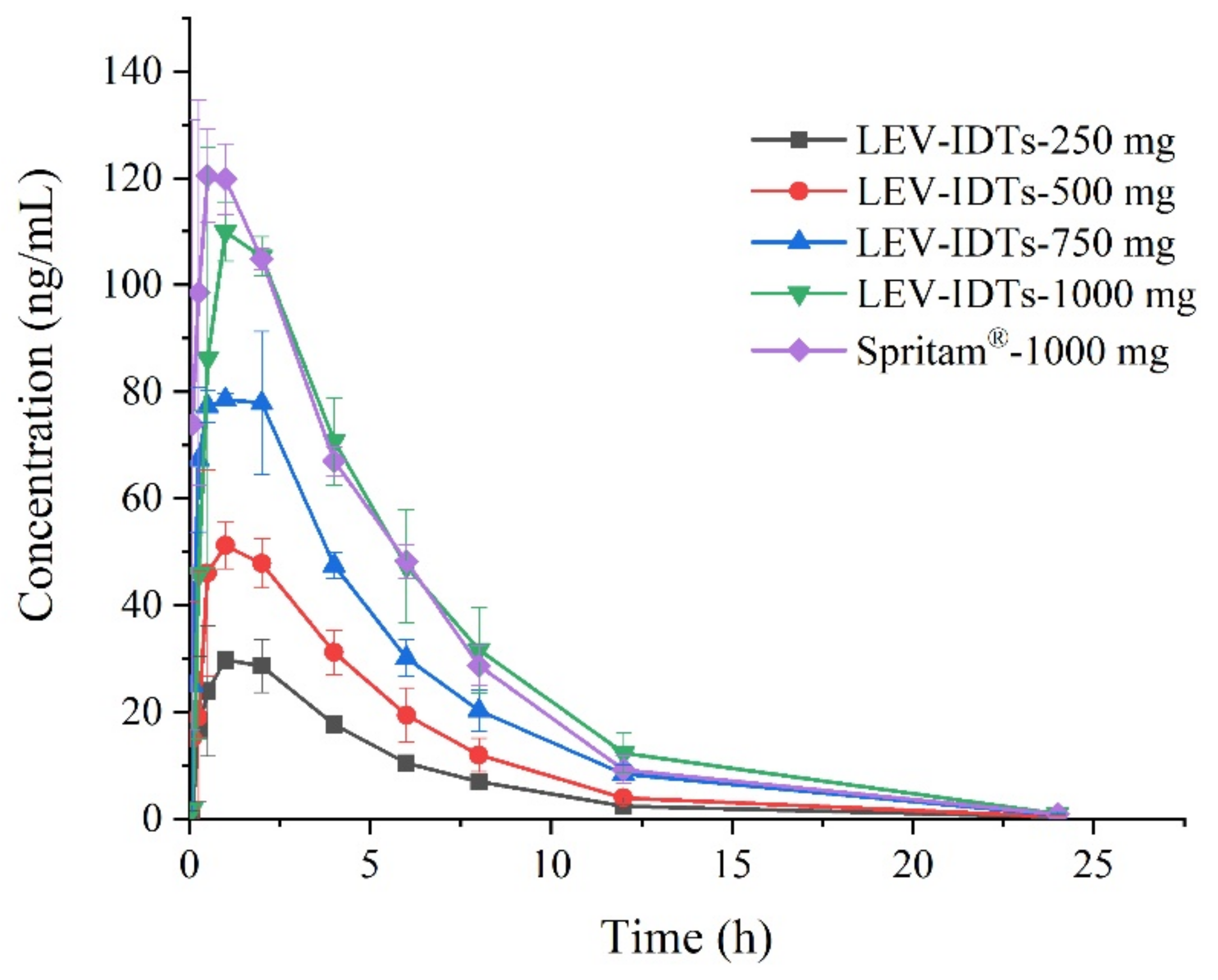
3.2. Pharmacokinetic Study Results in Dogs
3.3. Pharmacokinetic Study Results in Dogs
3.4. Pharmacokinetic Study Results in Dogs
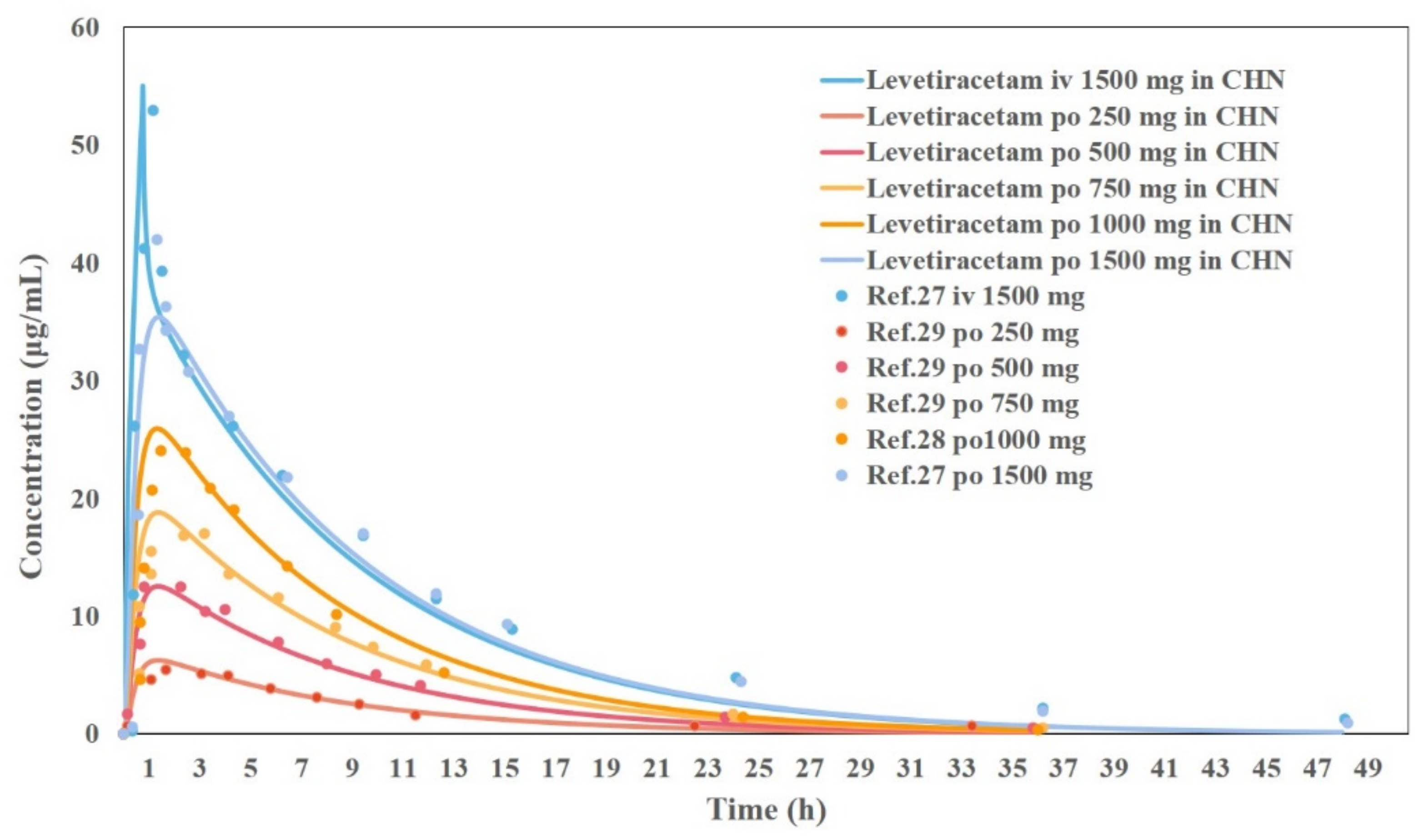
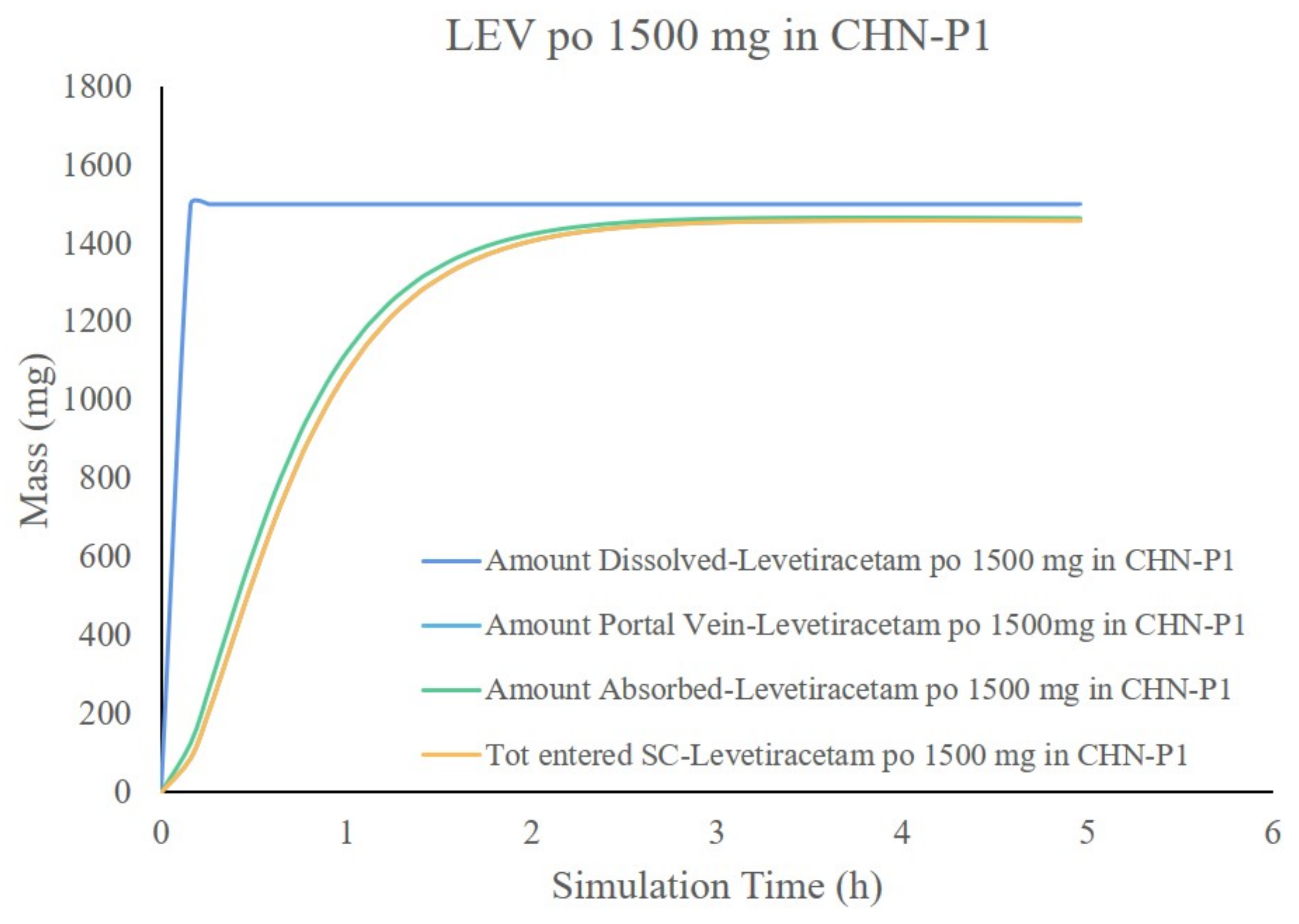
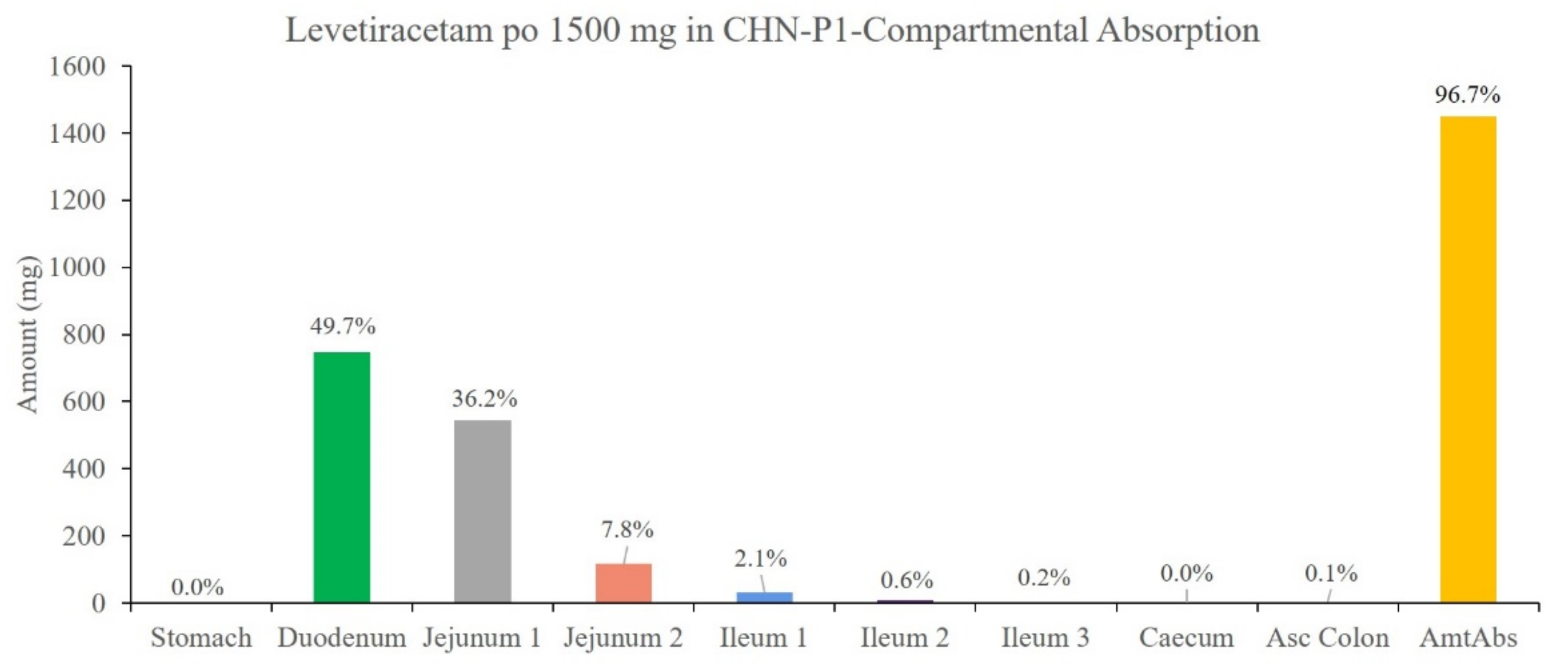
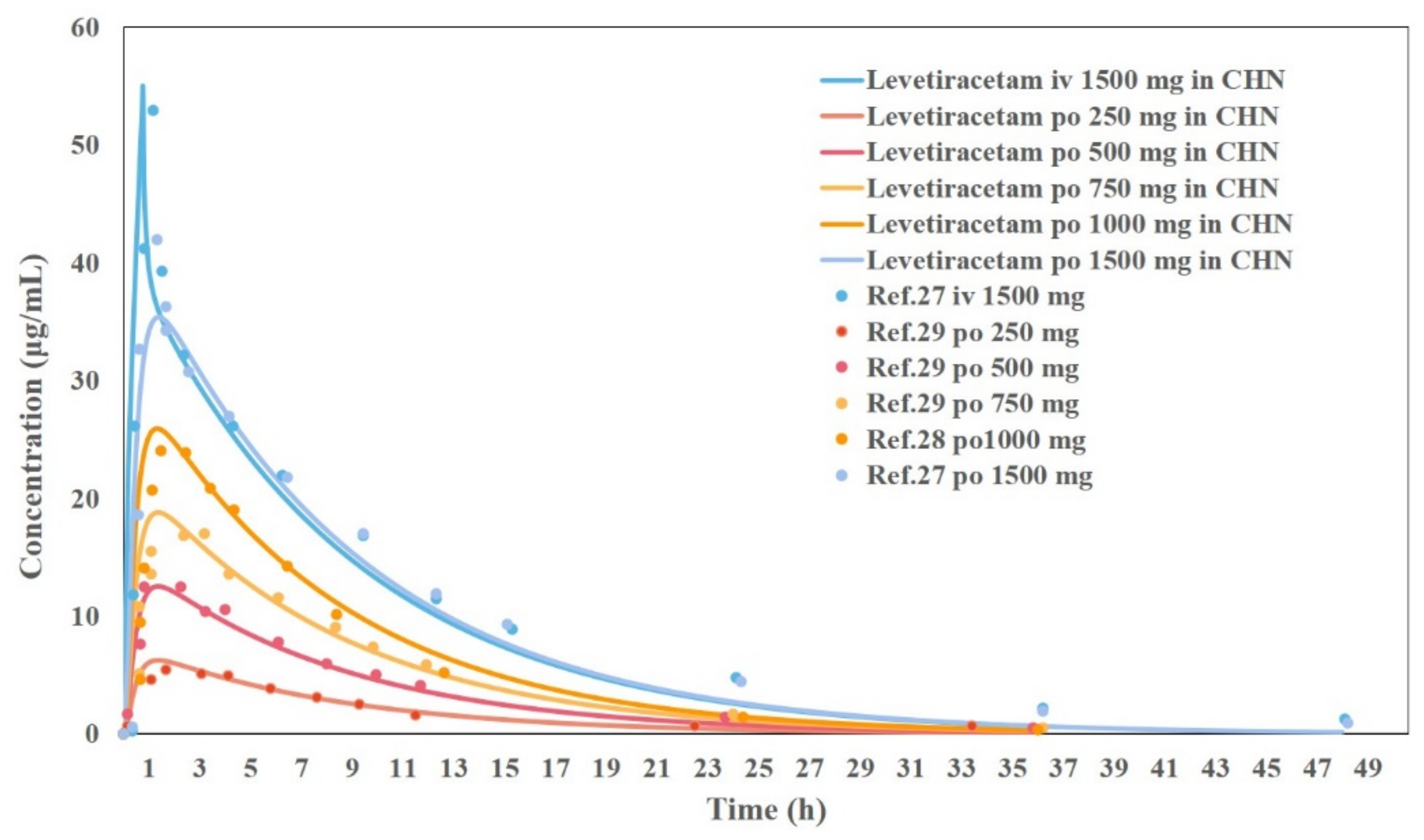
3.5. Results of the Development and Validation of the PBPK Model for Chinese Adults
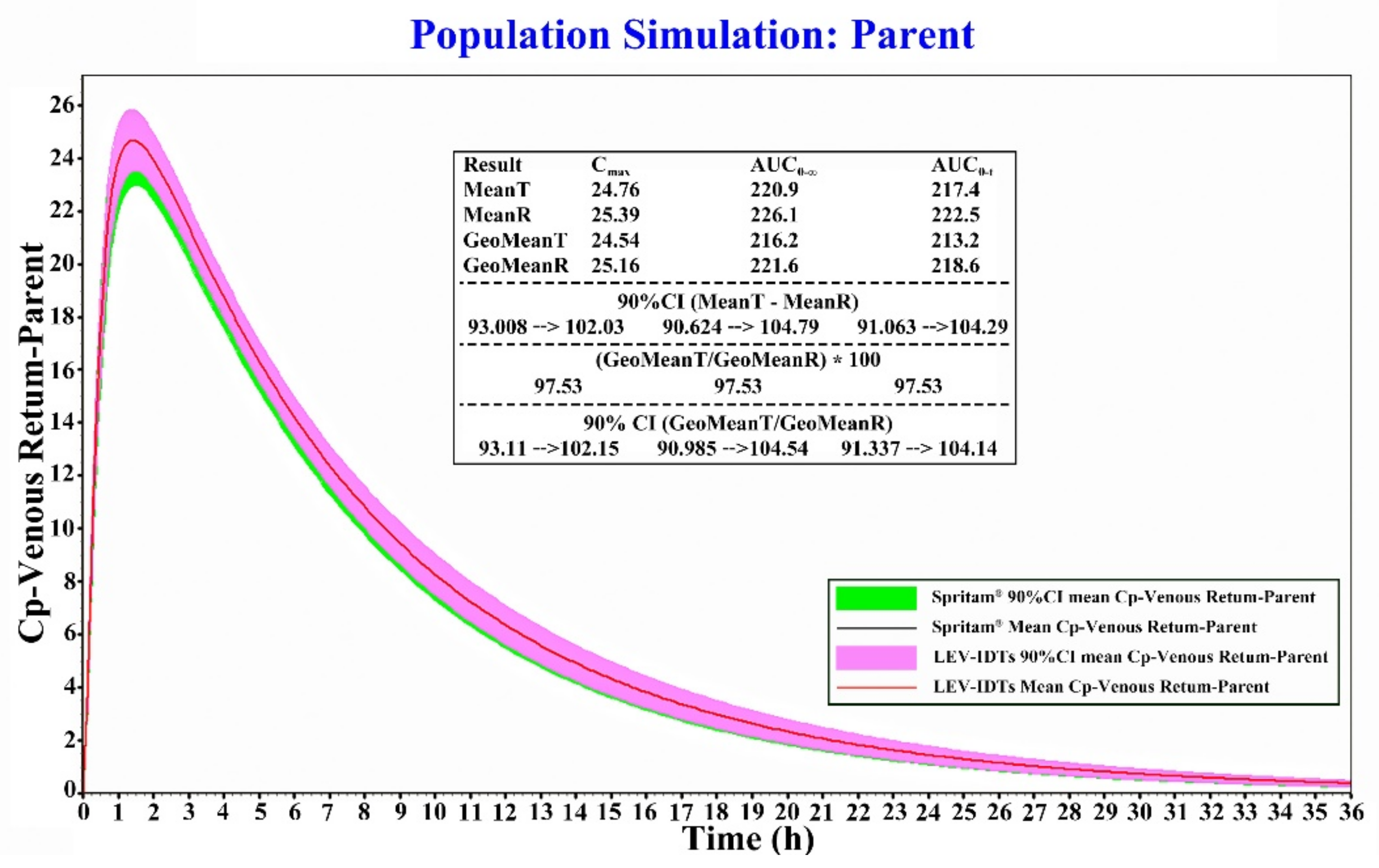
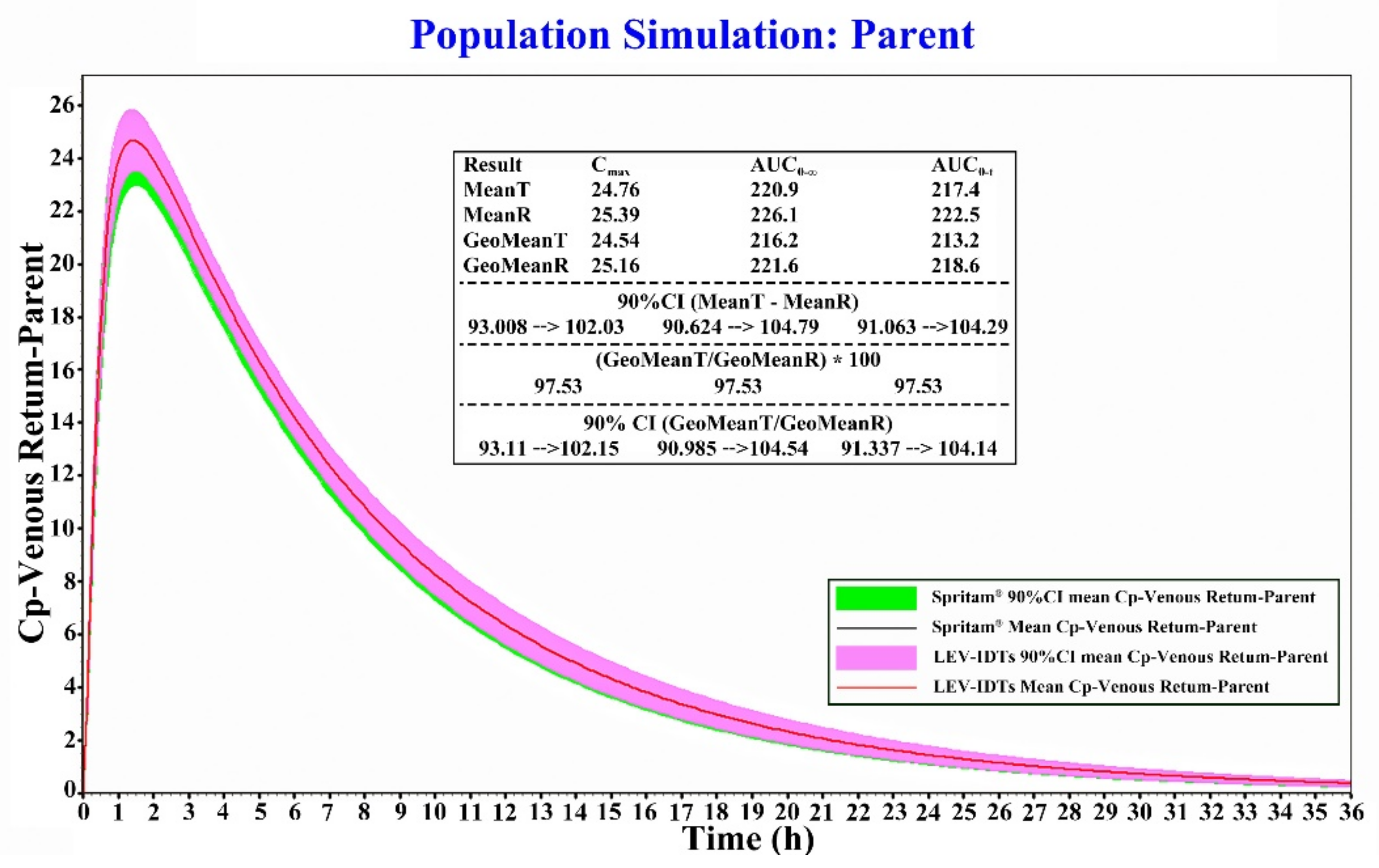
3.6. Results of Bioequivalence Evaluation Based on the PBPK Model
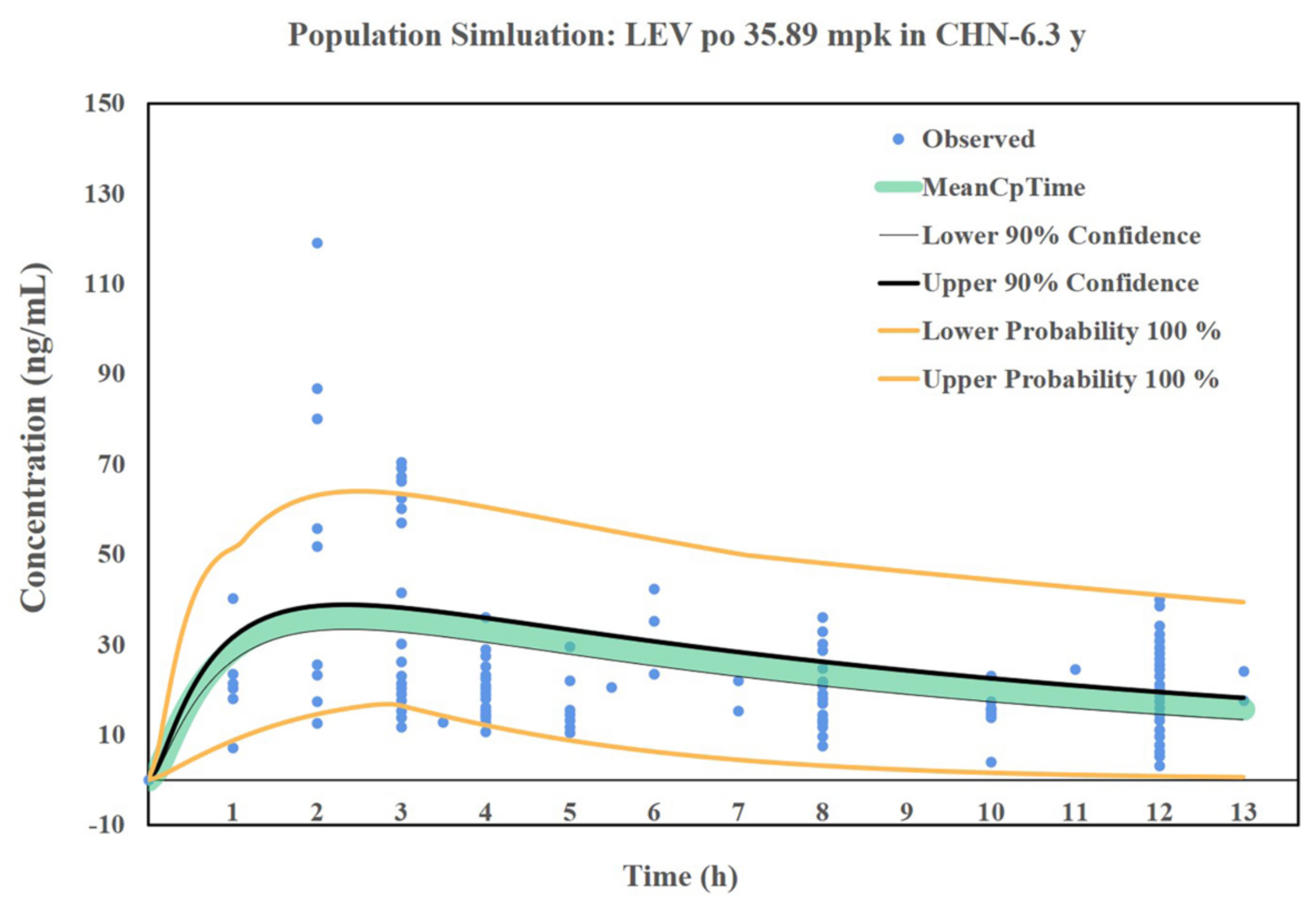
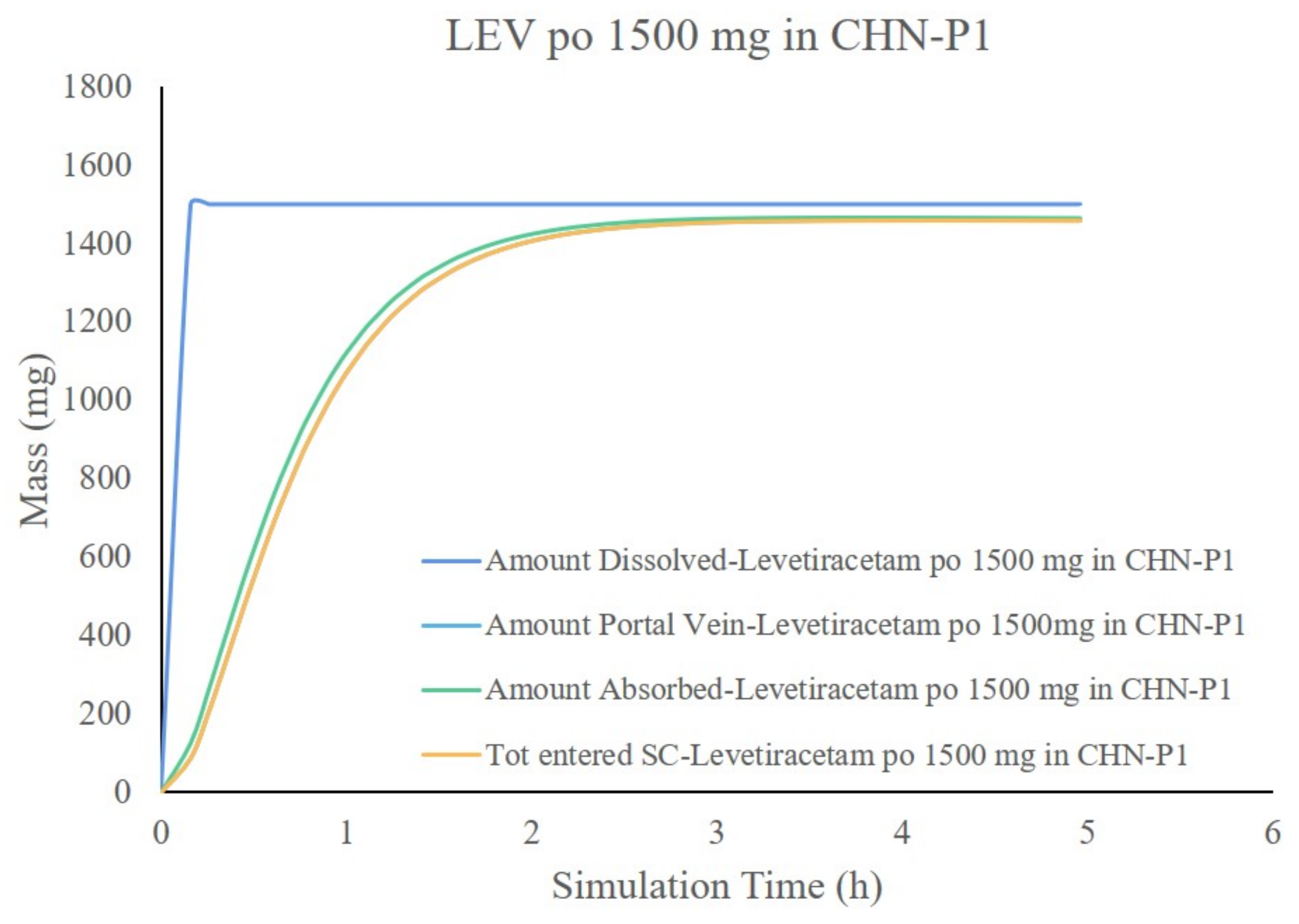
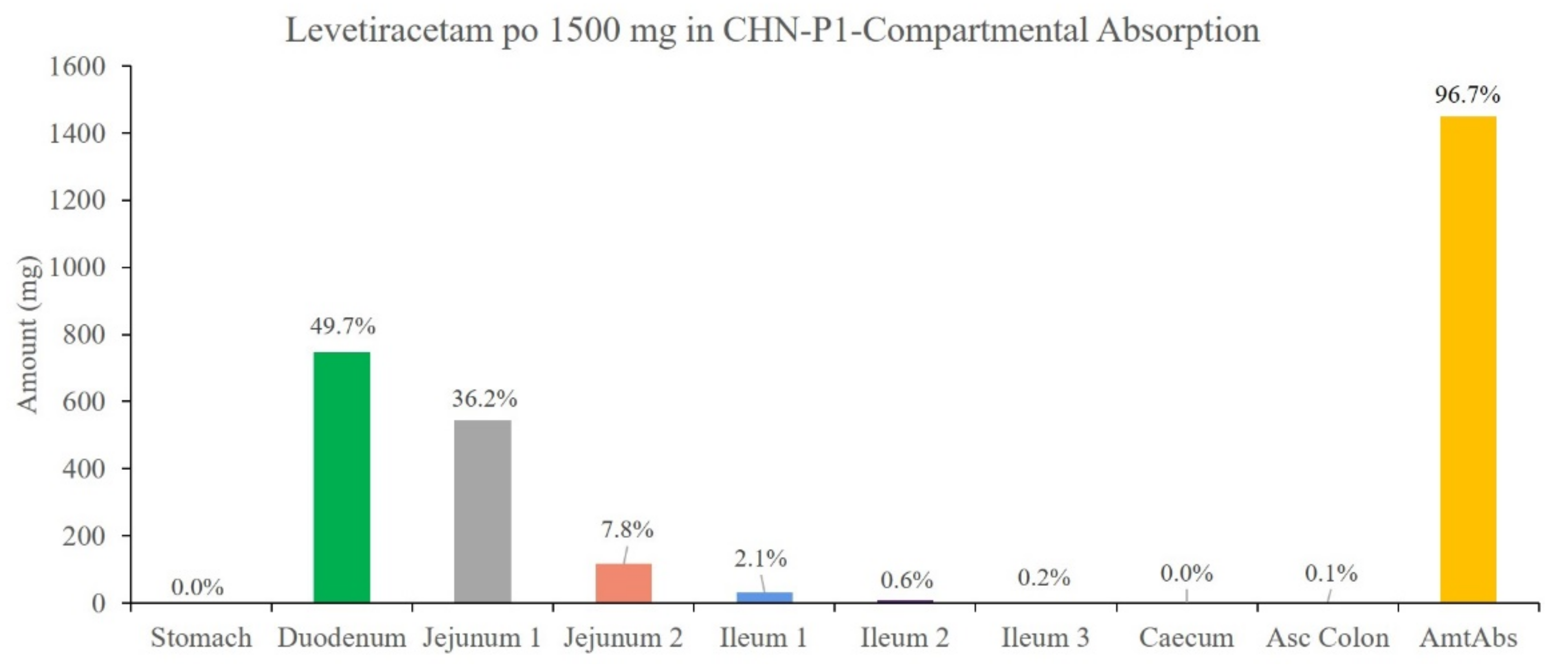
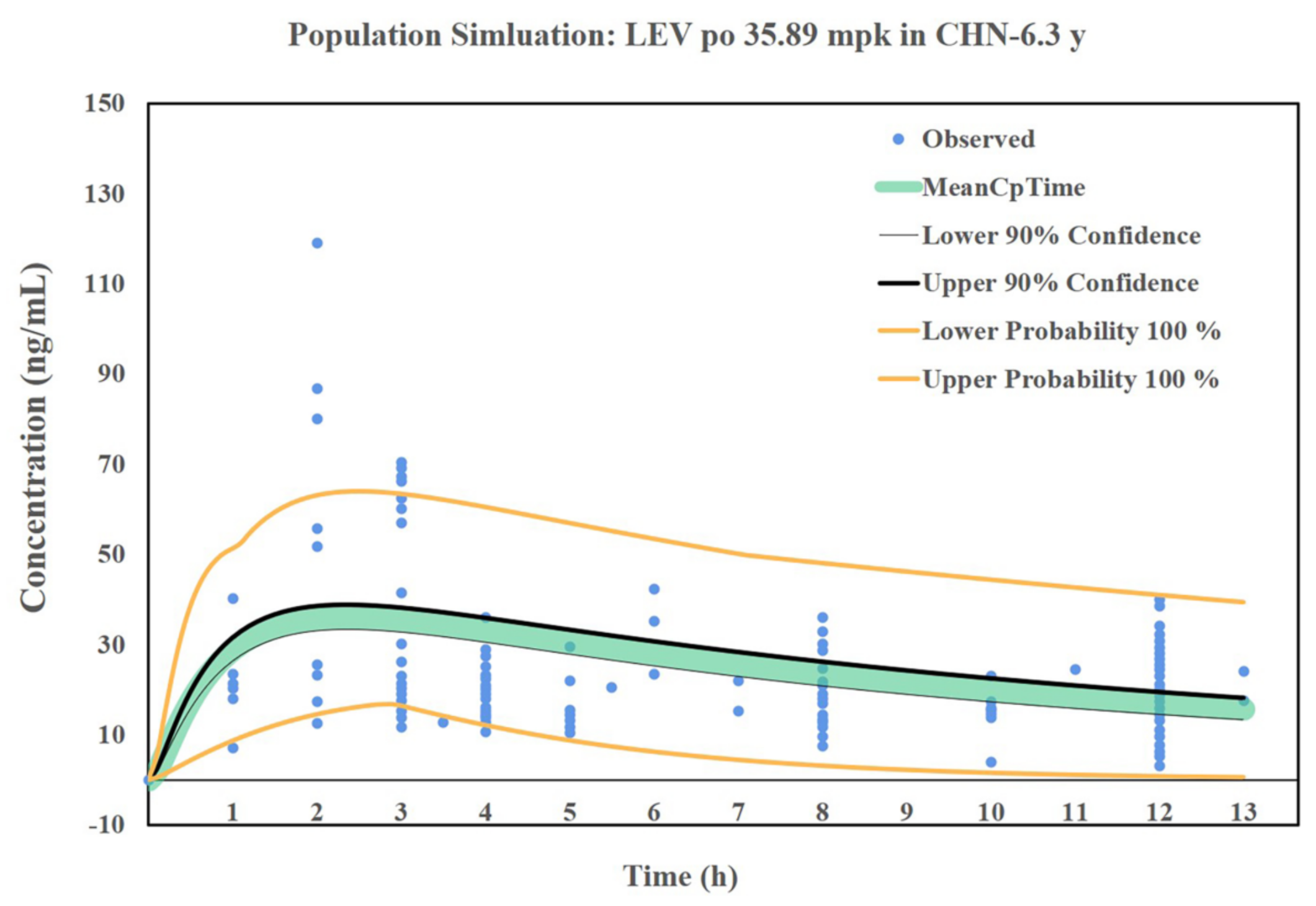
3.7. Simulation Results of the LEV PBPK Model for Chinese Children
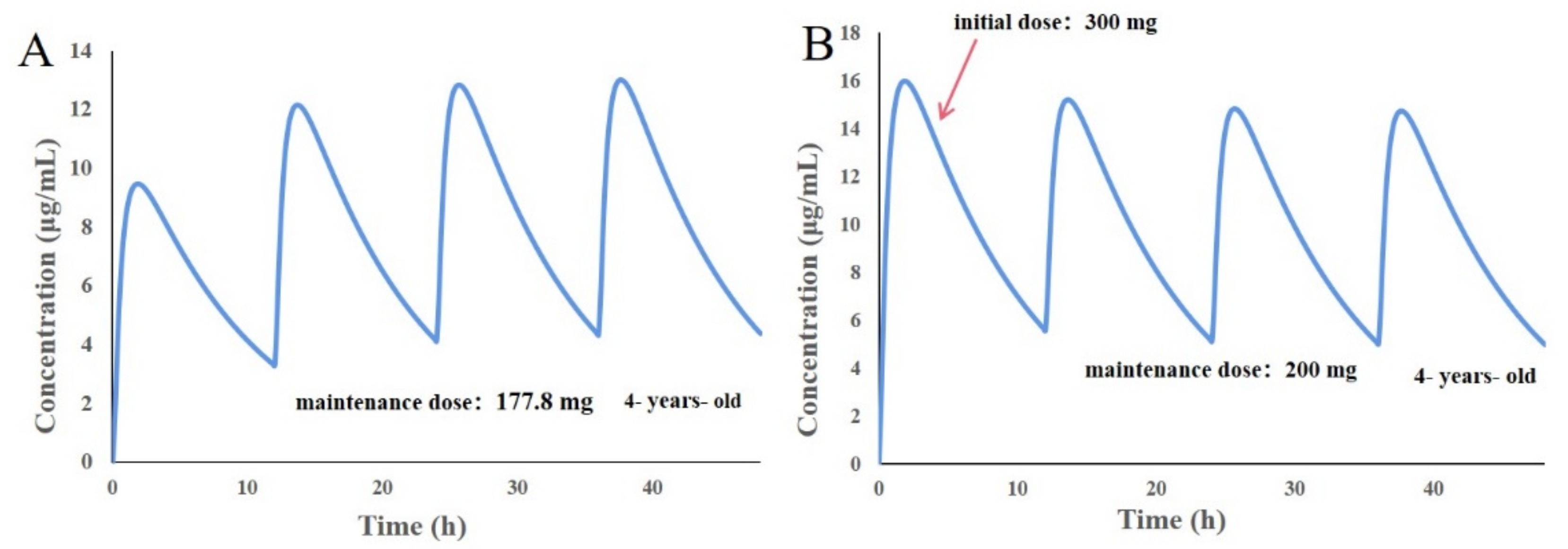
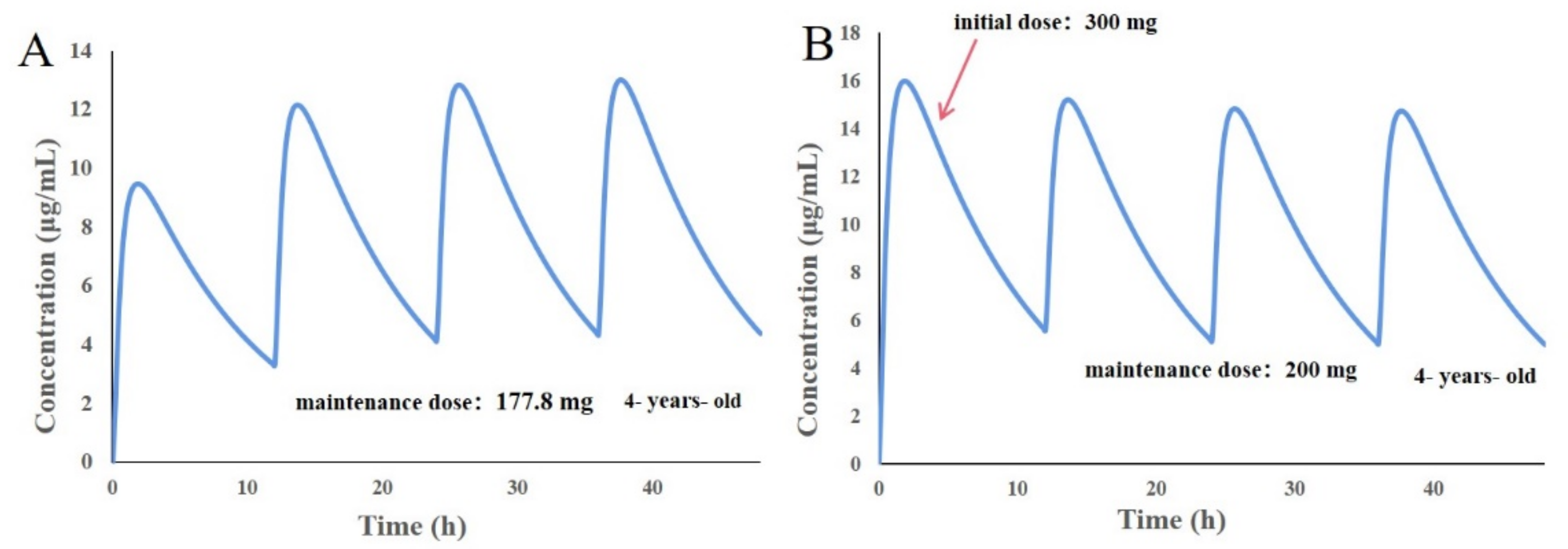
3.8. Design of Individualized Doses for 4-Year-Old Chinese Children
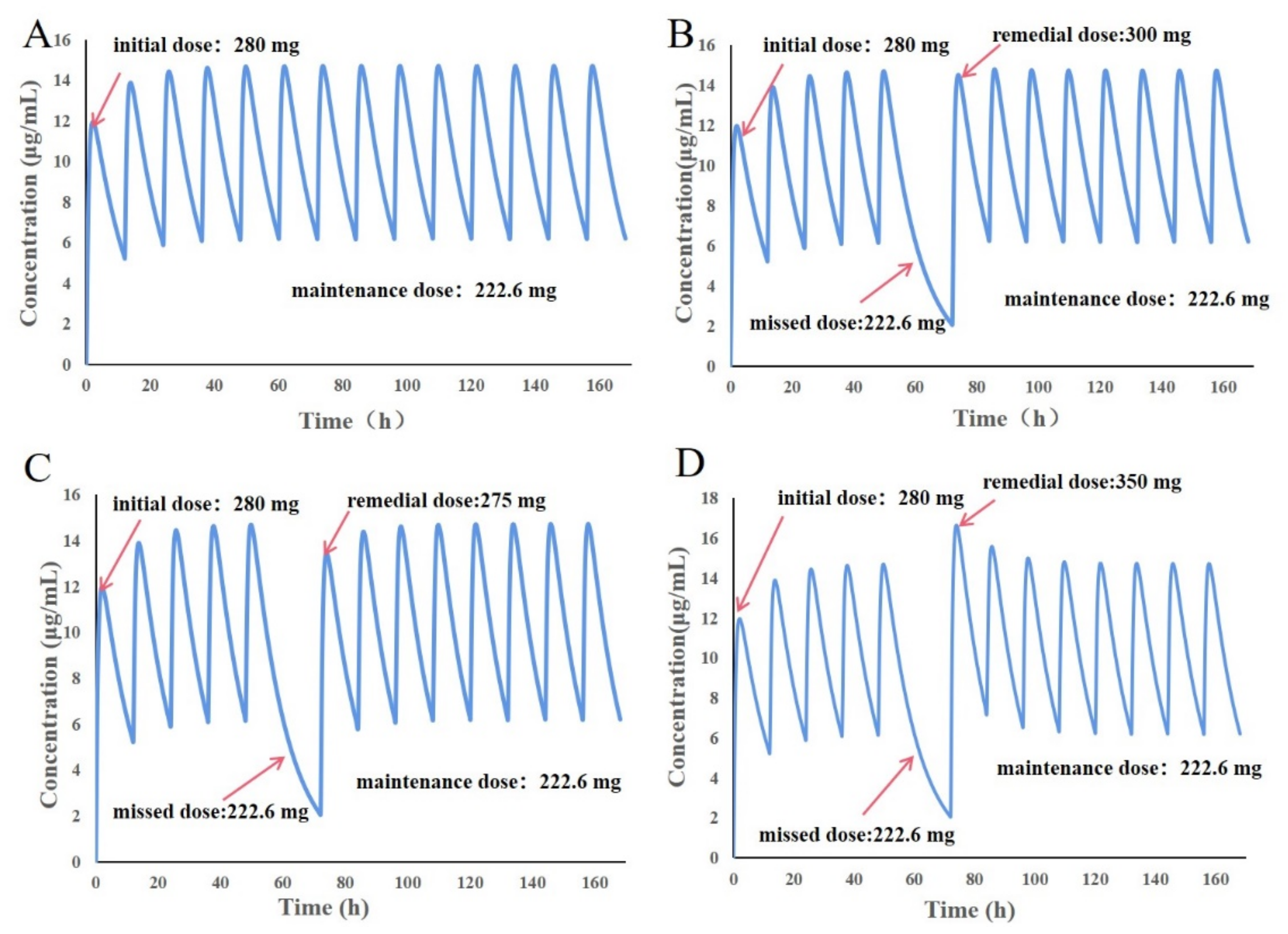
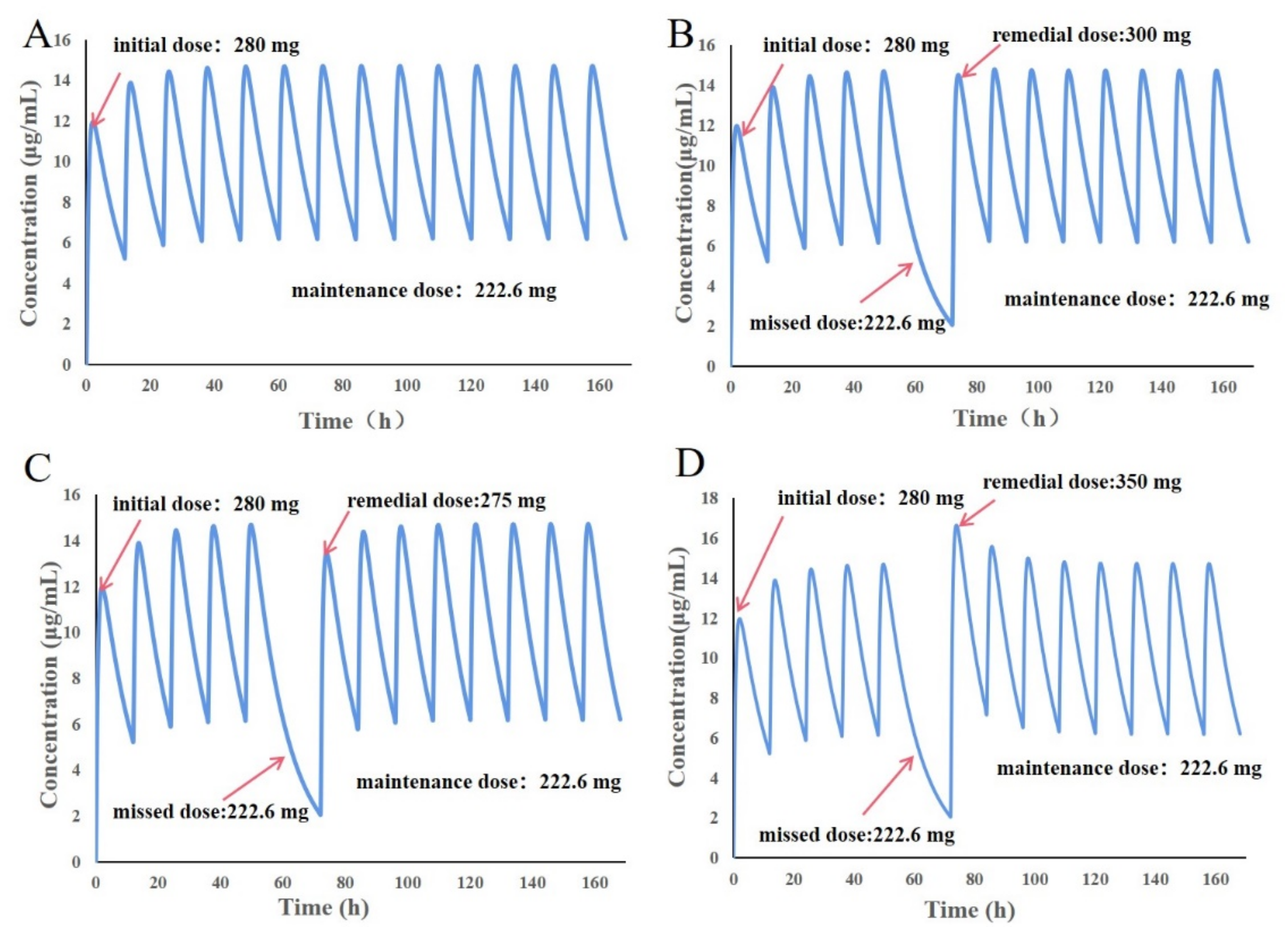
3.9. Design of Individualized Doses for 6-Year-Old Chinese Children
4. Discussion and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sen, K.; Mehta, T.; Sansare, S.; Sharifi, L.; Ma, A.W.; Chaudhuri, B. Pharmaceutical applications of powder-based binder jet 3D printing process—A review. Adv. Drug Deliv. Rev. 2021, 177, 113943. [Google Scholar] [CrossRef] [PubMed]
- Khaled, S.A.; Burley, J.C.; Alexander, M.; Yang, J.; Roberts, C.J. 3D printing of five-in-one dose combination polypill with defined immediate and sustained release profiles. J. Control. Release 2015, 217, 308–314. [Google Scholar] [CrossRef]
- Goyanes, A.; Scarpa, M.; Kamlow, M.; Gaisford, S.; Basit, A.W.; Orlu, M. Patient acceptability of 3D printed medicines. Int. J. Pharm. 2017, 530, 71–78. [Google Scholar] [CrossRef]
- Saeheng, T.; Na-Bangchang, K.; Karbwang, J. Utility of physiologically based pharmacokinetic (PBPK) modeling in oncology drug development and its accuracy: A systematic review. Eur. J. Clin. Pharmacol. 2018, 74, 1365–1376. [Google Scholar] [CrossRef]
- Kostewicz, E.S.; Aarons, L.; Bergstrand, M.; Bolger, M.B.; Galetin, A.; Hatley, O.; Jamei, M.; Lloyd, R.; Pepin, X.; Rostami-Hodjegan, A.; et al. PBPK models for the prediction of in vivo performance of oral dosage forms. Eur. J. Pharm. Sci. 2014, 57, 300–321. [Google Scholar] [CrossRef] [PubMed]
- Sager, J.E.; Yu, J.; Ragueneau-Majlessi, I.; Isoherranen, N. Physiologically Based Pharmacokinetic (PBPK) Modeling and Simulation Approaches: A Systematic Review of Published Models, Applications, and Model Verification. Drug Metab. Dispos. 2015, 43, 1823–1837. [Google Scholar] [CrossRef]
- Khalid, S.; Rasool, M.F.; Imran, I.; Majeed, A.; Saeed, H.; Rehman, A.U.; Ashraf, W.; Ahmad, T.; Bin Jardan, Y.A.; Alqahtani, F. A Physiologically Based Pharmacokinetic Model for Predicting Diazepam Pharmacokinetics after Intravenous, Oral, Intranasal, and Rectal Applications. Pharmaceutics 2021, 13, 1480. [Google Scholar] [CrossRef]
- Jones, H.M.; Gardner, I.B.; Collard, W.T.; Stanley, P.J.; Oxley, P.; Hosea, N.A.; Plowchalk, D.; Gernhardt, S.; Lin, J.; Dickins, M.; et al. Simulation of Human Intravenous and Oral Pharmacokinetics of 21 Diverse Compounds Using Physiologically Based Pharmacokinetic Modelling. Clin. Pharmacokinet. 2011, 50, 331–347. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Heimbach, T.; Lin, W.; Zhang, J.; He, H. Prospective Predictions of Human Pharmacokinetics for Eighteen Compounds. J. Pharm. Sci. 2015, 104, 2795–2806. [Google Scholar] [CrossRef] [Green Version]
- Jones, H.M.; Parrott, N.; Jorga, K.; Lavé, T. A Novel Strategy for Physiologically Based Predictions of Human Pharmacokinetics. Clin. Pharmacokinet. 2006, 45, 511–542. [Google Scholar] [CrossRef]
- Physiologically Based Pharmacokinetic Analyses-Format and Content Guidance for Industry. Available online: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/physiologically-based-pharmacokinetic-analyses-format-and-content-guidance-industry (accessed on 4 September 2018).
- EMA. Guideline on the Reporting of Physiologically Based Pharmacokinetic (PBPK) Modelling and Simulation. Available online: https://www.ema.europa.eu/en/reporting-physiologically-based-pharmacokinetic-pbpk-modelling-simulation (accessed on 13 December 2018).
- Pepin, X.J.H.; Flanagan, T.R.; Holt, D.J.; Eidelman, A.; Treacy, D.; Rowlings, C.E. Justification of Drug Product Dissolution Rate and Drug Substance Particle Size Specifications Based on Absorption PBPK Modeling for Lesinurad Immediate Release Tablets. Mol. Pharm. 2016, 13, 3256–3269. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.I.; Park, M.-H.; Shin, S.-H.; Byeon, J.-J.; Park, Y.; Kim, N.; Choi, J.; Shin, Y.G. Quantitative Analysis of Tozadenant Using Liquid Chromatography-Mass Spectrometric Method in Rat Plasma and Its Human Pharmacokinetics Prediction Using Physiologically Based Pharmacokinetic Modeling. Molecules 2019, 24, 1295. [Google Scholar] [CrossRef] [Green Version]
- Mitra, A.; Kesisoglou, F.; Dogterom, P. Application of Absorption Modeling to Predict Bioequivalence Outcome of Two Batches of Etoricoxib Tablets. AAPS PharmSciTech 2014, 16, 76–84. [Google Scholar] [CrossRef] [Green Version]
- De Buck, S.S.; Sinha, V.K.; Fenu, L.A.; Nijsen, M.J.; Mackie, C.E.; Gilissen, R.A.H.J. Prediction of Human Pharmacokinetics Using Physiologically Based Modeling: A Retrospective Analysis of 26 Clinically Tested Drugs. Drug Metab. Dispos. 2007, 35, 1766–1780. [Google Scholar] [CrossRef]
- Zhang, F.; Zhou, Y.; Wu, N.; Jia, R.; Liu, A.; Liu, B.; Zhou, Z.; Hu, H.; Han, Z.; Ye, X.; et al. In silico prediction of bioequivalence of Isosorbide Mononitrate tablets with different dissolution profiles using PBPK modeling and simulation. Eur. J. Pharm. Sci. 2021, 157, 105618. [Google Scholar] [CrossRef]
- Loisios-Konstantinidis, I.; Cristofoletti, R.; Fotaki, N.; Turner, D.B.; Dressman, J. Establishing virtual bioequivalence and clinically relevant specifications using in vitro biorelevant dissolution testing and physiologically-based population pharmacokinetic modeling. case example: Naproxen. Eur. J. Pharm. Sci. 2020, 143, 105170. [Google Scholar] [CrossRef] [PubMed]
- Mitra, A.; Petek, B.; Bajc, A.; Velagapudi, R.; Legen, I. Physiologically based absorption modeling to predict bioequivalence of controlled release and immediate release oral products. Eur. J. Pharm. Biopharm. 2019, 134, 117–125. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Li, J.; Hong, X.; Han, X.; Liu, B.; Li, X.; Zhang, H.; Gao, J.; Liu, N.; Gao, X.; et al. Taste Masking Study Based on an Electronic Tongue: The Formulation Design of 3D Printed Levetiracetam Instant-Dissolving Tablets. Pharm. Res. 2021, 38, 831–842. [Google Scholar] [CrossRef]
- Wang, Z.; Han, X.; Chen, R.; Li, J.; Gao, J.; Zhang, H.; Liu, N.; Gao, X.; Zheng, A. Innovative color jet 3D printing of levetiracetam personalized paediatric preparations. Asian J. Pharm. Sci. 2021, 16, 374–386. [Google Scholar] [CrossRef] [PubMed]
- Patsalos, P. Pharmacokinetic profile of levetiracetam: Toward ideal characteristics. Pharmacol. Ther. 2000, 85, 77–85. [Google Scholar] [CrossRef]
- Li, Z.-R.; Wang, C.-Y.; Zhu, X.; Jiao, Z. Population Pharmacokinetics of Levetiracetam: A Systematic Review. Clin. Pharmacokinet. 2021, 60, 305–318. [Google Scholar] [CrossRef]
- Odi, R.; Bibi, D.; Wager, T.; Bialer, M. A perspective on the physicochemical and biopharmaceutic properties of marketed antiseizure drugs—From phenobarbital to cenobamate and beyond. Epilepsia 2020, 61, 1543–1552. [Google Scholar] [CrossRef]
- Beasley, M.; Boothe, D.M. Disposition of Extended Release Levetiracetam in Normal Healthy Dogs After Single Oral Dosing. J. Vet. Intern. Med. 2015, 29, 1348–1353. [Google Scholar] [CrossRef]
- Hernández-Mitre, M.P.; Medellín-Garibay, S.E.; Rodríguez-Leyva, I.; Rodríguez-Pinal, C.J.; Zarazúa, S.; Jung-Cook, H.H.; Roberts, J.A.; Romano-Moreno, S.; Milán-Segovia, R.d.C. Population Pharmacokinetics and Dosing Recommendations of Levetiracetam in Adult and Elderly Patients With Epilepsy. J. Pharm. Sci. 2020, 109, 2070–2078. [Google Scholar] [CrossRef] [PubMed]
- Nicolas, J.; Hannestad, J.; Holden, D.; Kervyn, S.; Nabulsi, N.; Tytgat, D.; Huang, Y.; Chanteux, H.; Staelens, L.; Matagne, A.; et al. Brivaracetam, a selective high-affinity synaptic vesicle protein 2A (SV 2A) ligand with preclinical evidence of high brain permeability and fast onset of action. Epilepsia 2016, 57, 201–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.-H.; Wang, L.; Lu, W.; Shang, D.-W.; Wei, M.-J.; Wu, Y. Population pharmacokinetics modeling of levetiracetam in Chinese children with epilepsy. Acta Pharmacol. Sin. 2012, 33, 845–851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.J.; Chen, D.; Yin, J.G.; Chu, J.H.; Xu, M.J.; Liu, F.; Dai, G.L.; Xiong, N.N. Comparative pharmacokinetics of levetiracetam injection and levetiracetam tablets in healthy subjects. Chin. J. Clin. Pharm. Ther. 2017, 22, 294–298. [Google Scholar]
- Liu, W.X.; Ding, Y.; Jia, Y.Y.; Lu, C.T.; Ding, L.K.; Yang, J.; Liang, H.; Yang, L. Pharmacokinetics of levetiracetam tablets in healthy volunteers. Chin. J. Clin. Pharmacol. 2012, 28, 272–274. [Google Scholar] [CrossRef]
- Ma, T.; Qian, J.H.; Zhu, X.G.; Tong, X.H.; Gao, S. Pharmacokinetics of Levetiracetam Tablets in Healthy Humans. Chin. J. Clin. Pharm. Ther. 2007, 27, 858–861. [Google Scholar]
- Wang, Y.H.; Wang, L.; Lu, W.; Wei, M.J.; Wu, Y.; Shang, D.W.; Wang, Y.X. Population pharmacokinetic analysis of levetiracetam in Chinese children with epilepsy. Chin. J. Prat Pediatr. 2012, 27, 517–521. [Google Scholar]
- Wang, J.; Zhang, H.N.; Chen, Y.J.; Wang, Y.J.; Wang, Y.; Li, S.C.; Xu, Q. Relationship between serum concentration and clinical efficacy of levetiracetam monotherapy in children. Chin. Hosp. Pharm. J. 2016, 36, 476–480. [Google Scholar] [CrossRef]
- Berezhkovskiy, L.M. Volume of Distribution at Steady State for a Linear Pharmacokinetic System with Peripheral Elimination. J. Pharm. Sci. 2004, 93, 1628–1640. [Google Scholar] [CrossRef] [PubMed]
- Barrett, J.S.; Della Casa Alberighi, O.; Läer, S.; Meibohm, B. Physiologically based pharmacokinetic (PBPK) modeling in children. Clin. Pharmacol. Ther. 2012, 92, 40. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | Values | References |
|---|---|---|
| Relative molecular mass | 170.21 g/mol | Gatroplus |
| LogP | 0.8 | [24] |
| pKa | 16.1 | [24] |
| LogD | 0.9 | [24] |
| Rbp | 1.11 | Gatroplus predict |
| Water solubility | 1.04 g/mol | [25] |
| Fup | 97% | [26] |
| Intestinal permeability coefficient | 9.6 × 10−6 cm/s | [27] |
| Adult clearance rate | 0.96 mL/min/kg | [22] |
| Clearance rate of children in China | 0.88 mL/min/kg | [28] |
| Vd | 0.5~0.7 L/kg | [22] |
| Administration | Dose (mg) | Quantities | Age | Weight (kg) | BMI | References |
|---|---|---|---|---|---|---|
| IV | 1500 mg | 24 | 23.71 | 64.19 ± 7.38 | -- | [29] |
| Oral tablet | 1500 mg | 24 | 23.71 | 64.19 ± 7.38 | -- | [29] |
| Oral tablet | 1000 mg | 8 | 33.8 | 57.4 ± 3.70 | 22.1 ± 0.8 | [30] |
| Oral tablet | 750 mg | 8 | 21.0 | 58.8 ± 6.50 | 19~24 | [31] |
| Oral tablet | 500 mg | 8 | 21.0 | 58.8 ± 6.50 | 19~24 | [31] |
| Oral tablet | 250 mg | 8 | 21.0 | 58.8 ± 6.50 | 19~24 | [31] |
| Parameters | Formulation | ||||
|---|---|---|---|---|---|
| LEV-IDTs-250 mg | LEV-IDTs-500 mg | LEV-IDTs-750 mg | LEV-IDTs-1000 mg | Spritam® | |
| HL_Lambda_z (h) | 3.54 ± 0.32 | 3.36 ± 0.27 | 3.30 ± 0.32 | 3.14 ± 0.31 | 3.13 ± 0.46 |
| Tmax (h) | 1.00 ± 0.87 | 1.00 ± 0.87 | 1.67 ± 0.58 | 0.83 ± 0.29 | 0.28 ± 0.21 |
| Cmax (μg/mL) | 32.01 ± 1.88 | 55.54 ± 4.83 | 82.81 ± 5.14 | 118.89 ± 11.07 | 127.85 ± 7.09 |
| AUC(0–t) (h × μg/mL) | 176.16 ± 1.09 | 304.95 ± 31.34 | 510.01 ± 10.39 | 718.05 ± 93.24 | 717.68 ± 48.66 |
| AUC(0–∞) (h × μg/mL) | 177.82 ± 0.66 | 307.17 ± 32.22 | 513.44 ± 9.60 | 722.43 ± 94.59 | 721.98 ± 51.03 |
| MRTlast (h) | 4.68 ± 0.08 | 4.58 ± 0.48 | 4.81 ± 0.43 | 5.02 ± 0.40 | 4.52 ± 0.33 |
| Parameters. | Lower Limit | Upper Limit | Standard |
|---|---|---|---|
| AUC(0–∞) | 93.42% | 107.85% | 80–125% |
| AUC(0–t) | 93.23% | 108.09% | |
| Cmax | 99.35% | 116.81% |
| IV 1500 mg | Oral 250 mg | Oral 500 mg | Oral 750 mg | Oral 1000 mg | Oral 1500 mg | ||
|---|---|---|---|---|---|---|---|
| Cmax | Observed | 50.80 | 5.51 | 12.50 | 17.00 | 24.10 | 35.30 |
| Predicted | 54.83 | 6.268 | 12.54 | 18.80 | 25.90 | 35.43 | |
| PE | 7.93 | 13.76 | 0.32 | 10.59 | 7.47 | 0.37 | |
| AUC0–t | Observed | 367.20 | 62.56 | 138.50 | 189.80 | 222.80 | 402.30 |
| Predicted | 357.40 | 58.96 | 117.90 | 176.90 | 236.60 | 354.70 | |
| PE | 2.67 | 5.75 | 14.87 | 6.80 | 6.19 | 11.83 | |
| AUC0–inf | Observed | 370.70 | 72.56 | 145.40 | 196.10 | 226.40 | 408.30 |
| Predicted | 358.90 | 59.75 | 119.50 | 179.30 | 239.40 | 356.30 | |
| PE | 3.18 | 17.65 | 17.81 | 8.57 | 5.74 | 12.74 | |
| Tmax | Observed | 0.711 | 1.67 | 0.80 | 3.16 | 1.45 | 0.646 |
| Predicted | 0.75 | 1.32 | 1.32 | 1.32 | 1.30 | 1.38 | |
| PE | 5.49 | 20.96 | 65.00 | 58.23 | 10.34 | 113.62 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, X.; Liang, E.; Hong, X.; Han, X.; Li, C.; Wang, Y.; Wang, Z.; Zheng, A. In Vitro and In Vivo Bioequivalence Study of 3D-Printed Instant-Dissolving Levetiracetam Tablets and Subsequent Personalized Dosing for Chinese Children Based on Physiological Pharmacokinetic Modeling. Pharmaceutics 2022, 14, 20. https://doi.org/10.3390/pharmaceutics14010020
Li X, Liang E, Hong X, Han X, Li C, Wang Y, Wang Z, Zheng A. In Vitro and In Vivo Bioequivalence Study of 3D-Printed Instant-Dissolving Levetiracetam Tablets and Subsequent Personalized Dosing for Chinese Children Based on Physiological Pharmacokinetic Modeling. Pharmaceutics. 2022; 14(1):20. https://doi.org/10.3390/pharmaceutics14010020
Chicago/Turabian StyleLi, Xianfu, En Liang, Xiaoxuan Hong, Xiaolu Han, Conghui Li, Yuxi Wang, Zengming Wang, and Aiping Zheng. 2022. "In Vitro and In Vivo Bioequivalence Study of 3D-Printed Instant-Dissolving Levetiracetam Tablets and Subsequent Personalized Dosing for Chinese Children Based on Physiological Pharmacokinetic Modeling" Pharmaceutics 14, no. 1: 20. https://doi.org/10.3390/pharmaceutics14010020
APA StyleLi, X., Liang, E., Hong, X., Han, X., Li, C., Wang, Y., Wang, Z., & Zheng, A. (2022). In Vitro and In Vivo Bioequivalence Study of 3D-Printed Instant-Dissolving Levetiracetam Tablets and Subsequent Personalized Dosing for Chinese Children Based on Physiological Pharmacokinetic Modeling. Pharmaceutics, 14(1), 20. https://doi.org/10.3390/pharmaceutics14010020