
Polyphosphazene-Based Nanocarriers for the Release of Camptothecin and Epirubicin

 ,
,  , and
, and 
Abstract
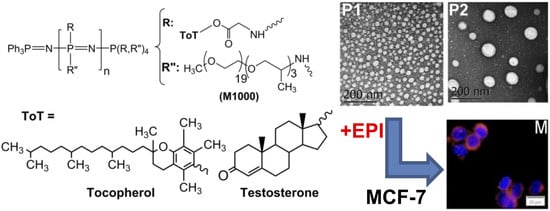
:
1. Introduction
2. Materials and Methods
2.1. Materials
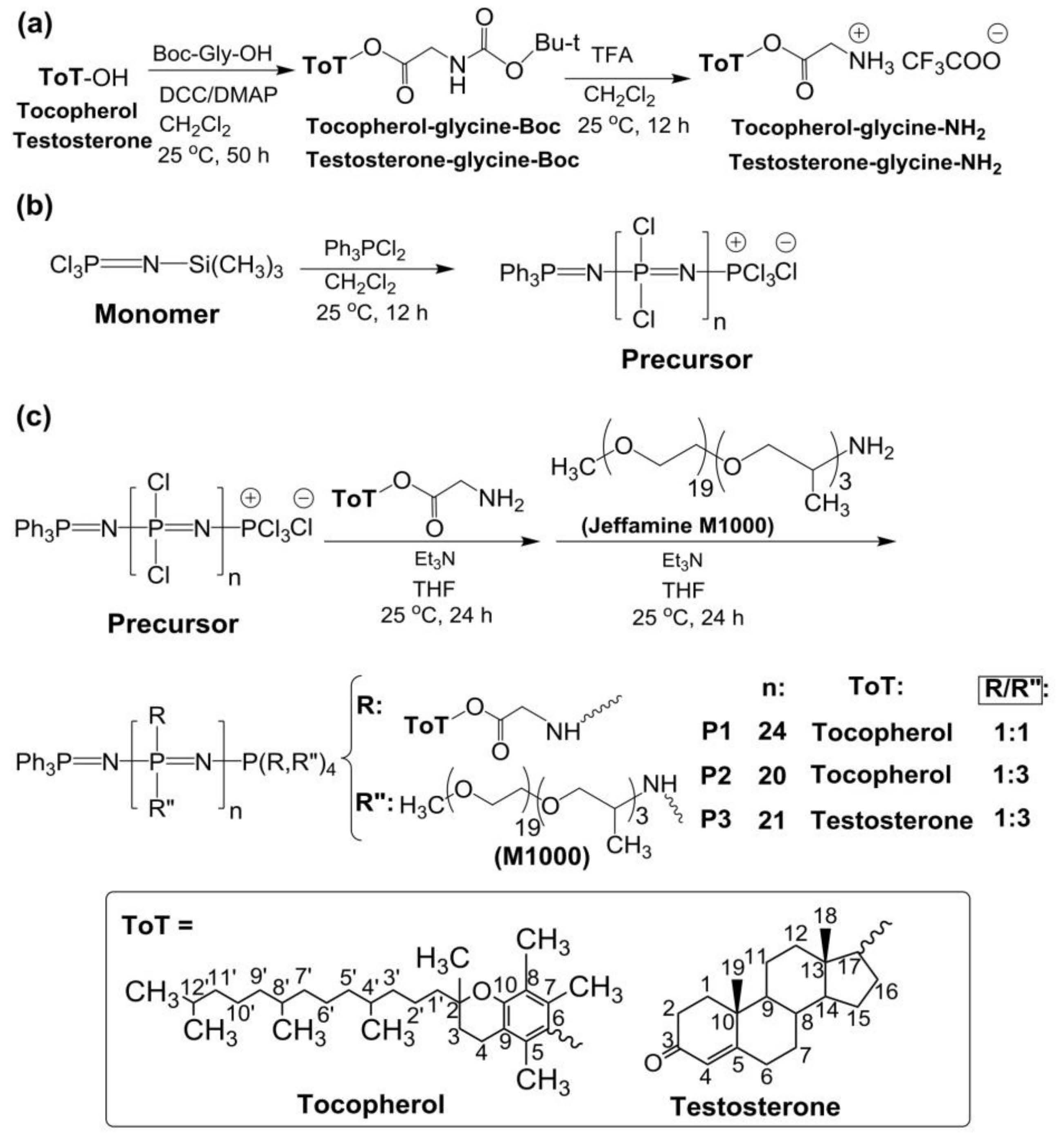
2.2. Synthesis of the Polymers
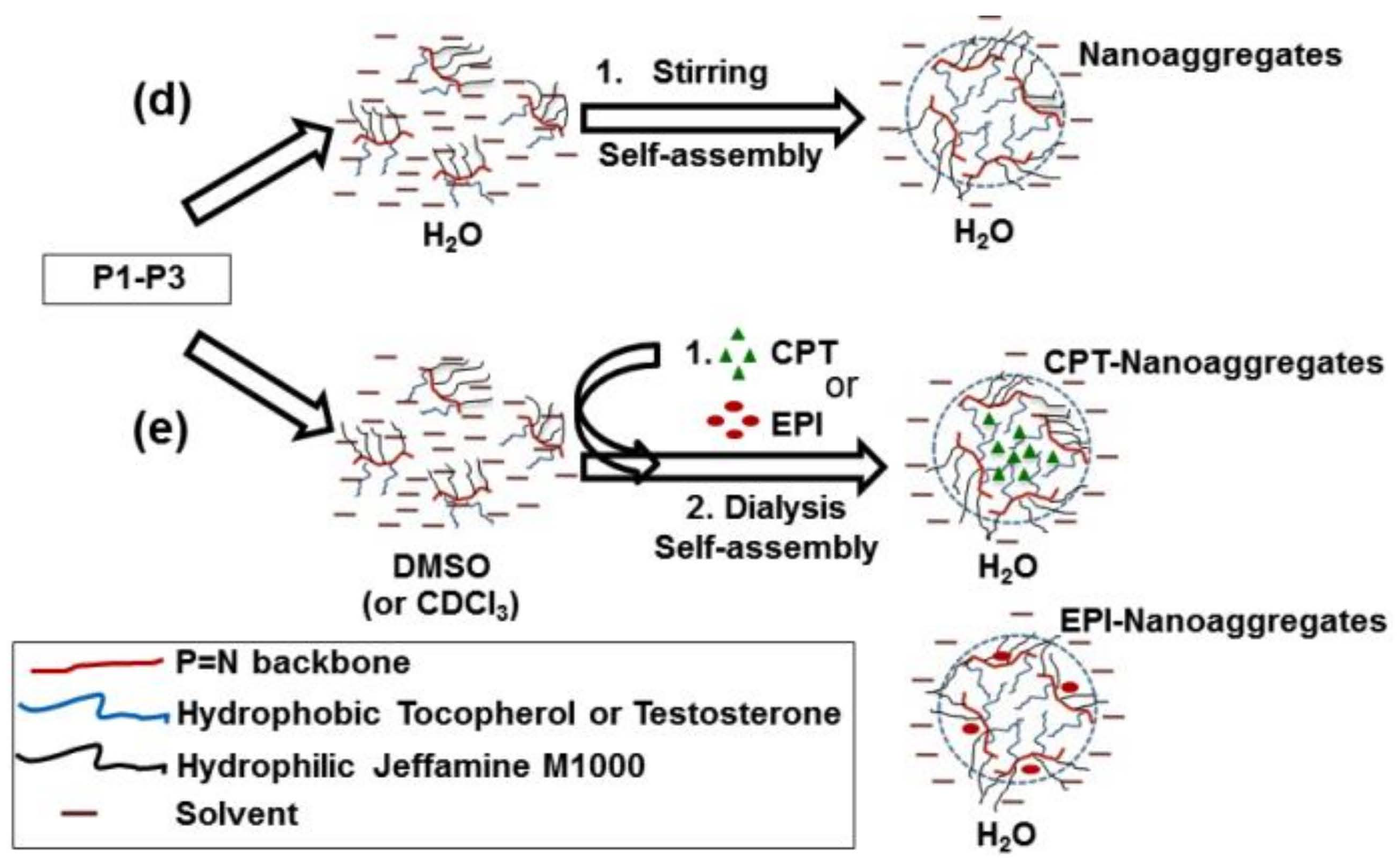
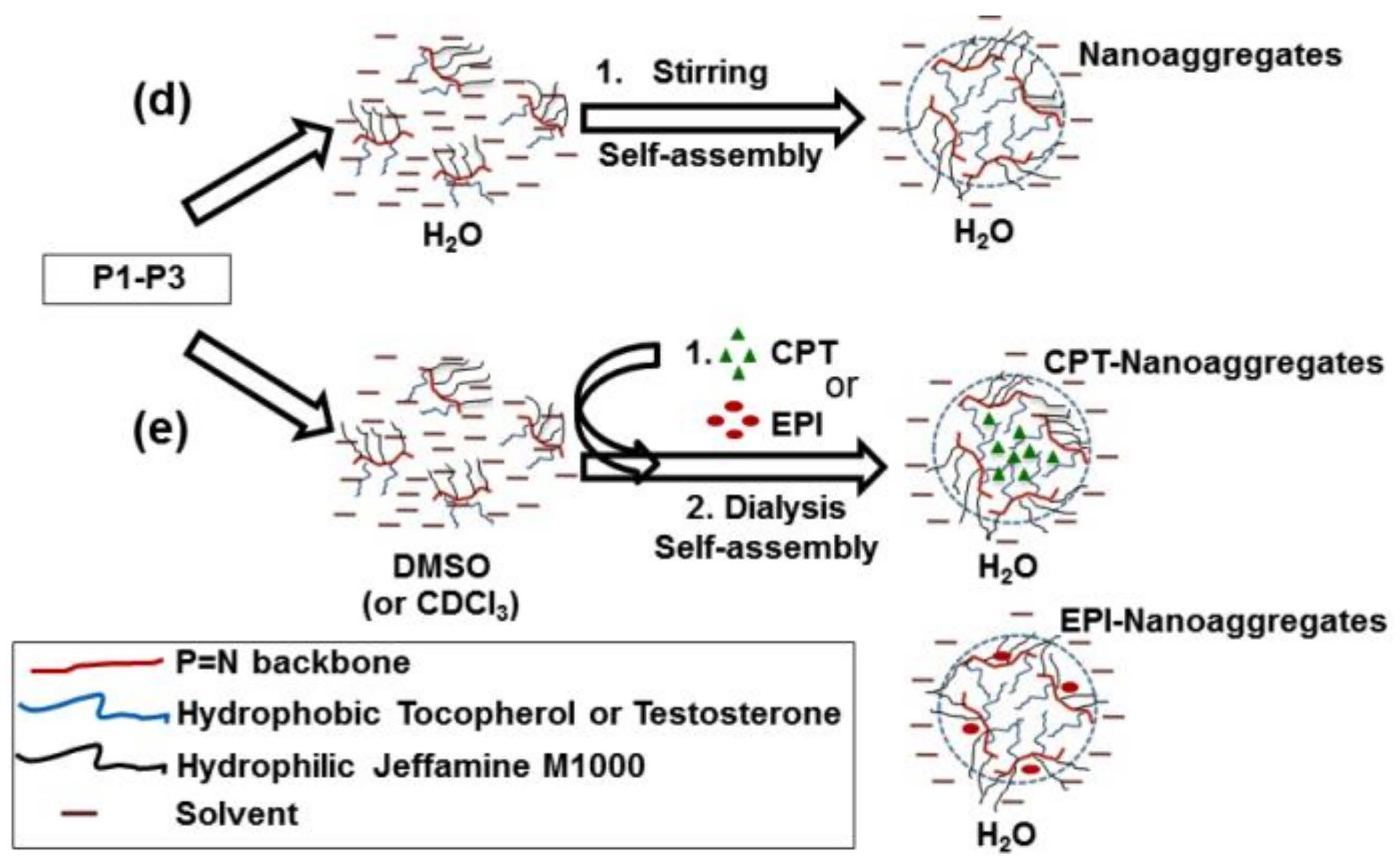
2.3. CPT and EPI Loading in Polyphosphazene Nanocarriers
2.4. Anticancer Drug Content and Drug Release Studies
2.5. Characterization
2.6. Cell Culture, Cell Uptake and Cytotoxicity Tests
2.7. Annexin V/PI Assay
2.8. Cell Cycle Analysis
2.9. Spheroid Generation
2.10. Compound Uptake into Spheroids
2.11. Cell Viability Analysis of Spheroids
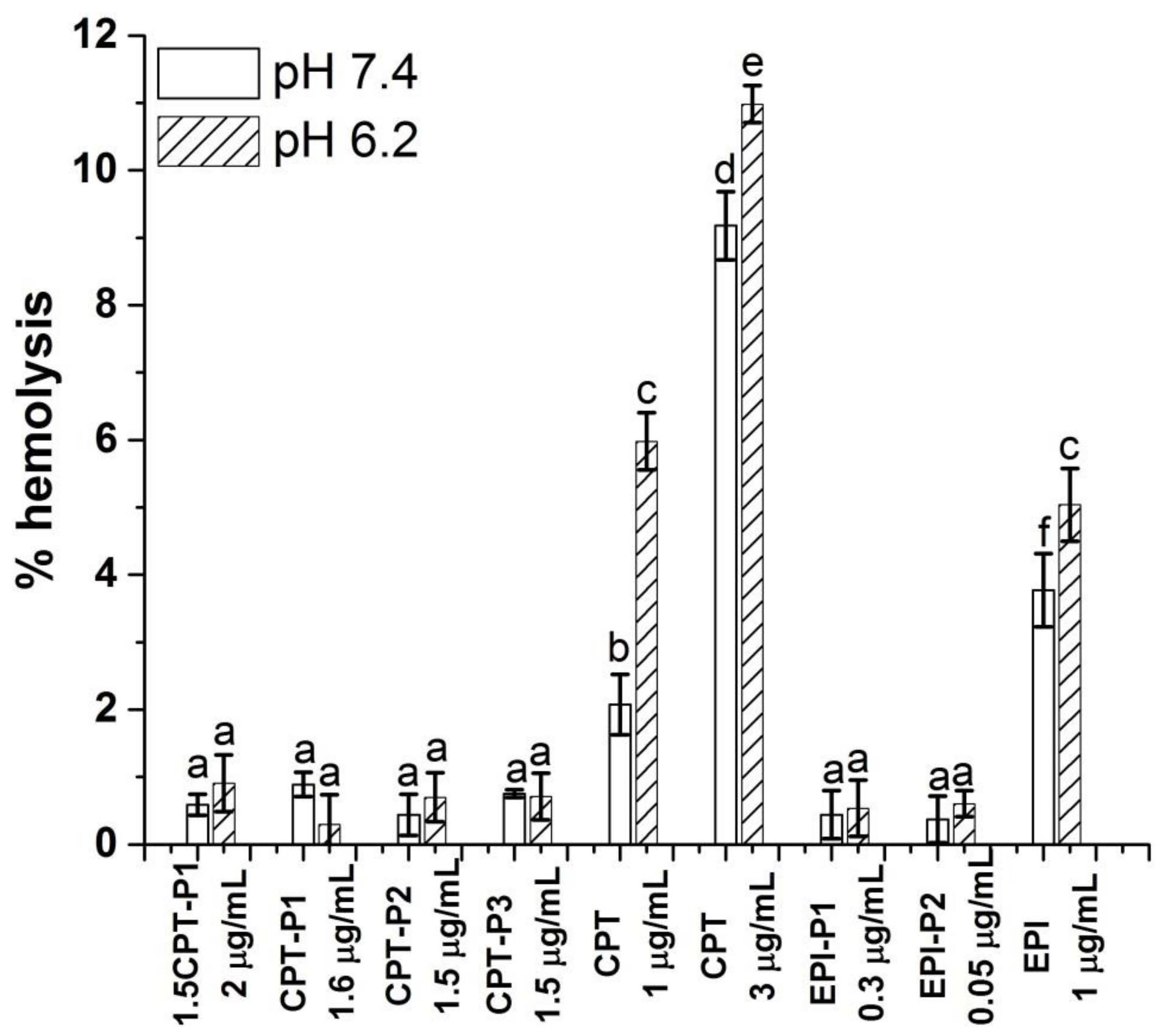
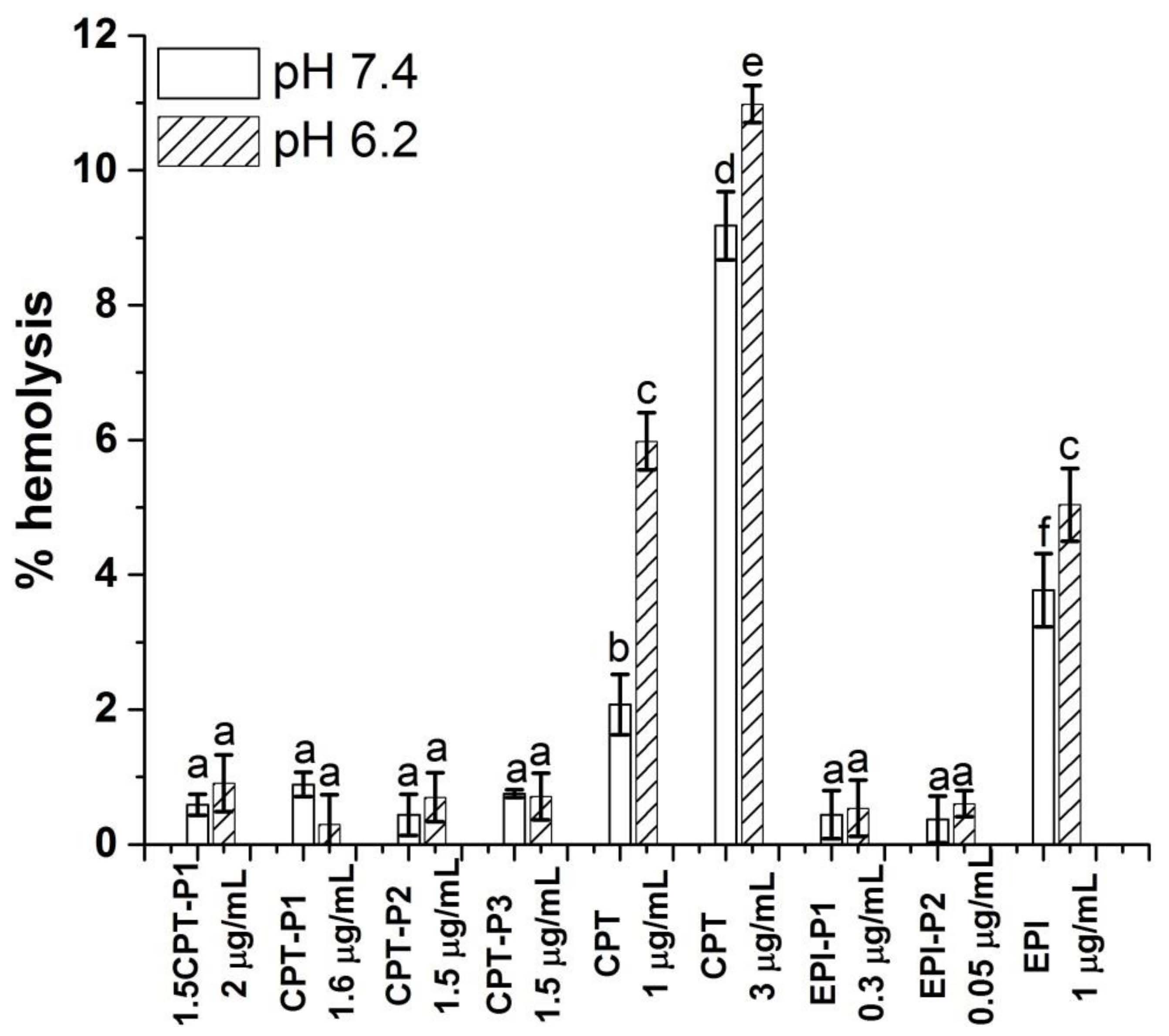
2.12. Hemolysis Assay
2.13. Statistical Analyses
3. Results and Discussion
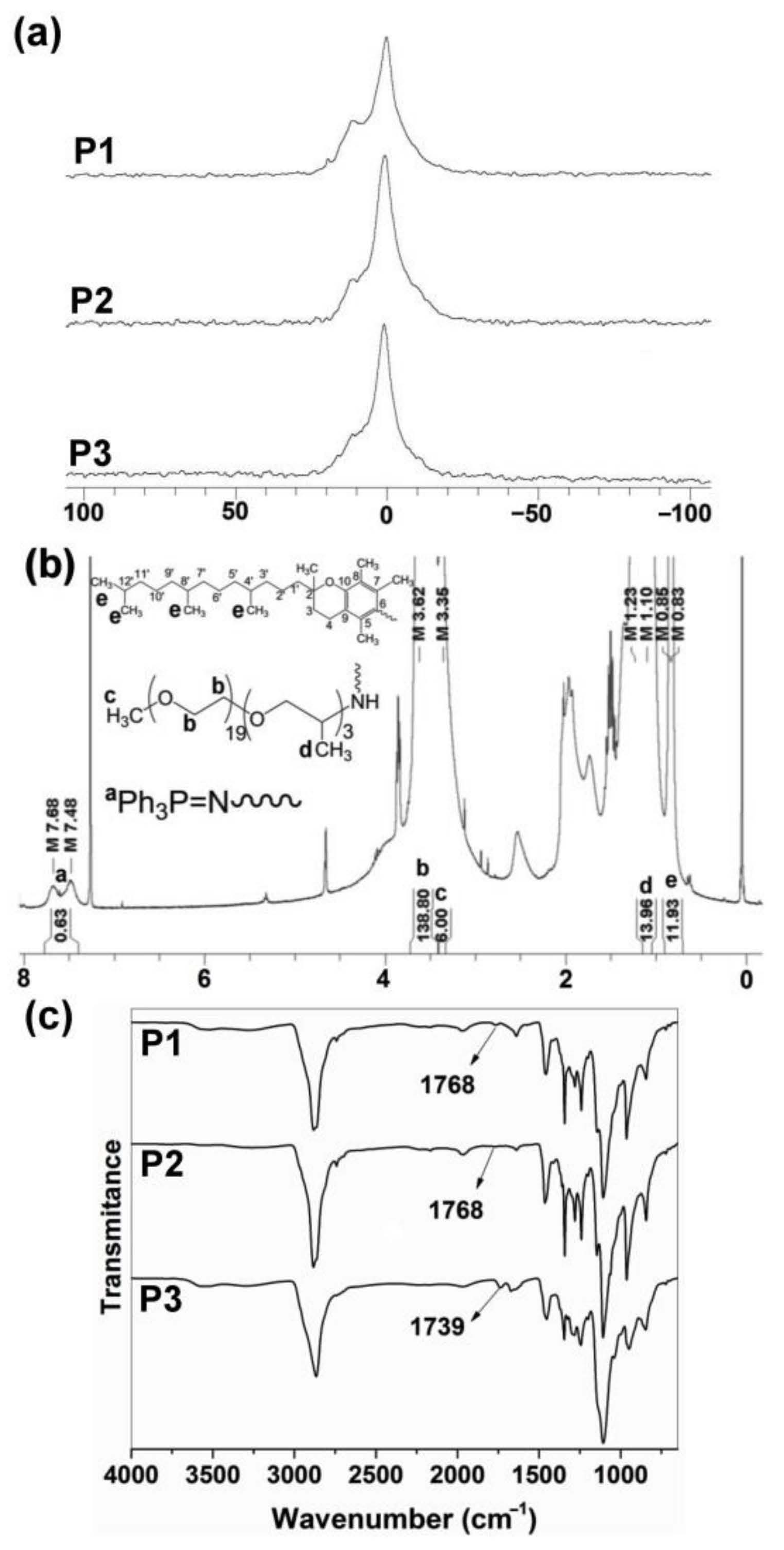
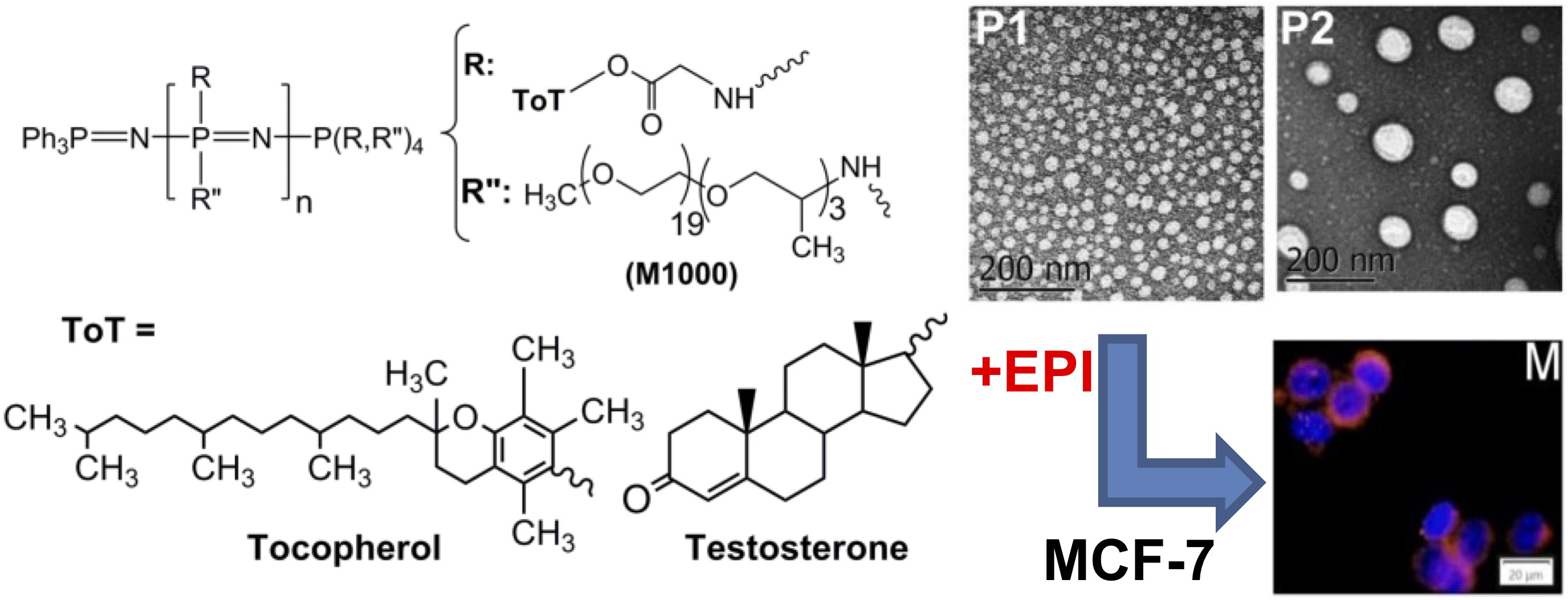
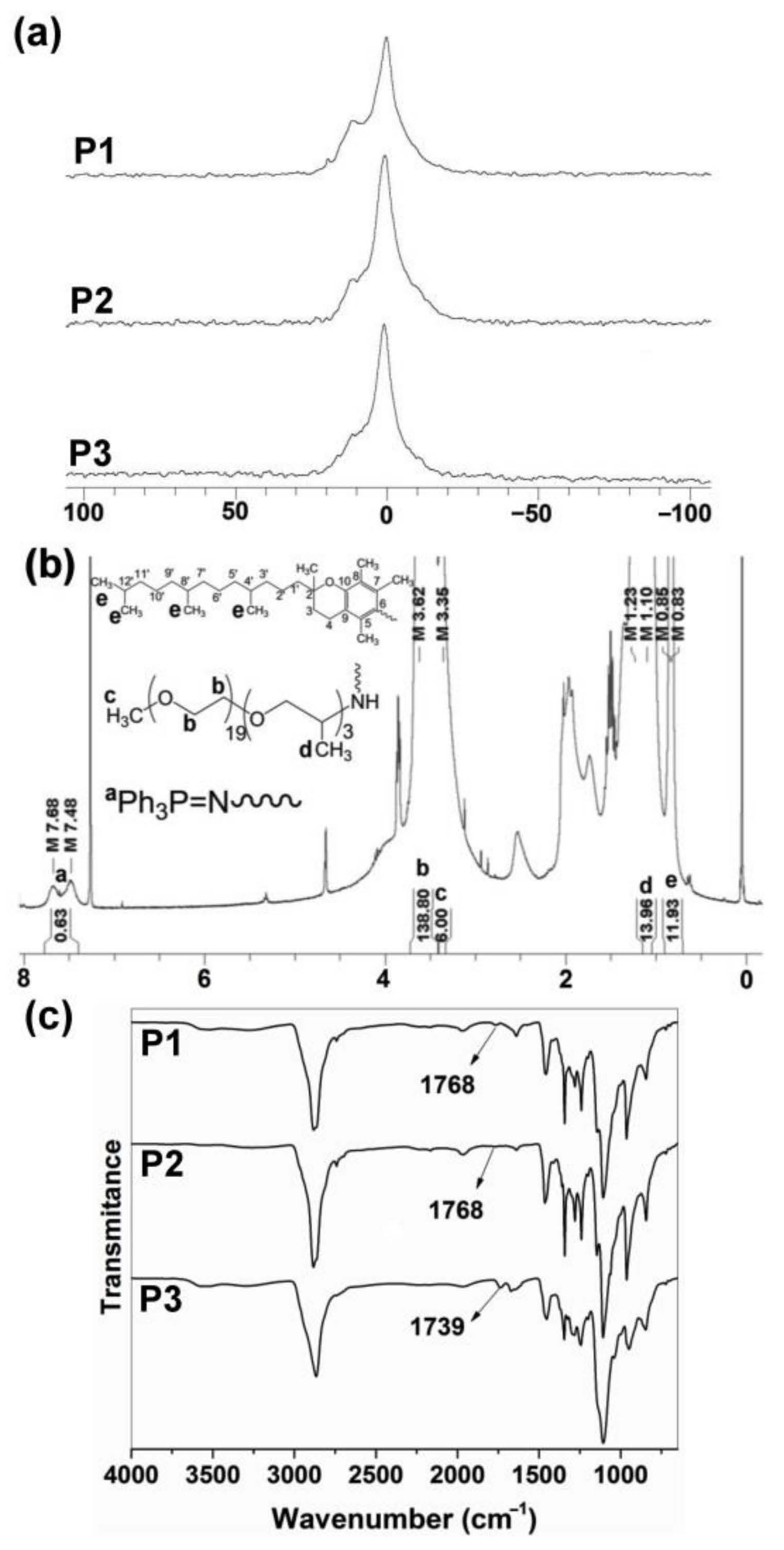
3.1. Synthesis and Characterization of Polyphosphazenes P1–P3
3.2. Structural Characterization and Morphologies
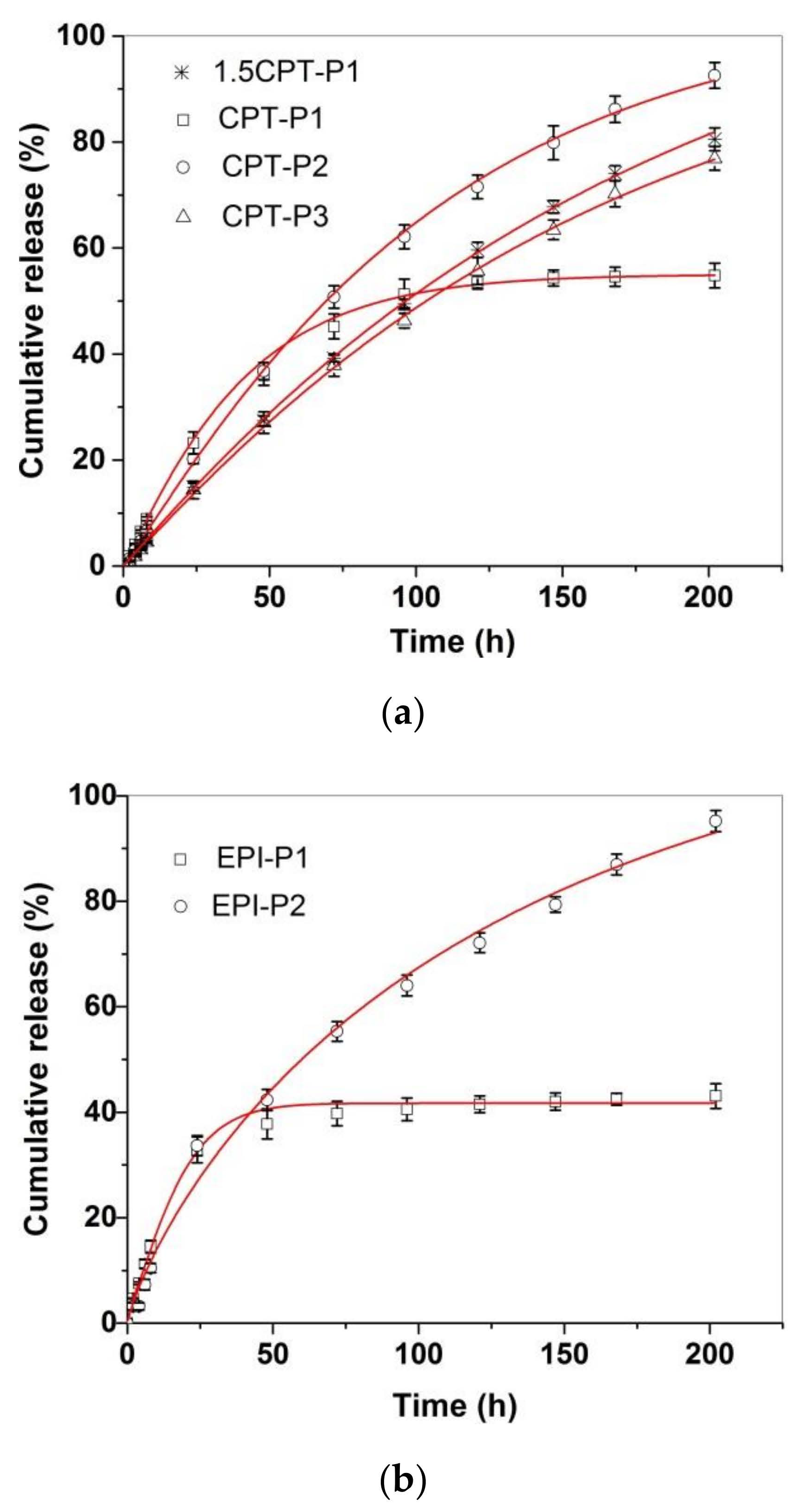
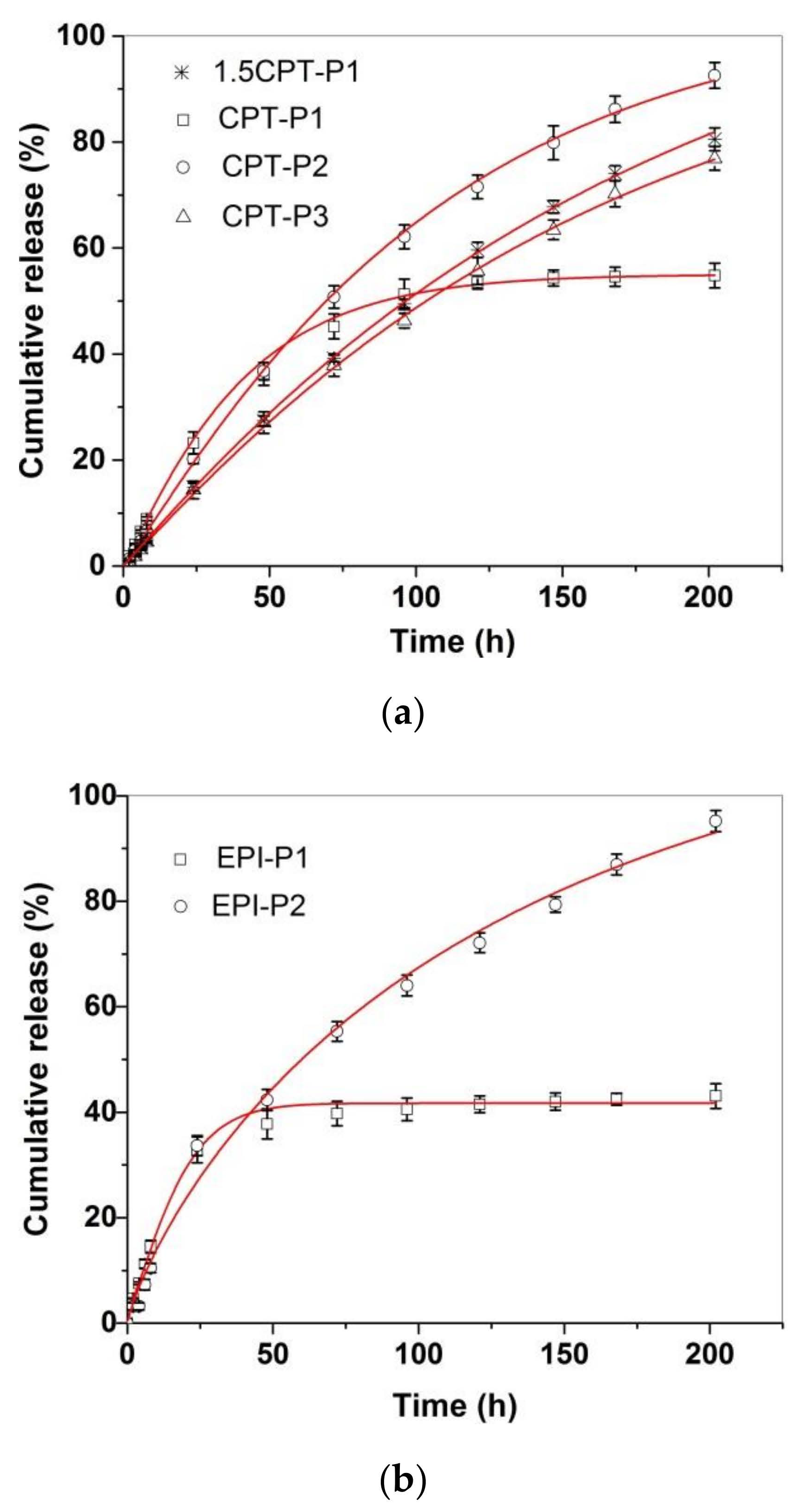
3.3. Drug Delivery Studies
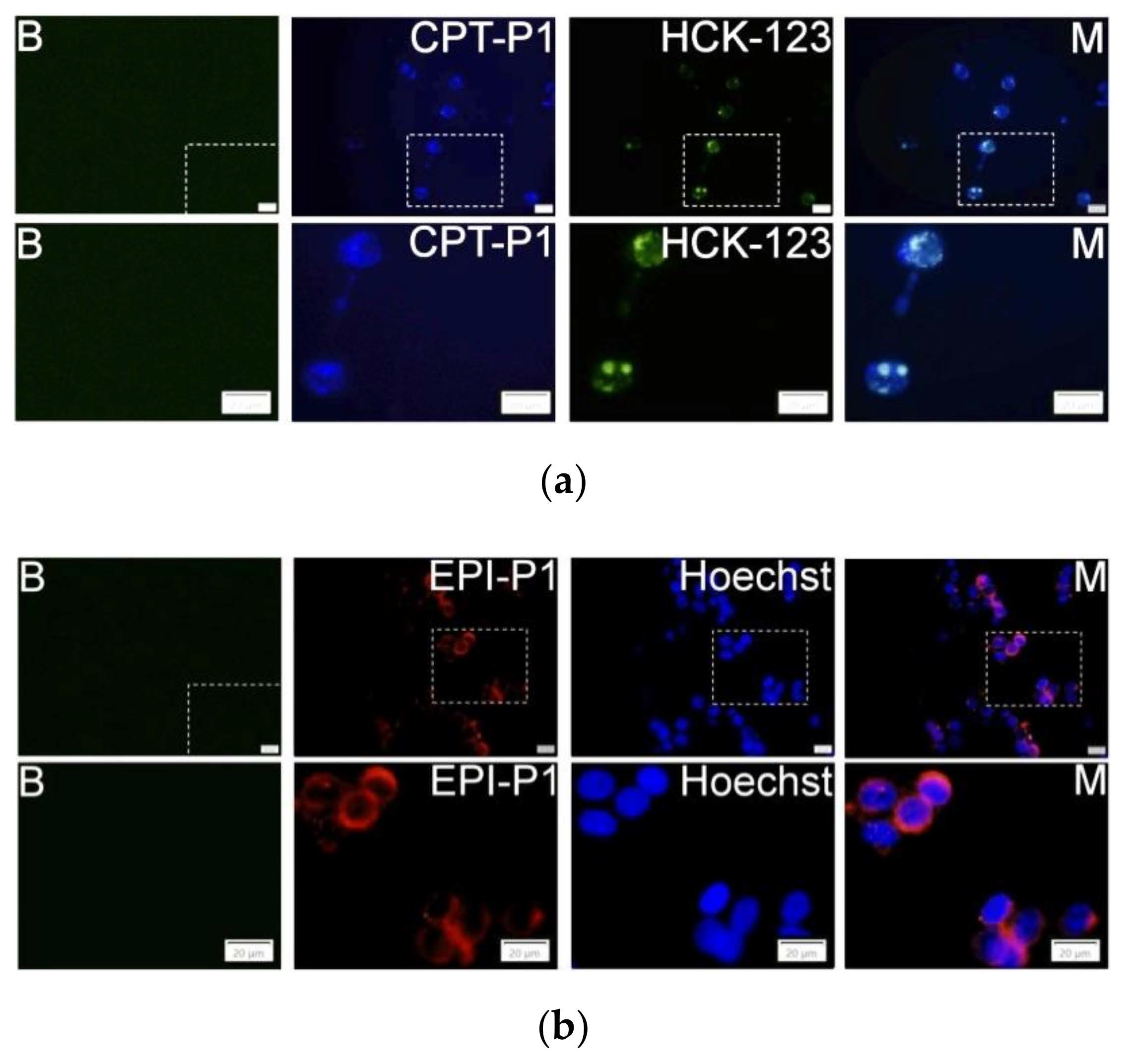
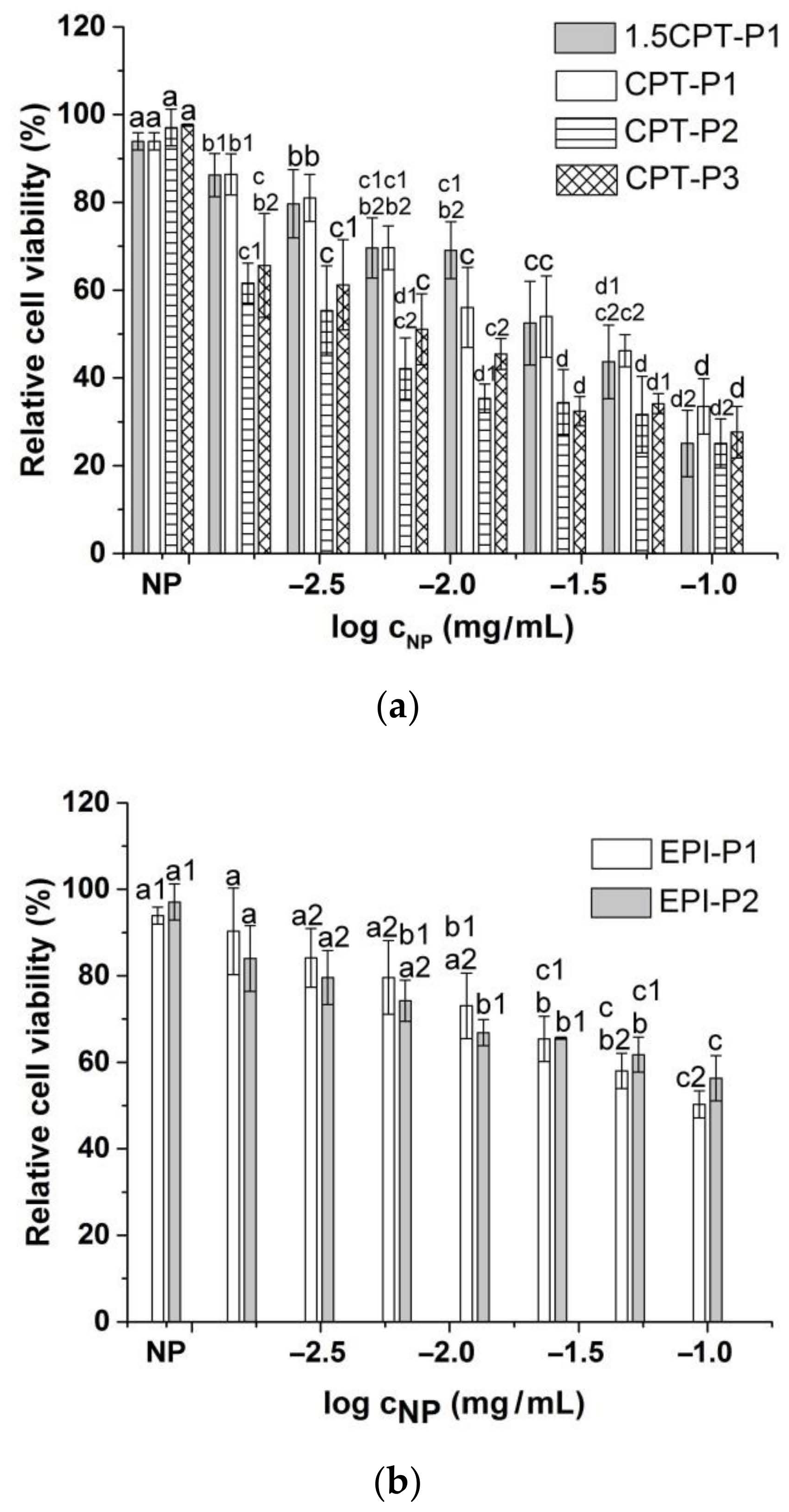
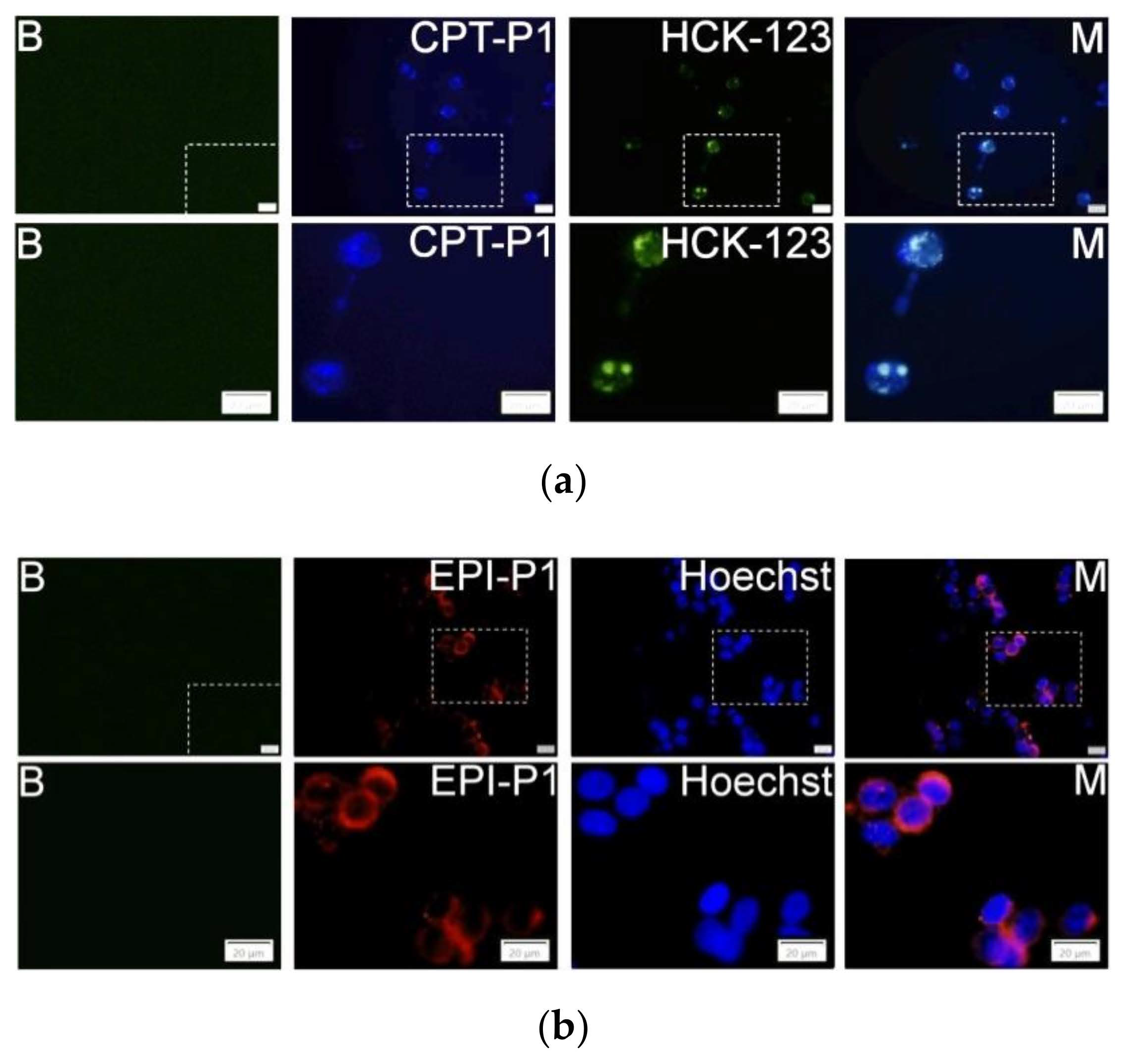
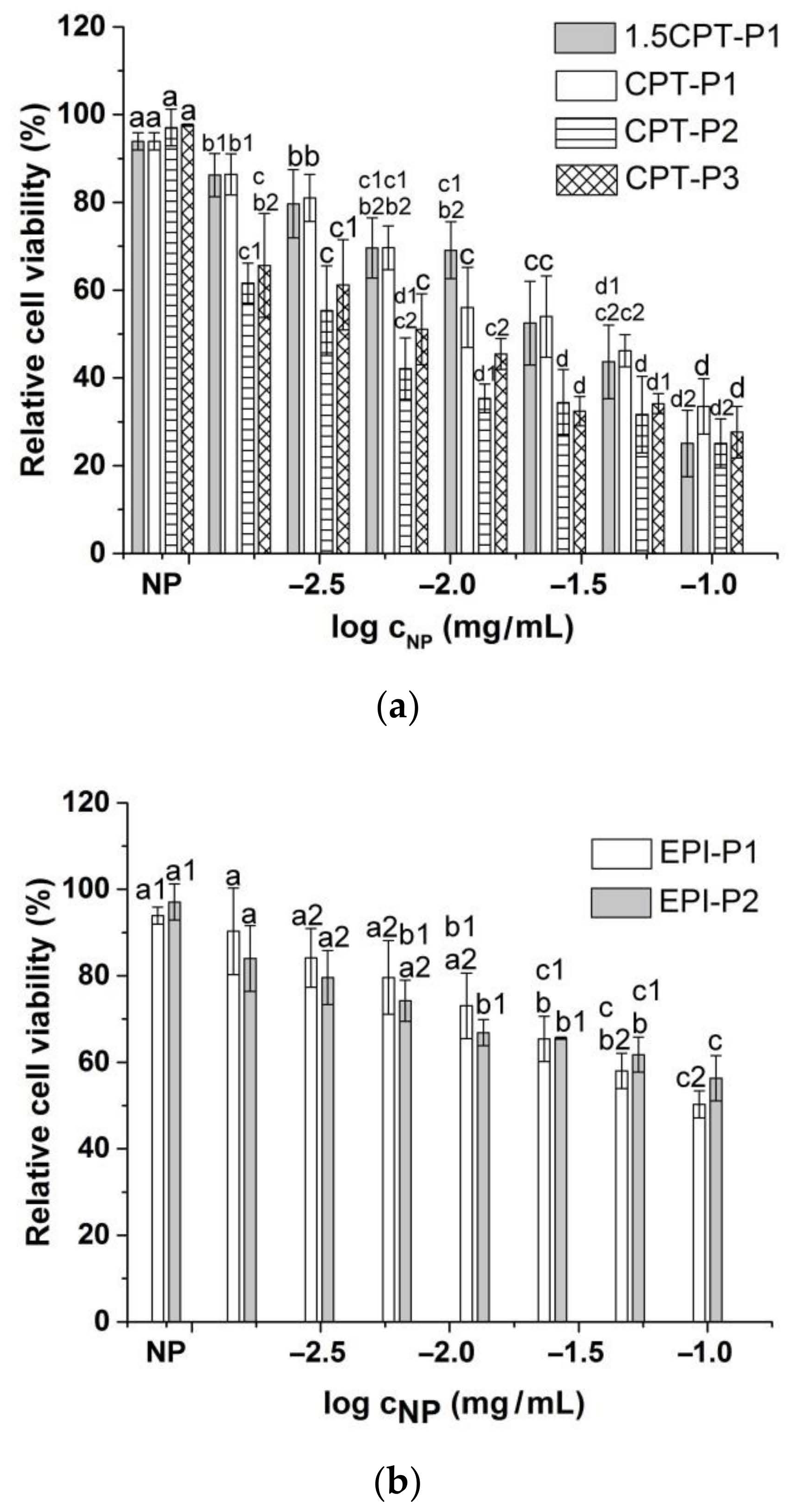
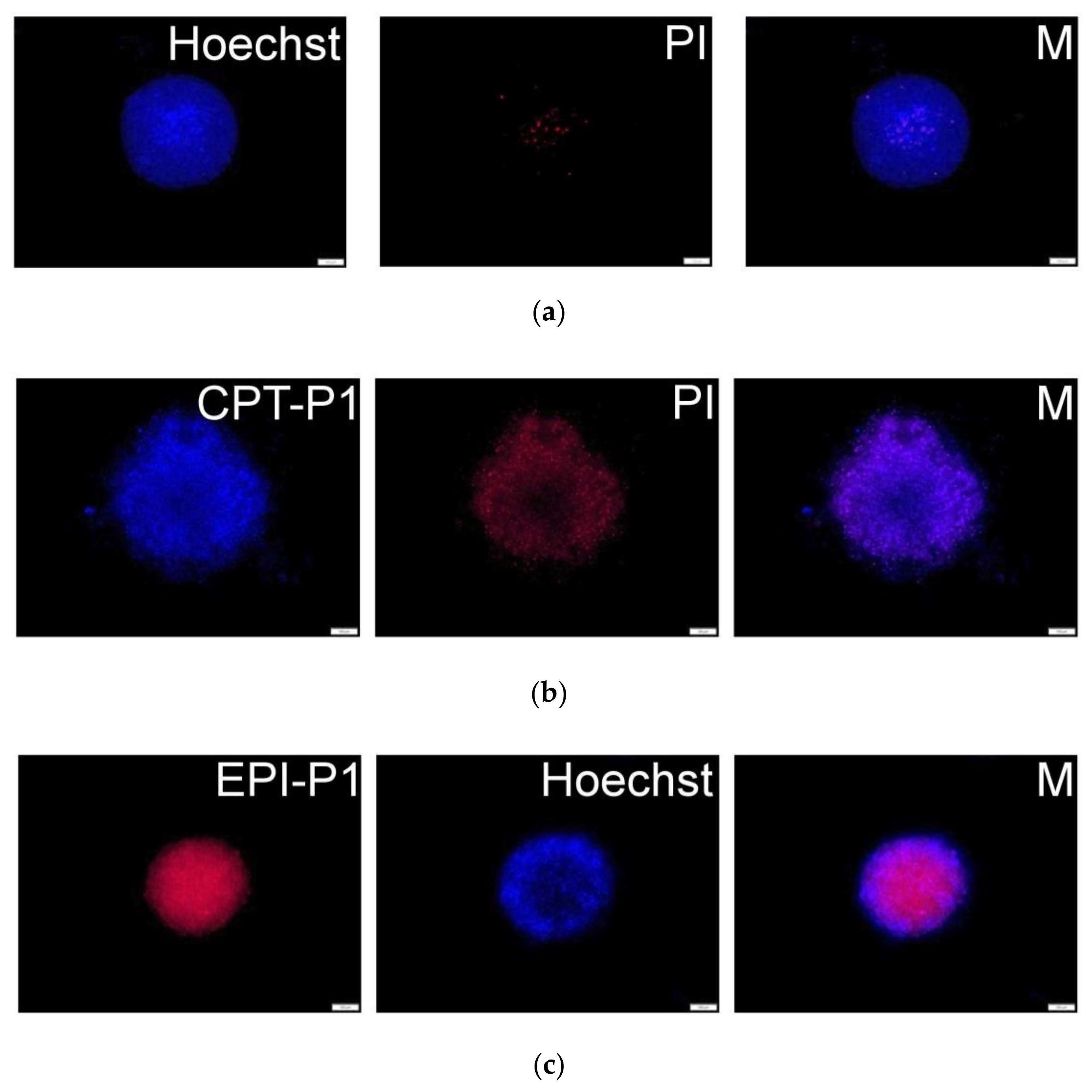
3.4. Cell Uptake and Cytotoxic Activity
3.5. Spheroid Uptake and Cytotoxic Activity
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jing, X.; Zhi, Z.; Jin, L.; Wang, F.; Wu, Y.; Wang, D.; Yan, K.; Shao, Y.; Meng, L. pH/redox dual-stimuli-responsive cross-linked polyphosphazene nanoparticles for multimodal imaging-guided chemo-photodynamic therapy. Nanoscale 2019, 11, 9457–9467. [Google Scholar] [CrossRef] [PubMed]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [Green Version]
- Rizvi, S.F.A.; Tariq, S.; Mehdi, M. Anthracyclines: Mechanism of Action, Classification, Pharmacokinetics and Future-A Mini Review. Int. J. Biotech. Bioeng. 2018, 4, 81–85. [Google Scholar]
- Pizzolato, J.F.; Saltz, L.B. The camptothecins. Lancet 2003, 361, 2235–2242. [Google Scholar] [CrossRef]
- Martino, E.; Volpe, S.D.; Terribile, E.; Benetti, E.; Sakaj, M.; Centamore, A.; Sala, A.; Collina, S. The long story of camptothecin: From traditional medicine to drugs. Bioorg. Med. Chem. Lett. 2017, 27, 701–707. [Google Scholar] [CrossRef] [PubMed]
- Botella, P.; Rivero-Buceta, E. Safe approaches for camptothecin delivery: Structural analogues and nanomedicines. J. Control. Release 2017, 247, 28–54. [Google Scholar] [CrossRef]
- Freitas, R.A. What is nanomedicine? Nanomed. Nanotechnol. Biol. Med. 2005, 1, 2–9. [Google Scholar] [CrossRef]
- Wiwanitkit, V. Advanced Nanomedicine and Nanobiotechnology; Nova Science Publishers, Inc.: New York, NY, USA, 2008. [Google Scholar]
- Cheng, C.J.; Tietjen, G.T.; Saucier-Sawyer, J.K.; Saltzman, W.M. A holistic approach to targeting disease with polymeric nanoparticles. Nat. Rev. Drug Discov. 2015, 14, 239–247. [Google Scholar] [CrossRef] [Green Version]
- Quiñones, J.P.; Jokinen, J.; Keinänen, S.; Covas, C.P.; Brüggemann, O.; Ossipov, D. Self-assembled hyaluronic acid-testosterone nanocarriers for delivery of anticancer drugs. Eur. Polym. J. 2018, 99, 384–393. [Google Scholar] [CrossRef]
- Quiñones, J.P.; Mardare, C.C.; Hassel, A.W.; Brüggemann, O. Testosterone- and vitamin-grafted cellulose ethers for sustained release of camptothecin. Carbohydr. Polym. 2019, 206, 641–652. [Google Scholar] [CrossRef] [PubMed]
- Sing, K.; Bhori, M.; Marar, T. α-Tocopherol mediated amelioration of camptothecin-induced free radical damage to avert cardiotoxicities. Hum. Exp. Toxicol. 2015, 34, 380–389. [Google Scholar] [CrossRef]
- Ingold, M.; Dapueto, R.; Victoria, S.; Galliusi, G.; Batthyany, C.; Bollati-Fogolín, M.; Tejedor, D.; García-Tellado, F.; Padrón, J.M.; Porcal, W.; et al. A green multicomponent synthesis of tocopherol analogues with antiproliferative activities. Eur. J. Med. Chem. 2018, 143, 1888–1902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glaser, R.; York, A.E.; Dimitrakis, C.D. Subcutaneous testosterone-letrozole therapy before and concurrent with neoadjuvant breast chemotherapy: Clinical response and therapeutic implications. Menopause 2017, 24, 859–864. [Google Scholar] [CrossRef] [Green Version]
- Teasdale, I.; Brüggemann, O. Polyphosphazenes for Medical Applications; Smithers Rapra: Shrewsbury, UK, 2014. [Google Scholar]
- Teasdale, I.; Brüggemann, O. Polyphosphazenes: Multifunctional, biodegradable vehicles for drug and gene delivery. Polymers 2013, 5, 161–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilfert, S.; Iturmendi, A.; Schoefberger, W.; Kryeziu, K.; Heffeter, P.; Berger, W.; Brüggemann, O.; Teasdale, I. Water-soluble, biocompatible polyphosphazenes with controllable and pH-promoted degradation behaviour. J. Polym. Sci. Part A Polym. Chem. 2014, 52, 287–294. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Zhao, Q.; Jin, Y.; Qiu, L. High loading of hydrophilic/hydrophobic doxorubicin into polyphosphazene polymersome for breast cancer therapy. Nanomedicine 2014, 10, 349–358. [Google Scholar] [CrossRef]
- Cho, J.-K.; Chun, C.; Kuh, H.-J.; Song, S.-C. Injectable poly (organophosphazene)-camptothecin conjugate hydrogels: Synthesis, characterization, and antitumor activities. Eur. J. Pharm. Biopharm. 2012, 81, 582–590. [Google Scholar] [CrossRef]
- Teasdale, I.; Wilfert, S.; Nischang, I.; Brüggemann, O. Multifunctional and biodegradable polyphosphazenes for use as macromolecular anti-cancer drug carriers. Polym. Chem. 2011, 2, 828–834. [Google Scholar]
- Schwach, G.; Pfragner, R.; Hoeger, H.; Schoefberger, W.; Wilfert, S.; Brüggemann, O.; Teasdale, I. Epirubicin Loaded Polyphosphazene; A New Agent against Small Intestinal Neuroendocrine Tumors and Medullary Thyroid Carcinomas. Neuroendocrinology 2015, 102, 161. [Google Scholar]
- Henke, H.; Kryeziu, K.; Banfic, J.; Theiner, S.; Koerner, W.; Brueggemann, O.; Berger, W.; Keppler, B.K.; Heffeter, P.; Teasdale, I. Macromolecular Pt (IV) prodrugs from poly (organo) phosphazenes. Macromol. Biosci. 2016, 16, 1239–1249. [Google Scholar] [CrossRef]
- Hackl, C.M.; Schoenhacker-Alte, B.; Klose, M.H.M.; Henke, H.; Legina, M.S.; Jakupec, M.A.; Berger, W.; Keppler, B.K.; Bruggemann, O.; Teasdale, I.; et al. Synthesis and in vivo anticancer evaluation of poly (organo) phosphazene-based metallodrug conjugates. Dalton Trans. 2017, 46, 12114–12124. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Rivard, E.; Manners, I. A new high-yield synthesis of Cl3PNSiMe3. Inorg. Chem. 2002, 41, 1690–1691. [Google Scholar] [CrossRef] [PubMed]
- Rothemund, S.; Teasdale, I. Preparation of polyphosphazenes: A tutorial review. Chem. Soc. Rev. 2016, 45, 5200–5215. [Google Scholar] [CrossRef] [Green Version]
- Wilfert, S.; Henke, H.; Schoefberger, W.; Brüggemann, O.; Teasdale, I. Chain-End-Functionalized Polyphosphazenes via a One-Pot Phosphine-Mediated Living Polymerization. Macromol. Rapid Commun. 2014, 35, 1135–1141. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, A.V. New light on Chonermorpha grandiflora, (Roth) M.R. and S.M. Almeida, a medicinal plant for Tribal health. J. Pharmacogn. Phytochem. 2020, 9, 127–129. [Google Scholar]
- Degambada, K.D.; Kumara, P.A.A.S.P.; Salim, N.; Abeysekera, A.M.; Chandrika, U.G. Diaporthe sp. F18; A new source of camptothecin-producing endophytic fungus from Nothapodytes nimmoniana growing in Sri Lanka. Nat. Prod. Res. 2021, 1–16. [Google Scholar] [CrossRef]
- Pashaei, Y.; Mehrabi, M.; Shekarchi, M. A review on various analytical methods for determination of anthracyclines and their metabolites as anti-cancer chemotherapy drugs in different matrices over the last four decades. TrAC Trends Anal. Chem. 2020, 130, 115991. [Google Scholar] [CrossRef]
- König, M.; Linhardt, A.; Brüggemann, O.; Teasdale, I. Phosphine functionalized polyphosphazenes: Soluble and re-usable polymeric reagents for highly efficient halogenations under Appel conditions. Monatsh. Chem. 2016, 147, 1575–1582. [Google Scholar] [CrossRef]
- Zhang, J.X.; Qiu, L.Y.; Jin, Y.; Zhu, K.J. Thermally responsive polymeric micelles self-assembled by amphiphilic polyphosphazene with poly(N-isopropylacrylamide) and ethyl glycinate as side groups: Polymer synthesis, characterization, and in vitro drug release study. J. Biomed. Mater. Res. Part A 2006, 76, 773–780. [Google Scholar] [CrossRef]
- Linhardt, A.; König, M.; Schofberger, W.; Brüggemann, O.; Andrianov, A.K.; Teasdale, I. Biodegradable polyphosphazene based peptide-polymer hybrids. Polymers 2016, 8, 161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quiñones, J.P.; Peniche, H.; Peniche, C. Chitosan based self-assembled nanoparticles in drug delivery. Polymers 2018, 10, 235. [Google Scholar] [CrossRef] [Green Version]
- Quiñones, J.P.; Brüggemann, O.; Kjems, J.; Shahavi, M.H.; Peniche, C. Novel Brassinosteroid-Modified Polyethylene Glycol Micelles for Controlled Release of agrochemicals. J. Agric. Food Chem. 2018, 66, 1612–1619. [Google Scholar] [CrossRef] [PubMed]
- Cortesi, R.; Esposito, E.; Meietti, A.; Menegatti, E.; Nastruzzi, C. Formulation study for the antitumor drug camptothecin: Liposomes, micellar solutions and a microemulsion. Int. J. Pharm. 1997, 159, 95–103. [Google Scholar] [CrossRef]
- Zhang, L.; Cao, Z.; Li, Y.; Ella-Menye, J.-R.; Bai, T.; Jiang, S. Softer zwitterionic nanogels for longer circulation and lower splenic accumulation. ACS Nano 2012, 6, 6681–6686. [Google Scholar] [CrossRef] [PubMed]
- Haag, R.; Kratz, F. Polymer therapeutics: Concepts and applications. Angew. Chem. Int. Ed. 2006, 45, 1198–1215. [Google Scholar] [CrossRef]
- Dash, S.; Murthy, P.N.; Nath, L.; Chowdury, P. Kinetic modelling on drug release from controlled delivery systems. Acta Pol. Pharm. 2010, 67, 217–223. [Google Scholar] [PubMed]
- Kondaveeti, S.; Damato, T.C.; Carmona-Ribeiro, A.M.; Sierakowski, M.R.; Petri, D.F.S. Sustainable hydroxypropyl methylcellulose/xyloglucan/gentamicin films with antimicrobial properties. Carbohydr. Polym. 2017, 165, 285–293. [Google Scholar] [CrossRef]
- Totten, J.D.; Wongpinyochit, T.; Seib, F.P. Silk nanoparticles: Proof of lysosomotropic anticancer drug delivery at single-cell resolution. J. Drug Target. 2017, 25, 865–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prabhakar, U.; Maeda, H.; Jain, R.K.; Sevick-Muraca, E.M.; Zamboni, W.; Farokzhad, O.C.; Barry, S.T.; Gabizon, A.; Grodzinski, P.; Blakey, C. Challenges and key considerations of the enhanced permeability and retention effect for nanomedicine drug delivery in oncology. Cancer Res. 2013, 73, 2412–2417. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | DP 1 | ToT:M1000 2 | Y 3 (%) | MnTheor(kg/mol) 4 | Mnexp(kg/mol) 5 | Mw/Mn 5 |
|---|---|---|---|---|---|---|
| P1 | 24 | 1:1 | 56 | 39 | 14 | 1.35 |
| P2 | 20 | 1:3 | 44 | 45 | 19 | 1.24 |
| P3 | 21 | 1:3 | 48 | 43 | 17 | 1.48 |
| Sample | dh1 nm (PDI) | dh2 nm (PDI) | dAFM3 nm | wt% 4 | EE% 5 |
|---|---|---|---|---|---|
| P1 | 21 ± 1 a (0.29) | 22 ± 3 a (0.30) | 27 ± 9 | - | - |
| P2 | 108 ± 8 (0.58) | 127 ± 3 (0.42) | 42 ± 5 | - | - |
| P3 | 134 ± 2 (0.55) | 96 ± 2 (0.61) | 58 ± 6 | - | - |
| 1.5CPT-P1 | - | 198 ± 11 b (0.54) | - | 13.6 | 83 |
| CPT-P1 | - | 142 ± 1 (0.69) | - | 10.7 | 94 |
| CPT-P2 | - | 158 ± 3 (0.54) | - | 9.2 | 91 |
| CPT-P3 | - | 194 ± 5 b (0.29) | - | 9.6 | 87 |
| EPI-P1 | - | 250 ± 9 c (0.65) | - | 2.4 | 23 |
| EPI-P2 | - | 253 ± 4 c (0.52) | - | 0.3 | 3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Quiñones, J.P.; Roschger, C.; Iturmendi, A.; Henke, H.; Zierer, A.; Peniche-Covas, C.; Brüggemann, O. Polyphosphazene-Based Nanocarriers for the Release of Camptothecin and Epirubicin. Pharmaceutics 2022, 14, 169. https://doi.org/10.3390/pharmaceutics14010169
Quiñones JP, Roschger C, Iturmendi A, Henke H, Zierer A, Peniche-Covas C, Brüggemann O. Polyphosphazene-Based Nanocarriers for the Release of Camptothecin and Epirubicin. Pharmaceutics. 2022; 14(1):169. https://doi.org/10.3390/pharmaceutics14010169
Chicago/Turabian StyleQuiñones, Javier Pérez, Cornelia Roschger, Aitziber Iturmendi, Helena Henke, Andreas Zierer, Carlos Peniche-Covas, and Oliver Brüggemann. 2022. "Polyphosphazene-Based Nanocarriers for the Release of Camptothecin and Epirubicin" Pharmaceutics 14, no. 1: 169. https://doi.org/10.3390/pharmaceutics14010169
APA StyleQuiñones, J. P., Roschger, C., Iturmendi, A., Henke, H., Zierer, A., Peniche-Covas, C., & Brüggemann, O. (2022). Polyphosphazene-Based Nanocarriers for the Release of Camptothecin and Epirubicin. Pharmaceutics, 14(1), 169. https://doi.org/10.3390/pharmaceutics14010169