
Targeted EV to Deliver Chemotherapy to Treat Triple-Negative Breast Cancers



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines and Media
2.2. Patient Tissue Microarray (TMA) and Immunohistochemistry (IHC) Staining
2.3. Anti-CD47 mAb Production
2.4. Construction of mAb-EV-Ver-A, -Cy5 or -Cy7
2.5. In Vitro Anti-TNBC Cytotoxicity Assay
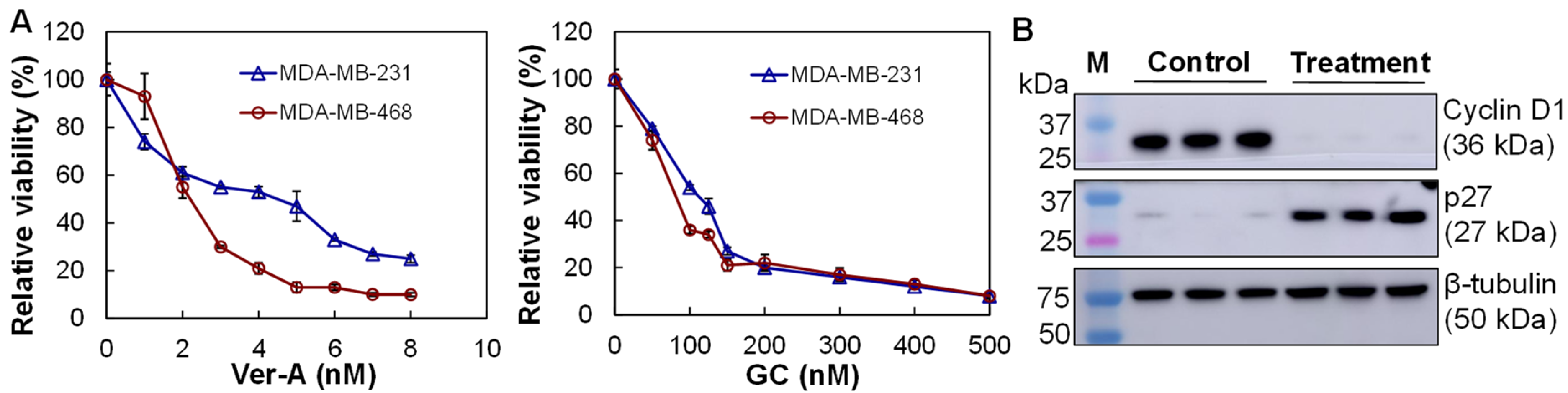
2.6. Western Blotting
2.7. Flow Cytometry
2.8. Live-Cell Confocal Imaging
2.9. In Vivo Imaging System (IVIS) Imaging
2.10. Primary TNBC Xenograft Models and In Vivo Treatment
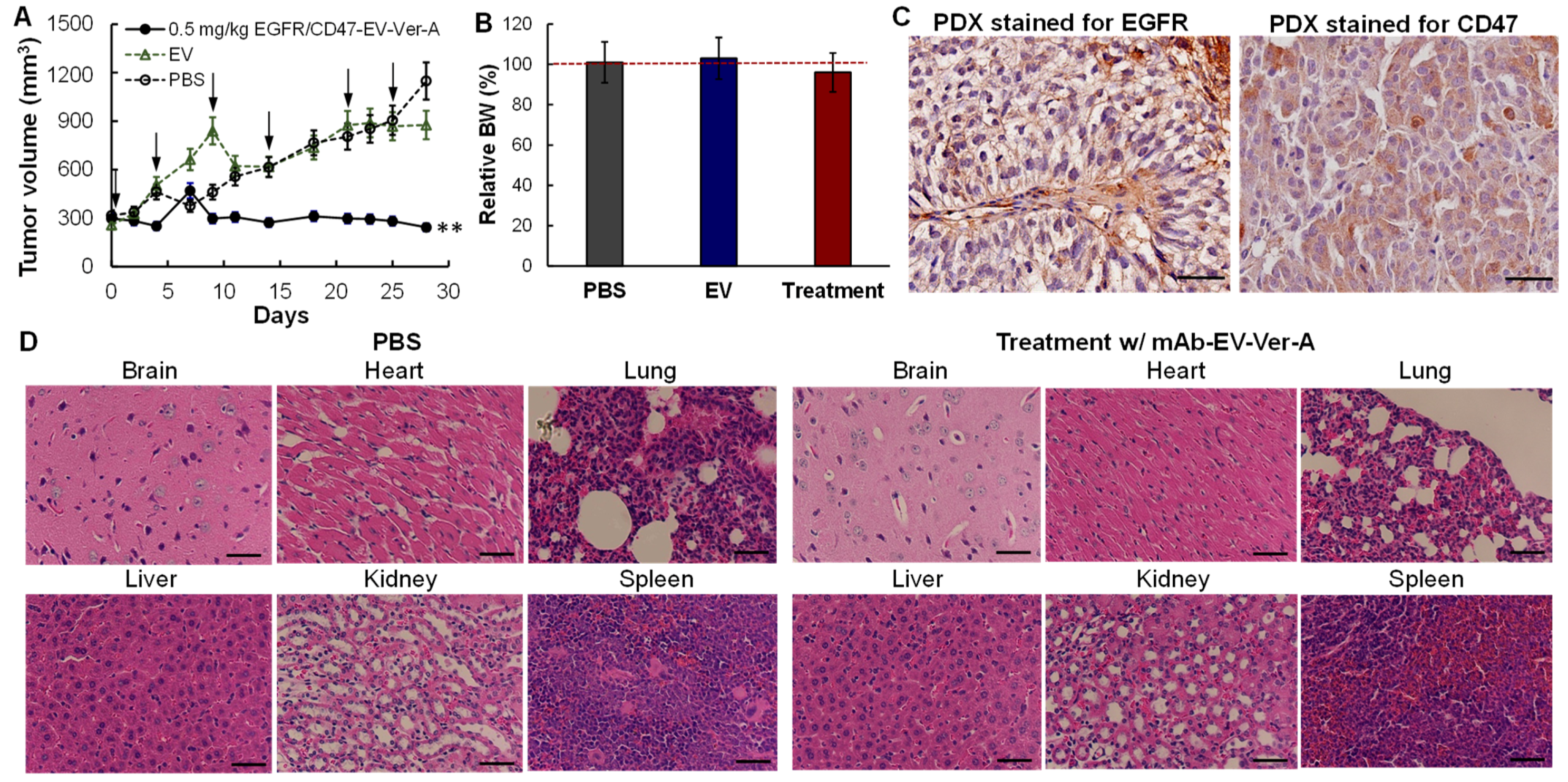
2.11. TNBC Patient-Derived Xenograft (PDX) Models and In Vivo Treatment
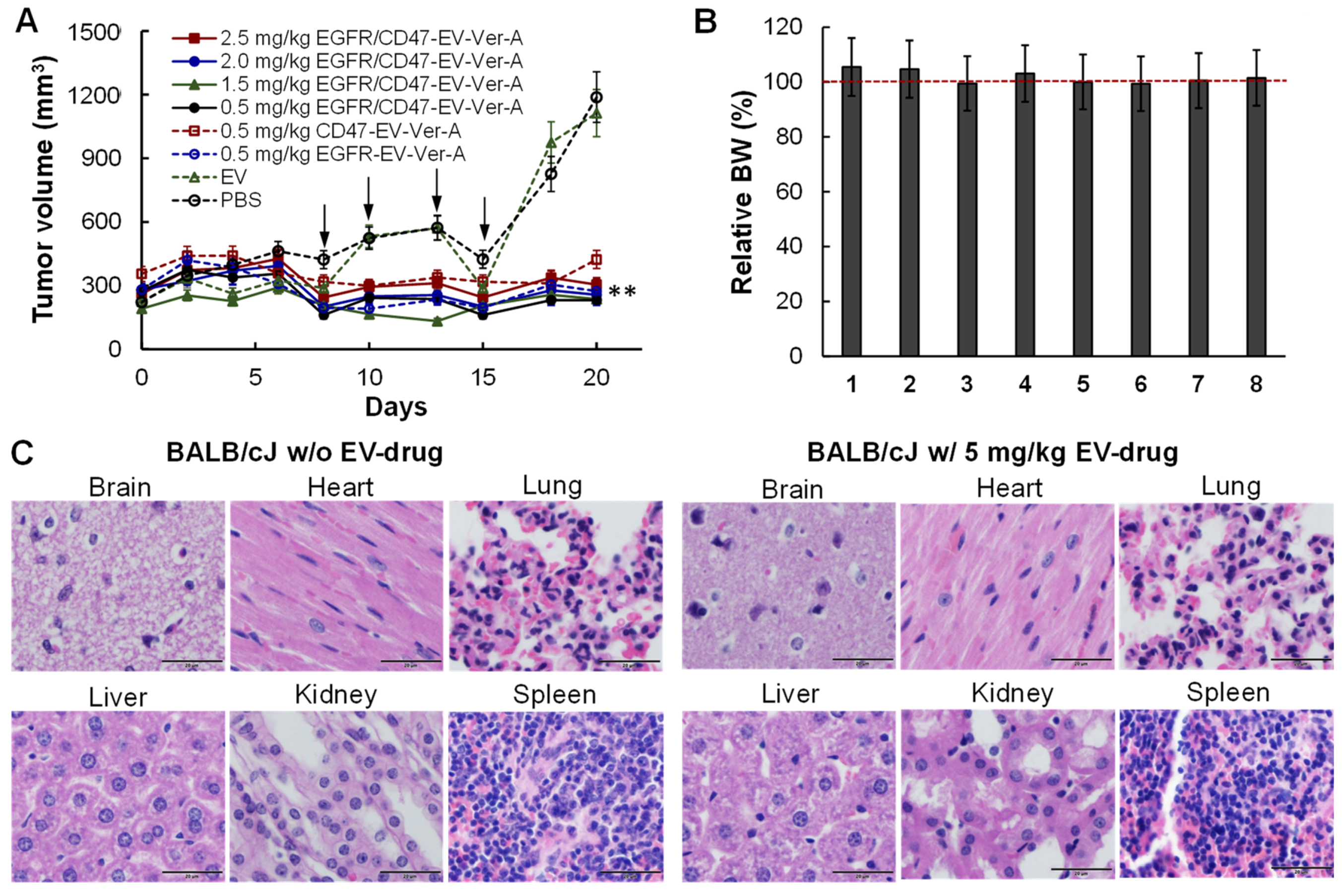
2.12. Hematoxylin and Eosin (H&E) Staining
2.13. Statistical Analysis
3. Results
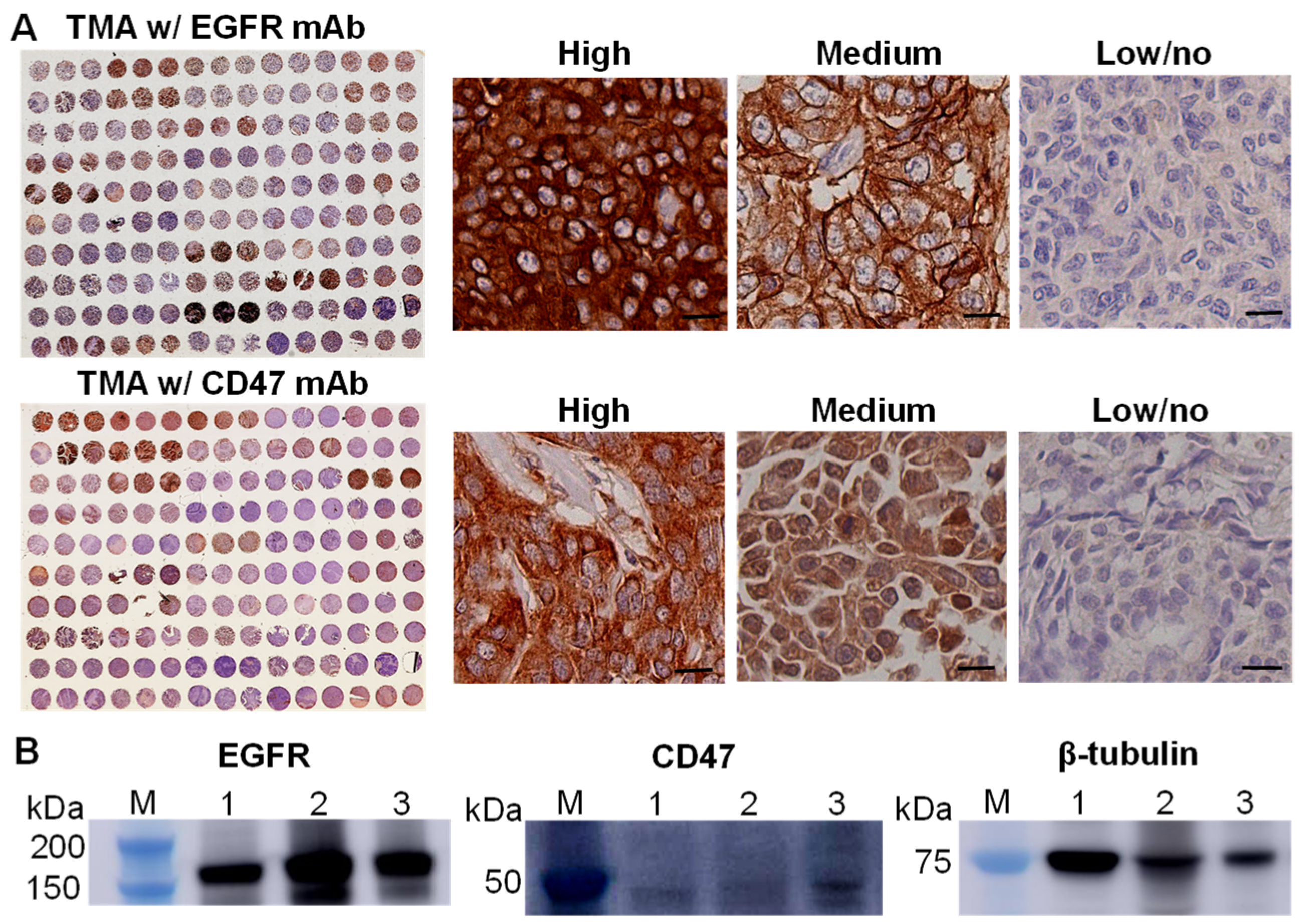
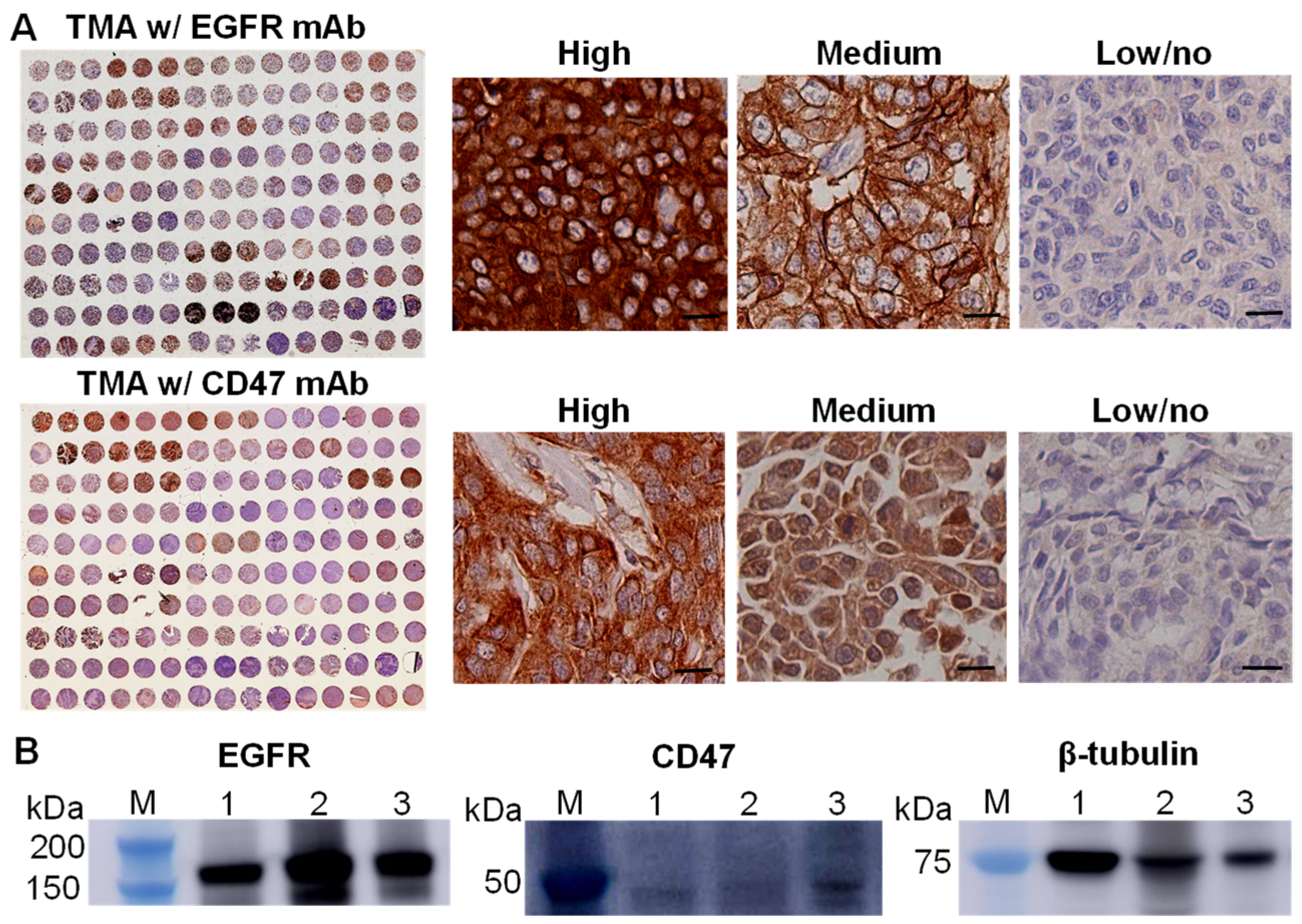
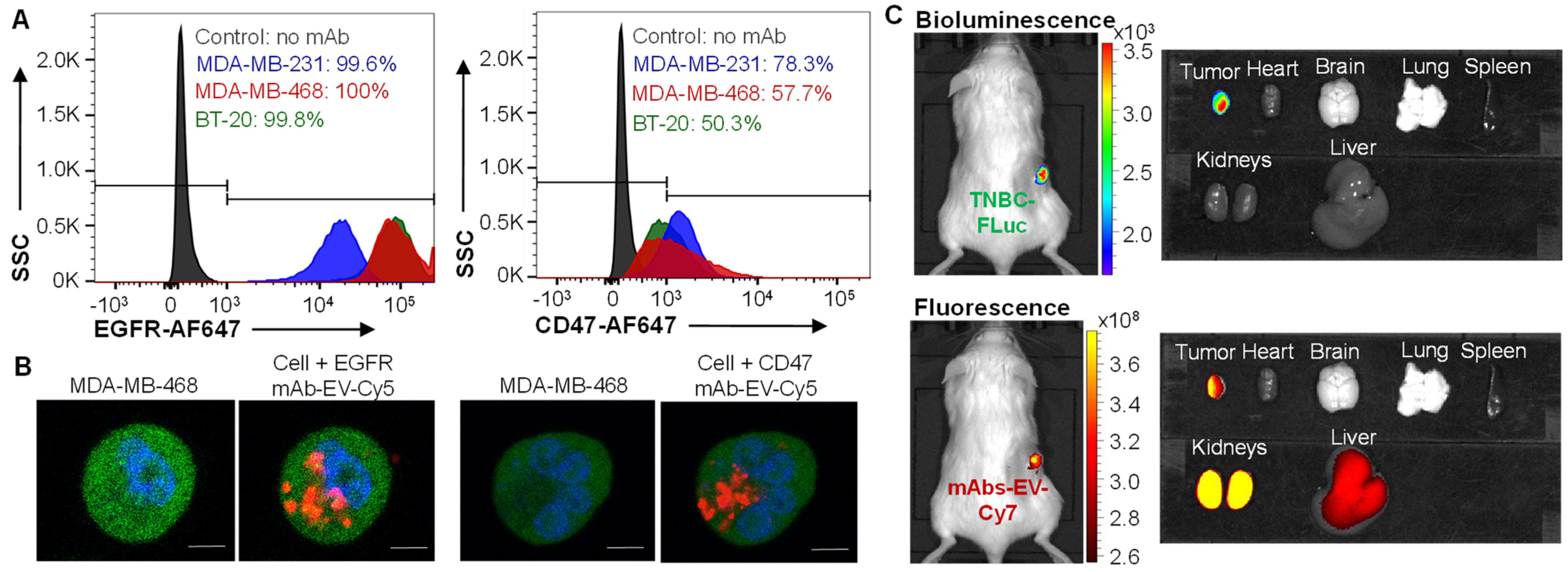
3.1. EGFR and CD47 Expression in TNBCs
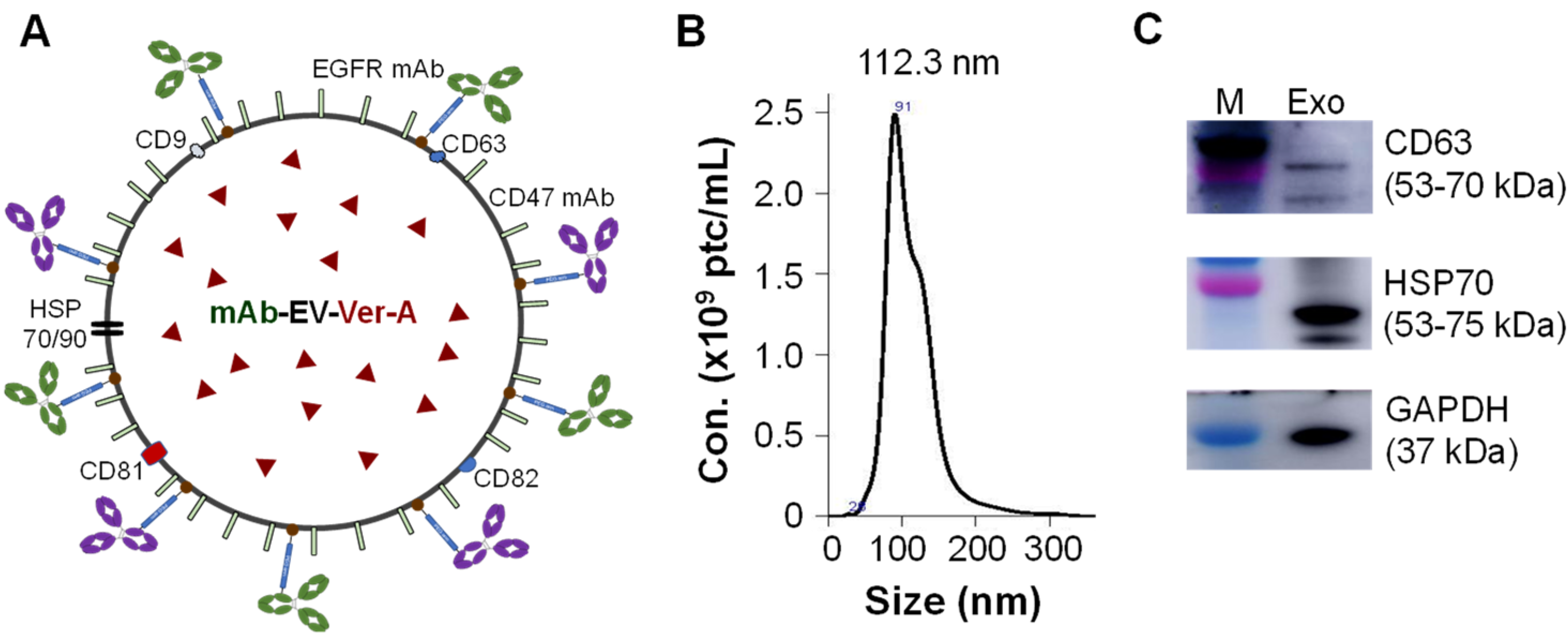
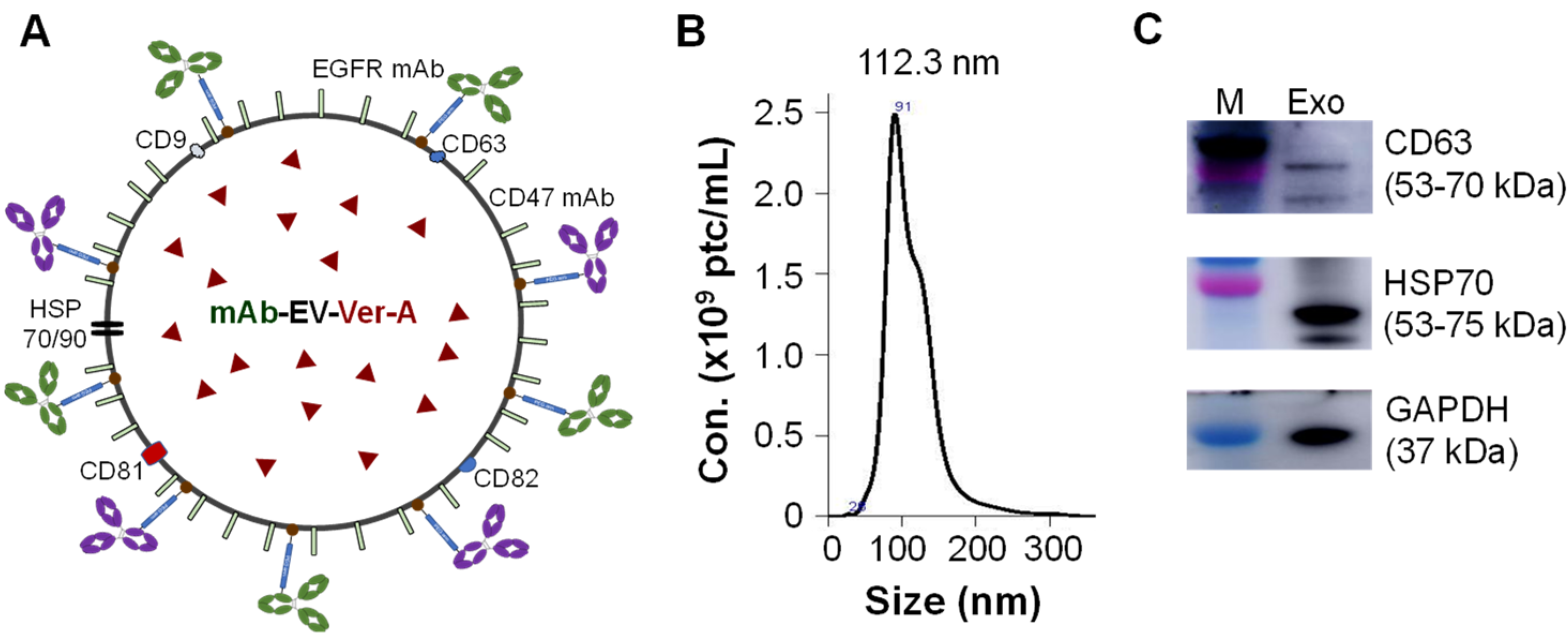
3.2. Construction of Dual-Targeted mAb-EV-Ver-A
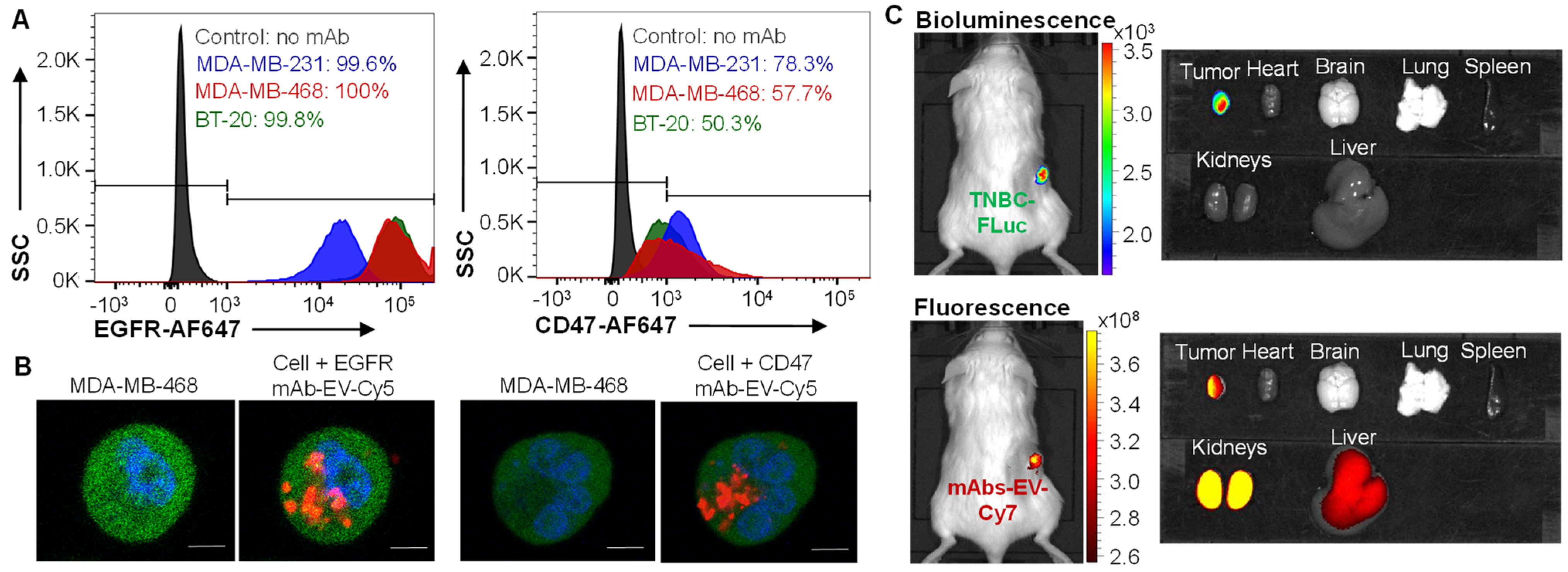
3.3. TNBC Targeting by EGFR/CD47 mAb-EV
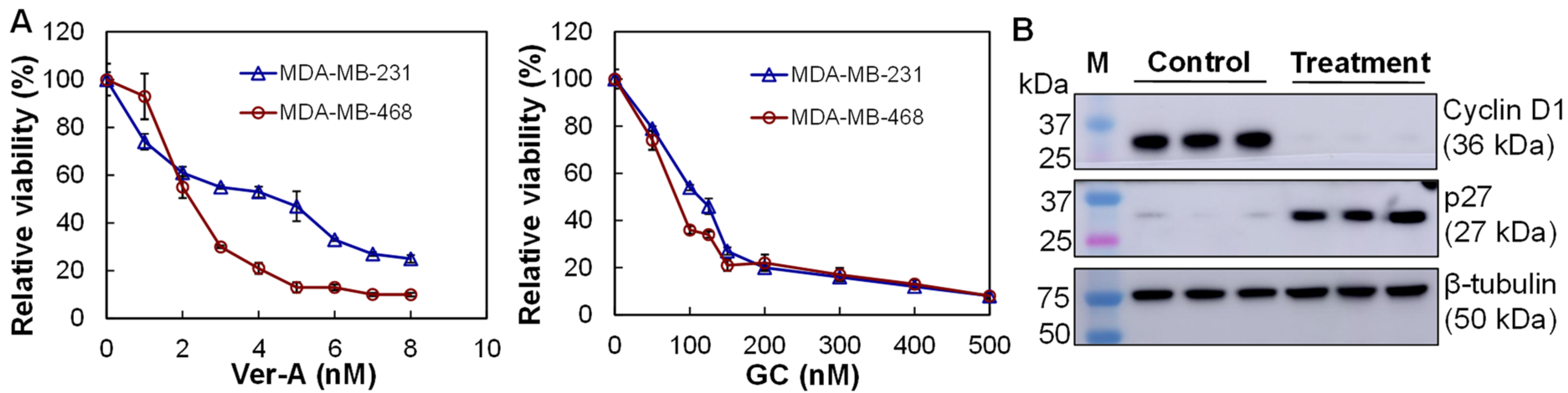
3.4. In Vitro Anti-TNBC Cytotoxicity
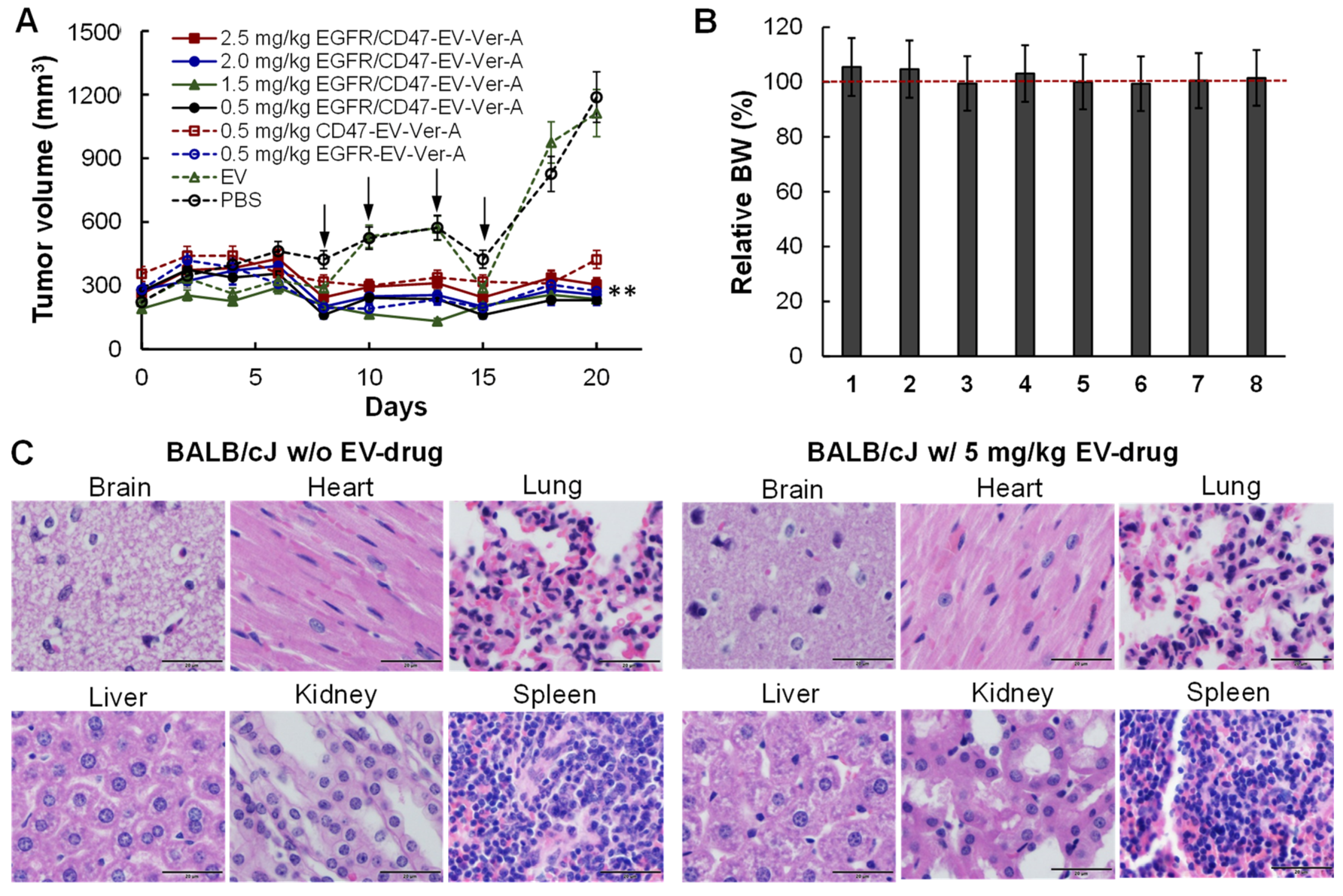
3.5. Tolerated Dosage (TD) and Anti-Tumor Efficacy in Immunocompetet Models
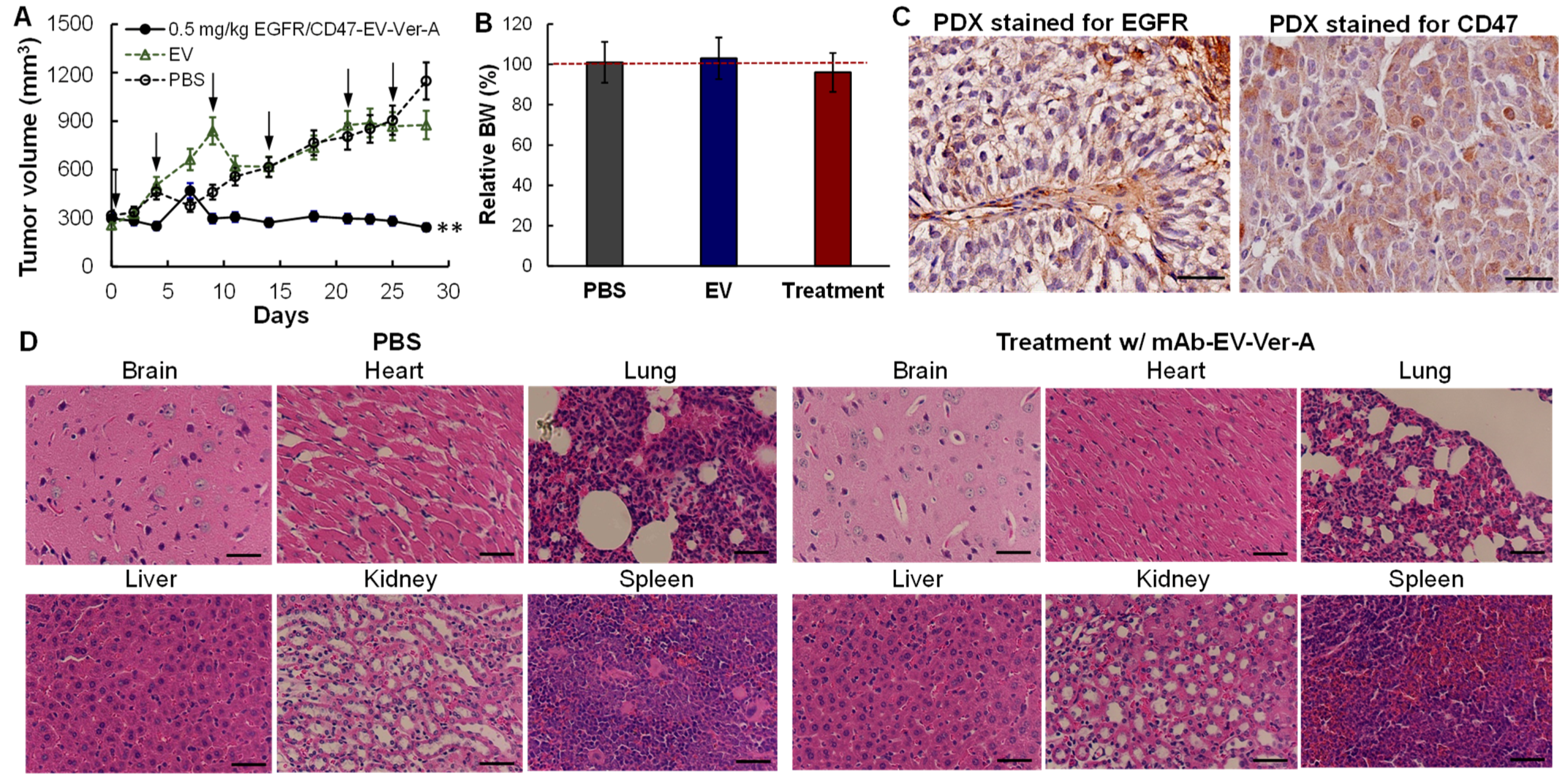
3.6. In Vivo Anti-Tumor Efficacy in PDX Models
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Espinosa Fernandez, J.R.; Eckhardt, B.L.; Lee, J.; Lim, B.; Pearson, T.; Seitz, R.S.; Hout, D.R.; Schweitzer, B.L.; Nielsen, T.J.; Lawrence, O.R.; et al. Identification of triple-negative breast cancer cell lines classified under the same molecular subtype using different molecular characterization techniques: Implications for translational research. PLoS ONE 2020, 15, e0231953. [Google Scholar] [CrossRef]
- Mayer, I.A.; Abramson, V.G.; Lehmann, B.D.; Pietenpol, J.A. New strategies for triple-negative breast cancer--deciphering the heterogeneity. Clin. Cancer Res. 2014, 20, 782–790. [Google Scholar] [CrossRef] [Green Version]
- Metzger-Filho, O.; Tutt, A.; de Azambuja, E.; Saini, K.S.; Viale, G.; Loi, S.; Bradbury, I.; Bliss, J.M.; Azim, H.A., Jr.; Ellis, P.; et al. Dissecting the heterogeneity of triple-negative breast cancer. J. Clin. Oncol. 2012, 30, 1879–1887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liedtke, C.; Mazouni, C.; Hess, K.R.; Andre, F.; Tordai, A.; Mejia, J.A.; Symmans, W.F.; Gonzalez-Angulo, A.M.; Hennessy, B.; Green, M.; et al. Response to neoadjuvant therapy and long-term survival in patients with triple-negative breast cancer. J. Clin. Oncol. 2008, 26, 1275–1281. [Google Scholar] [CrossRef]
- Silver, D.P.; Richardson, A.L.; Eklund, A.C.; Wang, Z.C.; Szallasi, Z.; Li, Q.; Juul, N.; Leong, C.O.; Calogrias, D.; Buraimoh, A.; et al. Efficacy of neoadjuvant Cisplatin in triple-negative breast cancer. J. Clin. Oncol. 2010, 28, 1145–1153. [Google Scholar] [CrossRef]
- Nedeljkovic, M.; Damjanovic, A. Mechanisms of Chemotherapy Resistance in Triple-Negative Breast Cancer-How We Can Rise to the Challenge. Cells 2019, 8, 957. [Google Scholar] [CrossRef] [Green Version]
- Wein, L.; Loi, S. Mechanisms of resistance of chemotherapy in early-stage triple negative breast cancer (TNBC). Breast 2017, 34 (Suppl. 1), S27–S30. [Google Scholar] [CrossRef]
- Romero, D. Benefit in patients with PD-L1-positive TNBC. Nat. Rev. Clin. Oncol. 2019, 16, 6. [Google Scholar] [CrossRef] [PubMed]
- Marra, A.; Viale, G.; Curigliano, G. Recent advances in triple negative breast cancer: The immunotherapy era. BMC Med. 2019, 17, 90. [Google Scholar] [CrossRef] [Green Version]
- Zhu, X.; Zhang, Q.; Wang, D.; Liu, C.; Han, B.; Yang, J.M. Expression of PD-L1 Attenuates the Positive Impacts of High-level Tumor-infiltrating Lymphocytes on Prognosis of Triple-negative Breast Cancer. Cancer Biol. Ther. 2019, 20, 1105–1112. [Google Scholar] [CrossRef]
- Bardia, A.; Hurvitz, S.A.; Tolaney, S.M.; Loirat, D.; Punie, K.; Oliveira, M.; Brufsky, A.; Sardesai, S.D.; Kalinsky, K.; Zelnak, A.B.; et al. Sacituzumab Govitecan in Metastatic Triple-Negative Breast Cancer. N. Engl. J. Med. 2021, 384, 1529–1541. [Google Scholar] [CrossRef]
- Bardia, A.; Mayer, I.A.; Vahdat, L.T.; Tolaney, S.M.; Isakoff, S.J.; Diamond, J.R.; O’Shaughnessy, J.; Moroose, R.L.; Santin, A.D.; Abramson, V.G.; et al. Sacituzumab Govitecan-hziy in Refractory Metastatic Triple-Negative Breast Cancer. N. Engl. J. Med. 2019, 380, 741–751. [Google Scholar] [CrossRef]
- McGuinness, J.E.; Kalinsky, K. Antibody-drug conjugates in metastatic triple negative breast cancer: A spotlight on sacituzumab govitecan, ladiratuzumab vedotin, and trastuzumab deruxtecan. Expert Opin. Biol. Ther. 2020, 21, 903–913. [Google Scholar] [CrossRef] [PubMed]
- Seligson, J.M.; Patron, A.M.; Berger, M.J.; Harvey, R.D.; Seligson, N.D. Sacituzumab Govitecan-hziy: An Antibody-Drug Conjugate for the Treatment of Refractory, Metastatic, Triple-Negative Breast Cancer. Ann. Pharmacother. 2021, 55, 921–931. [Google Scholar] [CrossRef] [PubMed]
- Wahby, S.; Fashoyin-Aje, L.; Osgood, C.L.; Cheng, J.; Fiero, M.H.; Zhang, L.; Tang, S.; Hamed, S.S.; Song, P.; Charlab, R.; et al. FDA Approval Summary: Accelerated Approval of Sacituzumab Govitecan-hziy for Third-line Treatment of Metastatic Triple-negative Breast Cancer. Clin. Cancer Res. 2021, 27, 1850–1854. [Google Scholar] [CrossRef] [PubMed]
- Carey, L.A. Directed therapy of subtypes of triple-negative breast cancer. Oncologist 2011, 16 (Suppl. 1), 71–78. [Google Scholar] [CrossRef]
- Bobrie, A.; Colombo, M.; Raposo, G.; Thery, C. Exosome secretion: Molecular mechanisms and roles in immune responses. Traffic 2011, 12, 1659–1668. [Google Scholar] [CrossRef]
- Thery, C.; Zitvogel, L.; Amigorena, S. Exosomes: Composition, biogenesis and function. Nat. Rev. Immunol. 2002, 2, 569–579. [Google Scholar] [CrossRef]
- Alvarez-Erviti, L.; Seow, Y.; Yin, H.; Betts, C.; Lakhal, S.; Wood, M.J. Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nat. Biotechnol. 2011, 29, 341–345. [Google Scholar] [CrossRef]
- Gyorgy, B.; Fitzpatrick, Z.; Crommentuijn, M.H.; Mu, D.; Maguire, C.A. Naturally enveloped AAV vectors for shielding neutralizing antibodies and robust gene delivery in vivo. Biomaterials 2014, 35, 7598–7609. [Google Scholar] [CrossRef] [Green Version]
- Tian, Y.; Li, S.; Song, J.; Ji, T.; Zhu, M.; Anderson, G.J.; Wei, J.; Nie, G. A doxorubicin delivery platform using engineered natural membrane vesicle exosomes for targeted tumor therapy. Biomaterials 2014, 35, 2383–2390. [Google Scholar] [CrossRef] [PubMed]
- Johnsen, K.B.; Gudbergsson, J.M.; Skov, M.N.; Pilgaard, L.; Moos, T.; Duroux, M. A comprehensive overview of exosomes as drug delivery vehicles—endogenous nanocarriers for targeted cancer therapy. Biochim. Biophys. Acta 2014, 1846, 75–87. [Google Scholar] [CrossRef]
- Ikarashi, Y.; Mikami, R.; Bendelac, A.; Terme, M.; Chaput, N.; Terada, M.; Tursz, T.; Angevin, E.; Lemonnier, F.A.; Wakasugi, H.; et al. Dendritic cell maturation overrules H-2D-mediated natural killer T (NKT) cell inhibition: Critical role for B7 in CD1d-dependent NKT cell interferon gamma production. J. Exp. Med. 2001, 194, 1179–1186. [Google Scholar] [CrossRef] [PubMed]
- Hao, S.; Bai, O.; Yuan, J.; Qureshi, M.; Xiang, J. Dendritic cell-derived exosomes stimulate stronger CD8+ CTL responses and antitumor immunity than tumor cell-derived exosomes. Cell. Mol. Immunol. 2006, 3, 205–211. [Google Scholar]
- Pitt, J.M.; Andre, F.; Amigorena, S.; Soria, J.C.; Eggermont, A.; Kroemer, G.; Zitvogel, L. Dendritic cell-derived exosomes for cancer therapy. J. Clin. Investg. 2016, 126, 1224–1232. [Google Scholar] [CrossRef] [PubMed]
- Ohno, S.; Takanashi, M.; Sudo, K.; Ueda, S.; Ishikawa, A.; Matsuyama, N.; Fujita, K.; Mizutani, T.; Ohgi, T.; Ochiya, T.; et al. Systemically injected exosomes targeted to EGFR deliver antitumor microRNA to breast cancer cells. Mol. Ther. 2013, 21, 185–191. [Google Scholar] [CrossRef] [Green Version]
- Hartman, Z.C.; Wei, J.; Glass, O.K.; Guo, H.; Lei, G.; Yang, X.Y.; Osada, T.; Hobeika, A.; Delcayre, A.; Le Pecq, J.B.; et al. Increasing vaccine potency through exosome antigen targeting. Vaccine 2011, 29, 9361–9367. [Google Scholar] [CrossRef] [Green Version]
- Si, Y.; Kim, S.; Zhang, E.; Tang, Y.; Jaskula-Sztul, R.; Markert, J.M.; Chen, H.; Zhou, L.; Liu, X.M. Targeted Exosomes for Drug Delivery: Biomanufacturing, Surface Tagging, and Validation. Biotechnol. J. 2020, 15, e1900163. [Google Scholar] [CrossRef]
- Si, Y.; Guan, J.; Xu, Y.; Chen, K.; Kim, S.; Zhou, L.; Jaskula-Sztul, R.; Liu, X.M. Dual-Targeted Extracellular Vesicles to Facilitate Combined Therapies for Neuroendocrine Cancer Treatment. Pharmaceutics 2020, 12, 1079. [Google Scholar] [CrossRef]
- Yu, Y.L.; Chou, R.H.; Liang, J.H.; Chang, W.J.; Su, K.J.; Tseng, Y.J.; Huang, W.C.; Wang, S.C.; Hung, M.C. Targeting the EGFR/PCNA signaling suppresses tumor growth of triple-negative breast cancer cells with cell-penetrating PCNA peptides. PLoS ONE 2013, 8, e61362. [Google Scholar] [CrossRef] [Green Version]
- Beniey, M.; Haque, T.; Hassan, S. Translating the role of PARP inhibitors in triple-negative breast cancer. Oncoscience 2019, 6, 287–288. [Google Scholar] [CrossRef]
- Nowsheen, S.; Cooper, T.; Stanley, J.A.; Yang, E.S. Synthetic Lethal Interactions between EGFR and PARP Inhibition in Human Triple Negative Breast Cancer Cells. PLoS ONE 2012, 7, e46614. [Google Scholar]
- Yang, E.S.; Nowsheen, S.; Rahman, M.A.; Cook, R.S.; Xia, F. Targeting BRCA1 localization to augment breast tumor sensitivity to poly(ADP-Ribose) polymerase inhibition. Cancer Res. 2012, 72, 5547–5555. [Google Scholar] [CrossRef] [Green Version]
- Shen, M.; Jiang, Y.Z.; Wei, Y.; Ell, B.; Sheng, X.; Esposito, M.; Kang, J.; Hang, X.; Zheng, H.; Rowicki, M.; et al. Tinagl1 Suppresses Triple-Negative Breast Cancer Progression and Metastasis by Simultaneously Inhibiting Integrin/FAK and EGFR Signaling. Cancer Cell 2019, 35, 64–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ali, R.; Wendt, M.K. The paradoxical functions of EGFR during breast cancer progression. Signal Transduct. Target. Ther. 2017, 2, 16042. [Google Scholar] [CrossRef] [PubMed]
- Masuda, H.; Zhang, D.; Bartholomeusz, C.; Doihara, H.; Hortobagyi, G.N.; Ueno, N.T. Role of epidermal growth factor receptor in breast cancer. Breast Cancer Res. Treat. 2012, 136, 331–345. [Google Scholar] [CrossRef] [Green Version]
- Nakai, K.; Hung, M.C.; Yamaguchi, H. A perspective on anti-EGFR therapies targeting triple-negative breast cancer. Am. J. Cancer Res. 2016, 6, 1609–1623. [Google Scholar] [PubMed]
- Bethune, G.; Bethune, D.; Ridgway, N.; Xu, Z. Epidermal growth factor receptor (EGFR) in lung cancer: An overview and update. J. Thorac. Dis. 2010, 2, 48–51. [Google Scholar] [PubMed]
- Charakidis, M.; Boyer, M. Targeting MET and EGFR in NSCLC-what can we learn from the recently reported phase III trial of onartuzumab in combination with erlotinib in advanced non-small cell lung cancer? Transl. Lung Cancer Res. 2014, 3, 395–396. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, T.O.; Hsu, F.D.; Jensen, K.; Cheang, M.; Karaca, G.; Hu, Z.; Hernandez-Boussard, T.; Livasy, C.; Cowan, D.; Dressler, L.; et al. Immunohistochemical and clinical characterization of the basal-like subtype of invasive breast carcinoma. Clin. Cancer Res. 2004, 10, 5367–5374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiskopf, K. Cancer immunotherapy targeting the CD47/SIRPalpha axis. Eur. J. Cancer 2017, 76, 100–109. [Google Scholar] [CrossRef]
- Huang, Y.; Ma, Y.; Gao, P.; Yao, Z. Targeting CD47: The achievements and concerns of current studies on cancer immunotherapy. J. Thorac. Dis. 2017, 9, E168–E174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samanta, D.; Park, Y.; Ni, X.; Li, H.; Zahnow, C.A.; Gabrielson, E.; Pan, F.; Semenza, G.L. Chemotherapy induces enrichment of CD47(+)/CD73(+)/PDL1(+) immune evasive triple-negative breast cancer cells. Proc. Natl. Acad. Sci. USA 2018, 115, E1239–E1248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nigro, A.; Ricciardi, L.; Salvato, I.; Sabbatino, F.; Vitale, M.; Crescenzi, M.A.; Montico, B.; Triggiani, M.; Pepe, S.; Stellato, C.; et al. Enhanced Expression of CD47 Is Associated With Off-Target Resistance to Tyrosine Kinase Inhibitor Gefitinib in NSCLC. Front. Immunol. 2019, 10, 3135. [Google Scholar] [CrossRef] [Green Version]
- Si, Y.; Zhang, Y.; Guan, J.-S.; Ngo, H.; Totoro, A.; Singh, A.; Chen, K.; Xu, Y.; Yang, E.; Zhou, L.; et al. Anti-CD47 Monoclonal Antibody-drug Conjugate: A Targeted Therapy to Treat Triple-negative Breast Cancers. Vaccines 2021, 9, 882. [Google Scholar] [CrossRef]
- Amagata, T.; Rath, C.; Rigot, J.F.; Tarlov, N.; Tenney, K.; Valeriote, F.A.; Crews, P. Structures and cytotoxic properties of trichoverroids and their macrolide analogues produced by saltwater culture of Myrothecium verrucaria. J. Med. Chem. 2003, 46, 4342–4350. [Google Scholar] [CrossRef] [PubMed]
- Palanivel, K.; Kanimozhi, V.; Kadalmani, B. Verrucarin A alters cell-cycle regulatory proteins and induces apoptosis through reactive oxygen species-dependent p38MAPK activation in the human breast cancer cell line MCF-7. Tumour Biol. 2014, 35, 10159–10167. [Google Scholar] [CrossRef]
- Woldemichael, G.M.; Turbyville, T.J.; Vasselli, J.R.; Linehan, W.M.; McMahon, J.B. Lack of a functional VHL gene product sensitizes renal cell carcinoma cells to the apoptotic effects of the protein synthesis inhibitor verrucarin A. Neoplasia 2012, 14, 771–777. [Google Scholar] [CrossRef] [Green Version]
- Yan, F.; Yu, Y.; Chow, D.C.; Palzkill, T.; Madoux, F.; Hodder, P.; Chase, P.; Griffin, P.R.; O’Malley, B.W.; Lonard, D.M. Identification of verrucarin a as a potent and selective steroid receptor coactivator-3 small molecule inhibitor. PLoS ONE 2014, 9, e95243. [Google Scholar] [CrossRef] [Green Version]
- Deeb, D.; Gao, X.; Liu, Y.; Zhang, Y.; Shaw, J.; Valeriote, F.A.; Gautam, S.C. The inhibition of cell proliferation and induction of apoptosis in pancreatic ductal adenocarcinoma cells by verrucarin A, a macrocyclic trichothecene, is associated with the inhibition of Akt/NF-small ka, CyrillicB/mTOR prosurvival signaling. Int. J. Oncol. 2016, 49, 1139–1147. [Google Scholar] [CrossRef] [Green Version]
- Si, Y.; Kim, S.; Ou, J.; Lu, Y.; Ernst, P.; Chen, K.; Whitt, J.; Carter, A.M.; Markert, J.M.; Bibb, J.A.; et al. Anti-SSTR2 antibody-drug conjugate for neuroendocrine tumor therapy. Cancer Gene Ther. 2020, 28, 799–812. [Google Scholar] [CrossRef] [PubMed]
- Ou, J.; Si, Y.; Goh, K.; Yasui, N.; Guo, Y.; Song, J.; Wang, L.; Jaskula-Sztul, R.; Fan, J.; Zhou, L.; et al. Bioprocess development of antibody-drug conjugate production for cancer treatment. PLoS ONE 2018, 13, e0206246. [Google Scholar] [CrossRef]
- Xu, N.; Ou, J.; Si, Y.; Goh, K.Y.; Flanigan, D.D.; Han, X.; Yang, Y.; Yang, S.-T.; Zhou, L.; Liu, X. Proteomics insight into the production of monoclonal antibody. Biochem. Eng. J. 2019, 145, 177–185. [Google Scholar] [CrossRef]
- Ji, X.; Xu, H.; Zhang, H.; Hillery, C.A.; Gao, H.Q.; Pritchard, K.A., Jr. Anion exchange HPLC isolation of high-density lipoprotein (HDL) and on-line estimation of proinflammatory HDL. PLoS ONE 2014, 9, e91089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ou, J.; Si, Y.; Tang, Y.; Salzer, G.E.; Lu, Y.; Kim, S.; Qin, H.; Zhou, L.; Liu, X. Novel biomanufacturing platform for large-scale and high-quality human T cells production. J. Biol. Eng. 2019, 13, 34. [Google Scholar] [CrossRef]
- Si, Y.; Xu, Y.; Guan, J.; Chen, K.; Kim, S.; Yang, E.S.; Zhou, L.; Liu, X.M. Anti-EGFR antibody-drug conjugate for triple-negative breast cancer therapy. Eng. Life Sci. 2021, 21, 37–44. [Google Scholar] [CrossRef]
- Si, Y.; Zhang, Y.; Ngo, H.G.; Guan, J.S.; Chen, K.; Wang, Q.; Singh, A.P.; Xu, Y.; Zhou, L.; Yang, E.S.; et al. Targeted Liposomal Chemotherapies to Treat Triple-Negative Breast Cancer. Cancers 2021, 13, 3749. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Guo, Y.D.; Li, X.N.; Zhang, Y.Q.; Gu, L.; Wu, P.P.; Bai, G.H.; Xiao, Y. B7-H3 expression in breast cancer and upregulation of VEGF through gene silence. Onco. Targets Ther. 2014, 7, 1979–1986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bachawal, S.V.; Jensen, K.C.; Wilson, K.E.; Tian, L.; Lutz, A.M.; Willmann, J.K. Breast Cancer Detection by B7-H3-Targeted Ultrasound Molecular Imaging. Cancer Res. 2015, 75, 2501–2509. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Liu, J.; Wang, J.; Liu, Y.; Zhang, F.; Lin, W.; Gao, A.; Sun, M.; Wang, Y.; Sun, Y. B7-H3 expression in ductal and lobular breast cancer and its association with IL-10. Mol. Med. Rep. 2013, 7, 134–138. [Google Scholar] [CrossRef] [Green Version]
- Seaman, S.; Zhu, Z.; Saha, S.; Zhang, X.M.; Yang, M.Y.; Hilton, M.B.; Morris, K.; Szot, C.; Morris, H.; Swing, D.A.; et al. Eradication of Tumors through Simultaneous Ablation of CD276/B7-H3-Positive Tumor Cells and Tumor Vasculature. Cancer Cell 2017, 31, 501–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Xu, L. GPR56 in cancer progression: Current status and future perspective. Future Oncol. 2012, 8, 431–440. [Google Scholar] [CrossRef] [PubMed]
- Lynch, J.R.; Wang, J.Y. G Protein-Coupled Receptor Signaling in Stem Cells and Cancer. Int. J. Mol. Sci. 2016, 17, 707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehtinen, J.; Raki, M.; Bergstrom, K.A.; Uutela, P.; Lehtinen, K.; Hiltunen, A.; Pikkarainen, J.; Liang, H.; Pitkanen, S.; Maatta, A.M.; et al. Pre-targeting and direct immunotargeting of liposomal drug carriers to ovarian carcinoma. PLoS ONE 2012, 7, e41410. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.K.; Huang, L. Nanoparticle delivery of a peptide targeting EGFR signaling. J. Control. Release 2012, 157, 279–286. [Google Scholar] [CrossRef] [Green Version]
- Masserini, M. Nanoparticles for brain drug delivery. ISRN Biochem. 2013, 2013, 238428. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Bertoni, H.; Kozielski, K.L.; Rui, Y.; Lal, B.; Vaughan, H.; Wilson, D.R.; Mihelson, N.; Eberhart, C.G.; Laterra, J.; Green, J.J. Bioreducible Polymeric Nanoparticles Containing Multiplexed Cancer Stem Cell Regulating miRNAs Inhibit Glioblastoma Growth and Prolong Survival. Nano Lett. 2018, 18, 4086–4094. [Google Scholar] [CrossRef]
- Qu, J.; Zhang, L.; Chen, Z.; Mao, G.; Gao, Z.; Lai, X.; Zhu, X.; Zhu, J. Nanostructured lipid carriers, solid lipid nanoparticles, and polymeric nanoparticles: Which kind of drug delivery system is better for glioblastoma chemotherapy? Drug Deliv. 2016, 23, 3408–3416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Si, Y.; Chen, K.; Ngo, H.G.; Guan, J.S.; Totoro, A.; Zhou, Z.; Kim, S.; Kim, T.; Zhou, L.; Liu, X. Targeted EV to Deliver Chemotherapy to Treat Triple-Negative Breast Cancers. Pharmaceutics 2022, 14, 146. https://doi.org/10.3390/pharmaceutics14010146
Si Y, Chen K, Ngo HG, Guan JS, Totoro A, Zhou Z, Kim S, Kim T, Zhou L, Liu X. Targeted EV to Deliver Chemotherapy to Treat Triple-Negative Breast Cancers. Pharmaceutics. 2022; 14(1):146. https://doi.org/10.3390/pharmaceutics14010146
Chicago/Turabian StyleSi, Yingnan, Kai Chen, Hanh Giai Ngo, Jia Shiung Guan, Angela Totoro, Zhuoxin Zhou, Seulhee Kim, Taehyun Kim, Lufang Zhou, and Xiaoguang Liu. 2022. "Targeted EV to Deliver Chemotherapy to Treat Triple-Negative Breast Cancers" Pharmaceutics 14, no. 1: 146. https://doi.org/10.3390/pharmaceutics14010146
APA StyleSi, Y., Chen, K., Ngo, H. G., Guan, J. S., Totoro, A., Zhou, Z., Kim, S., Kim, T., Zhou, L., & Liu, X. (2022). Targeted EV to Deliver Chemotherapy to Treat Triple-Negative Breast Cancers. Pharmaceutics, 14(1), 146. https://doi.org/10.3390/pharmaceutics14010146