
In Silico Evaluation of Binding of 2-Deoxy-D-Glucose with Mpro of nCoV to Combat COVID-19

 , , ,
, , ,  ,
,  and
and
Abstract
:1. Introduction
2. Materials and Methods
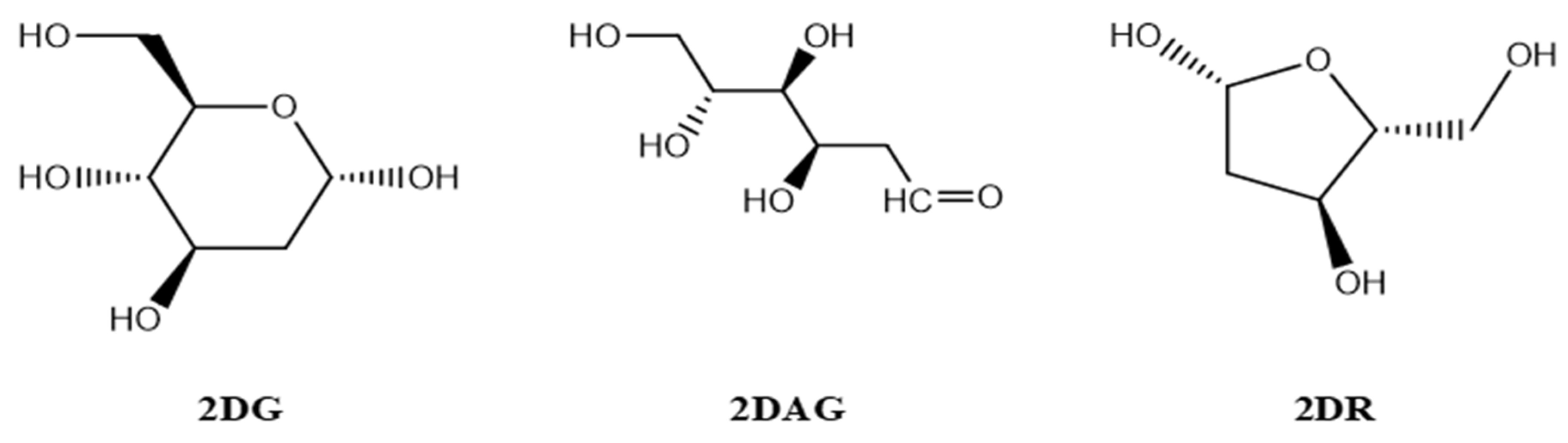
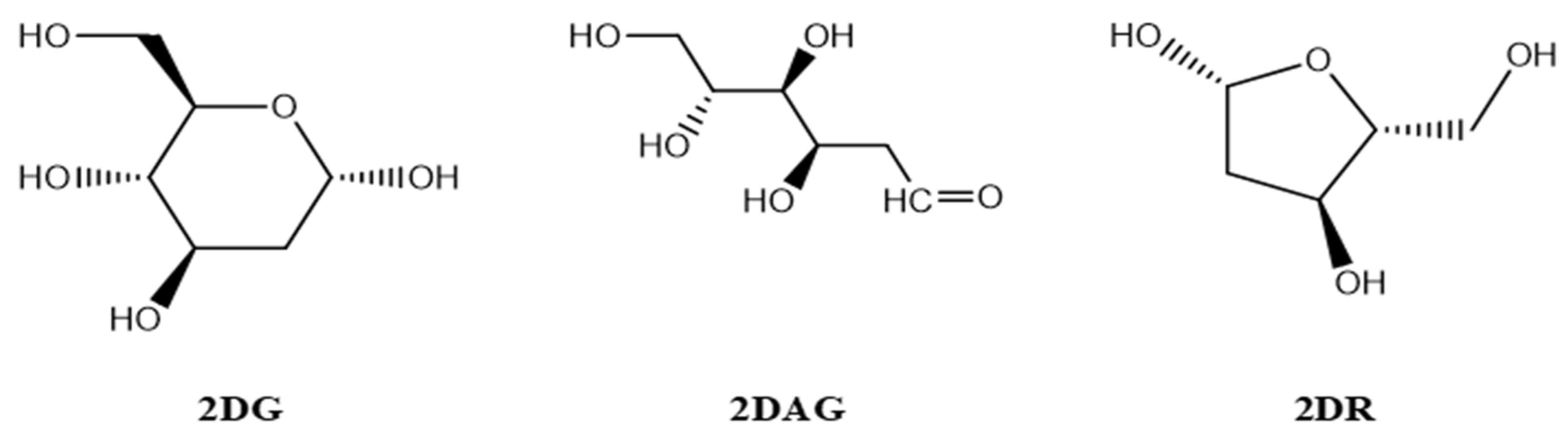
2.1. Designing of the Ligands
2.2. Molecular Docking
2.3. Molecular Dynamics (MD) Simulations
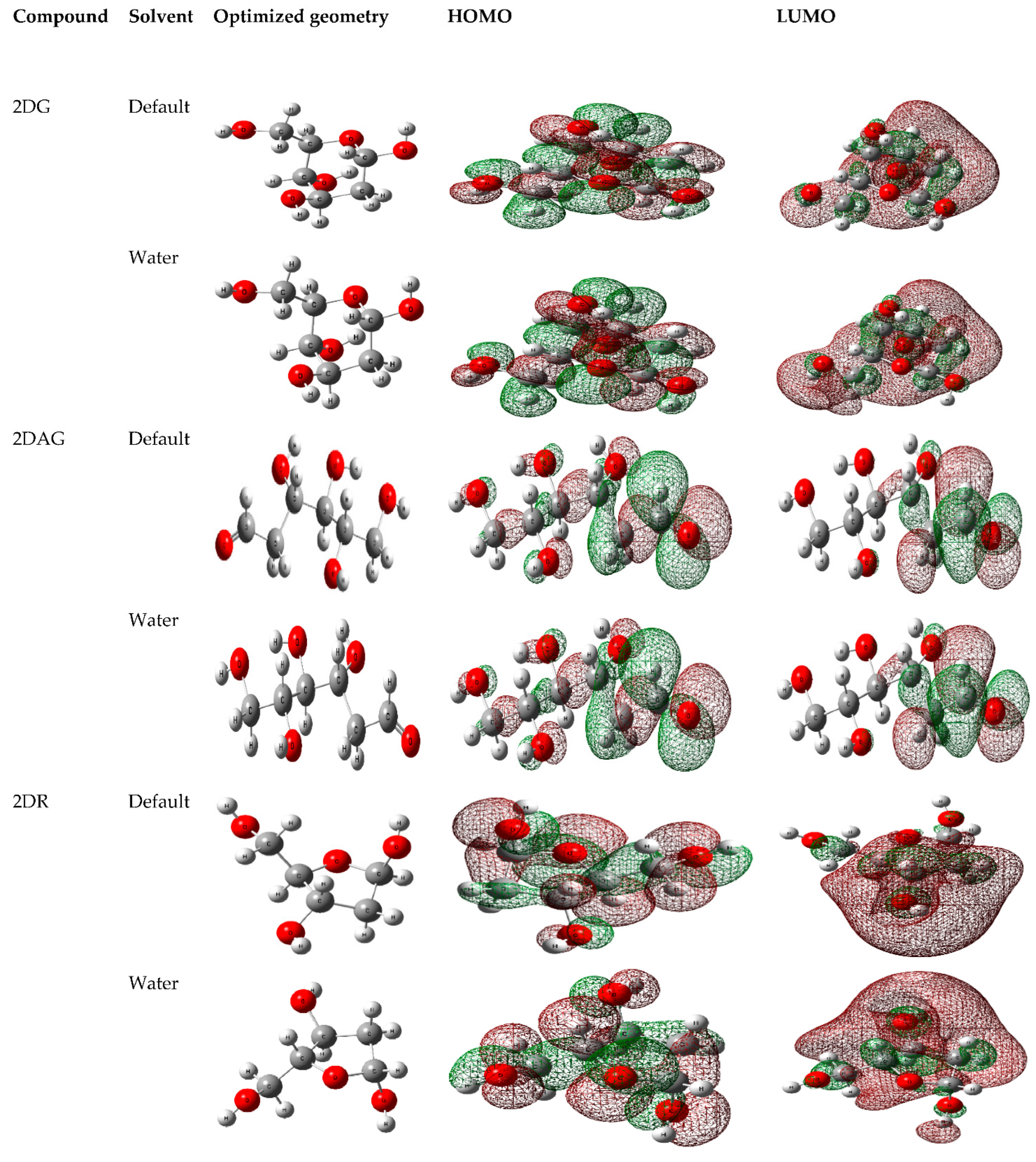
2.4. Density Functional Theory (DFT)-Based Calculations
3. Results
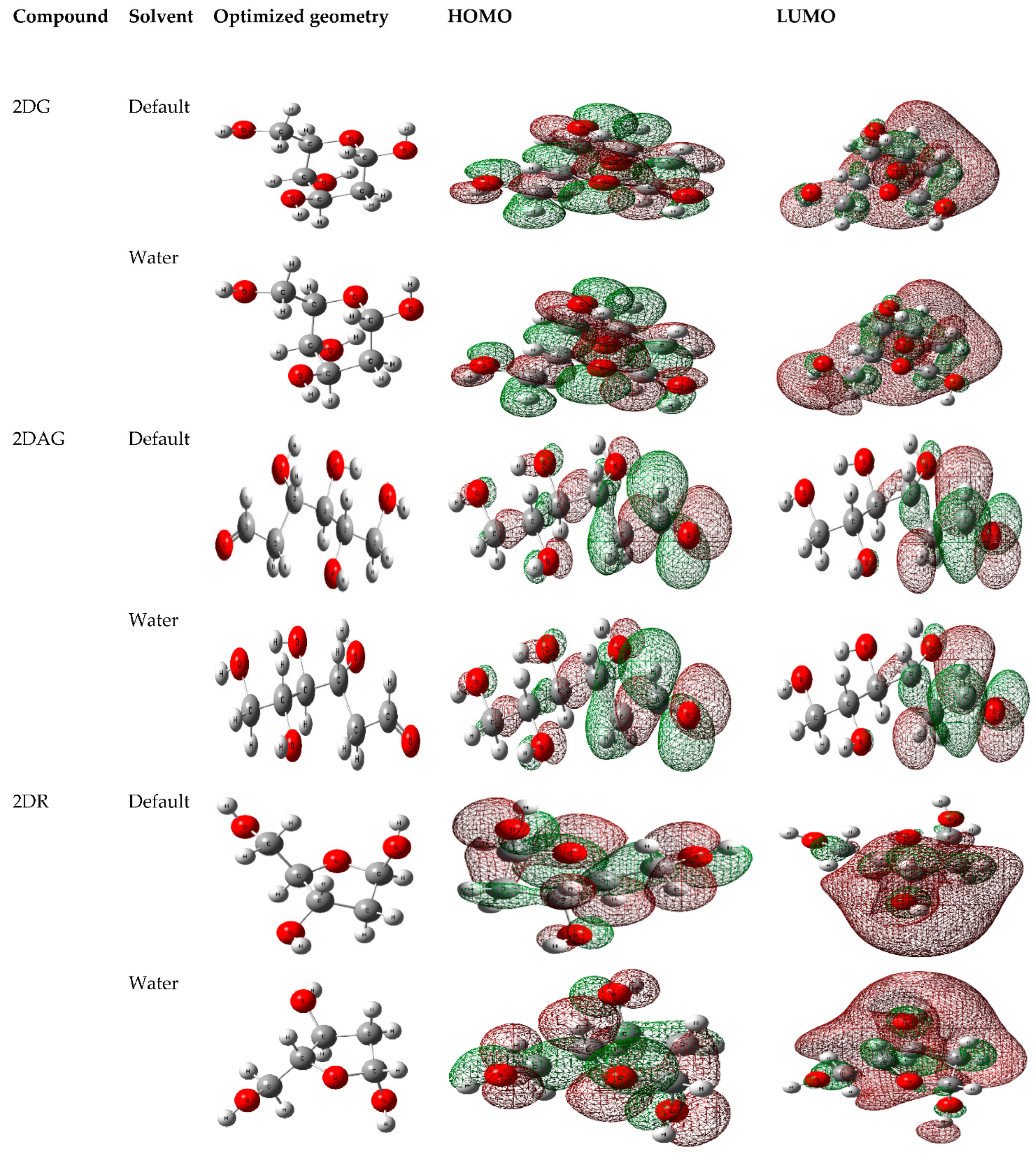
3.1. DFT Calculations
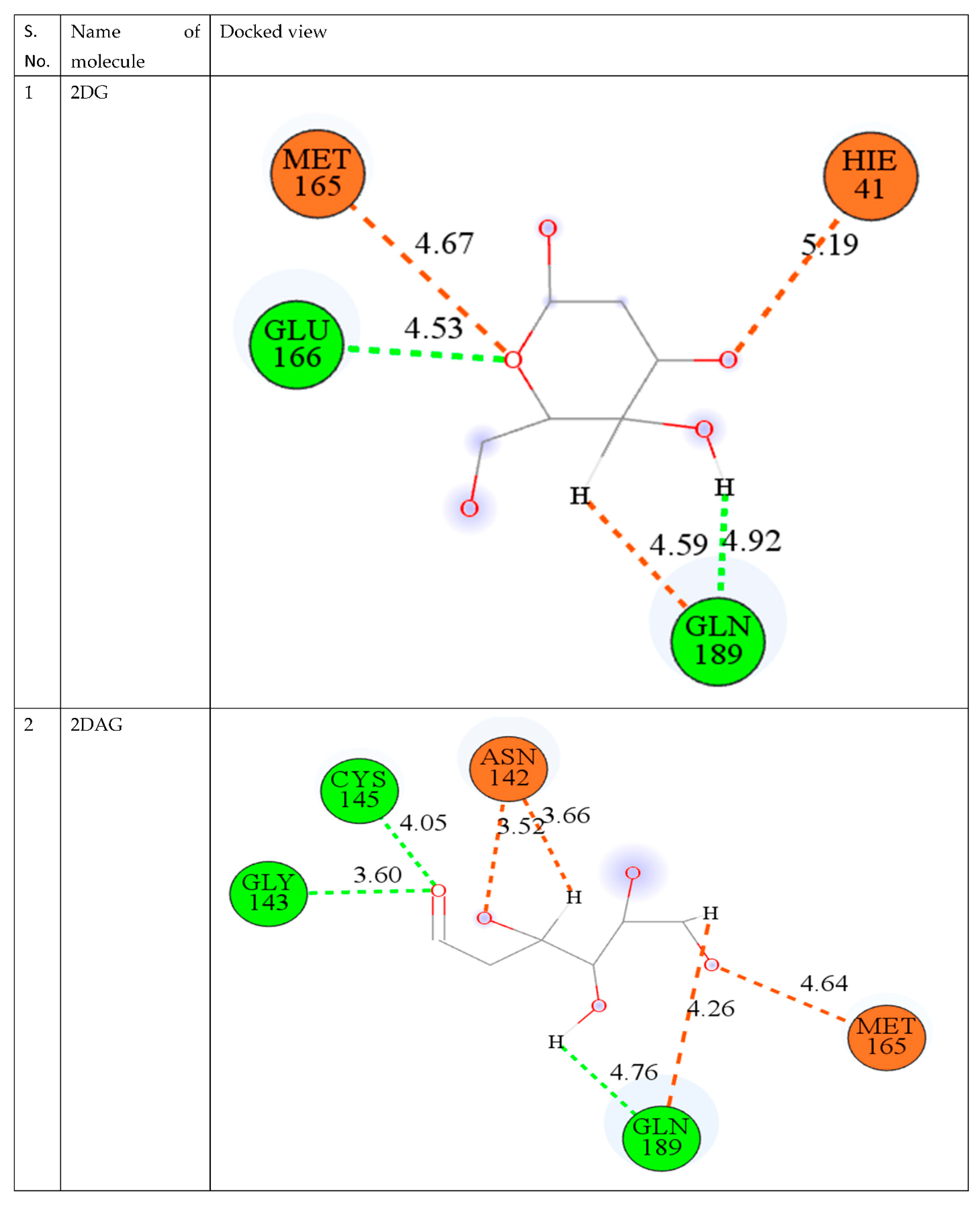
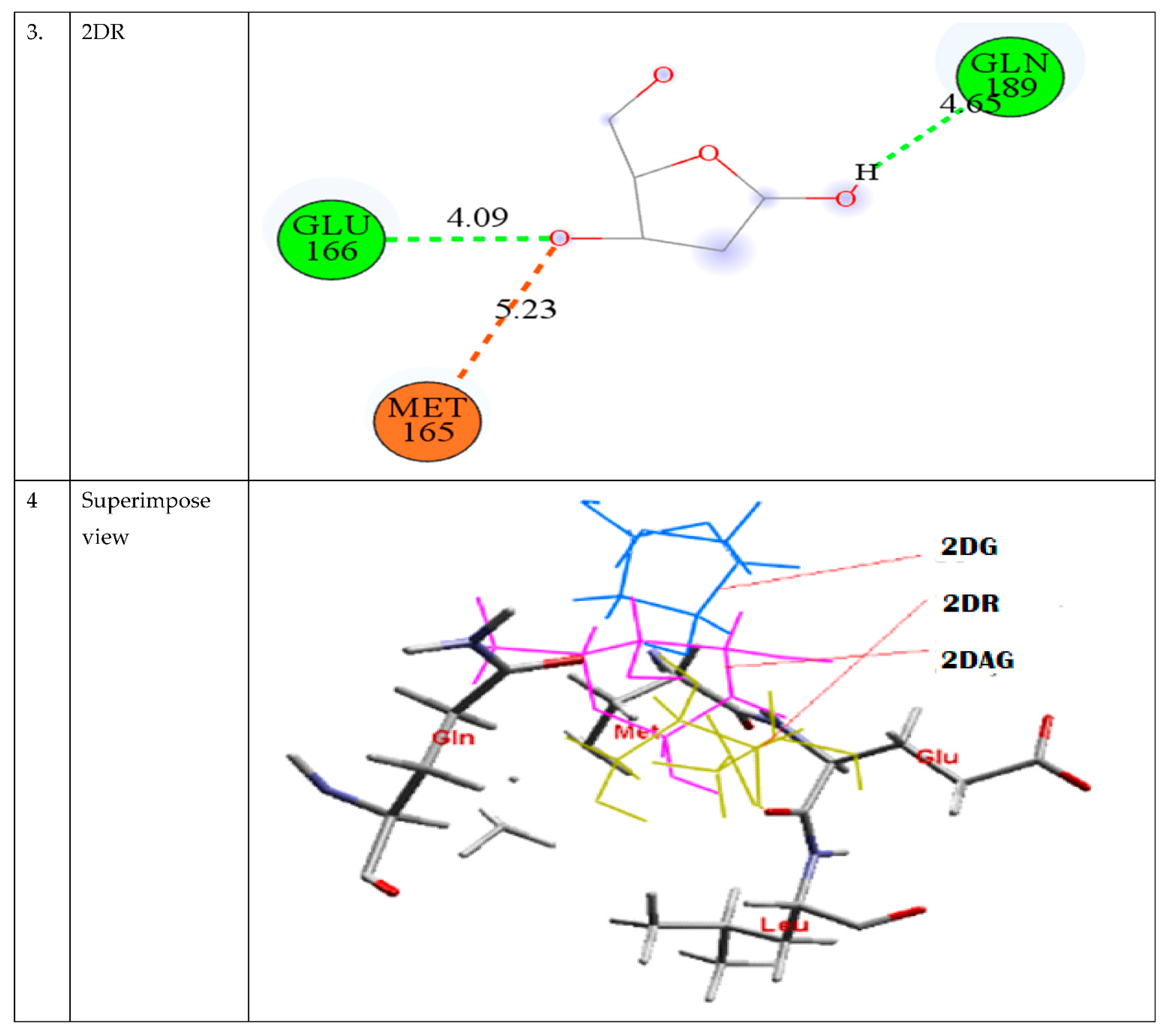
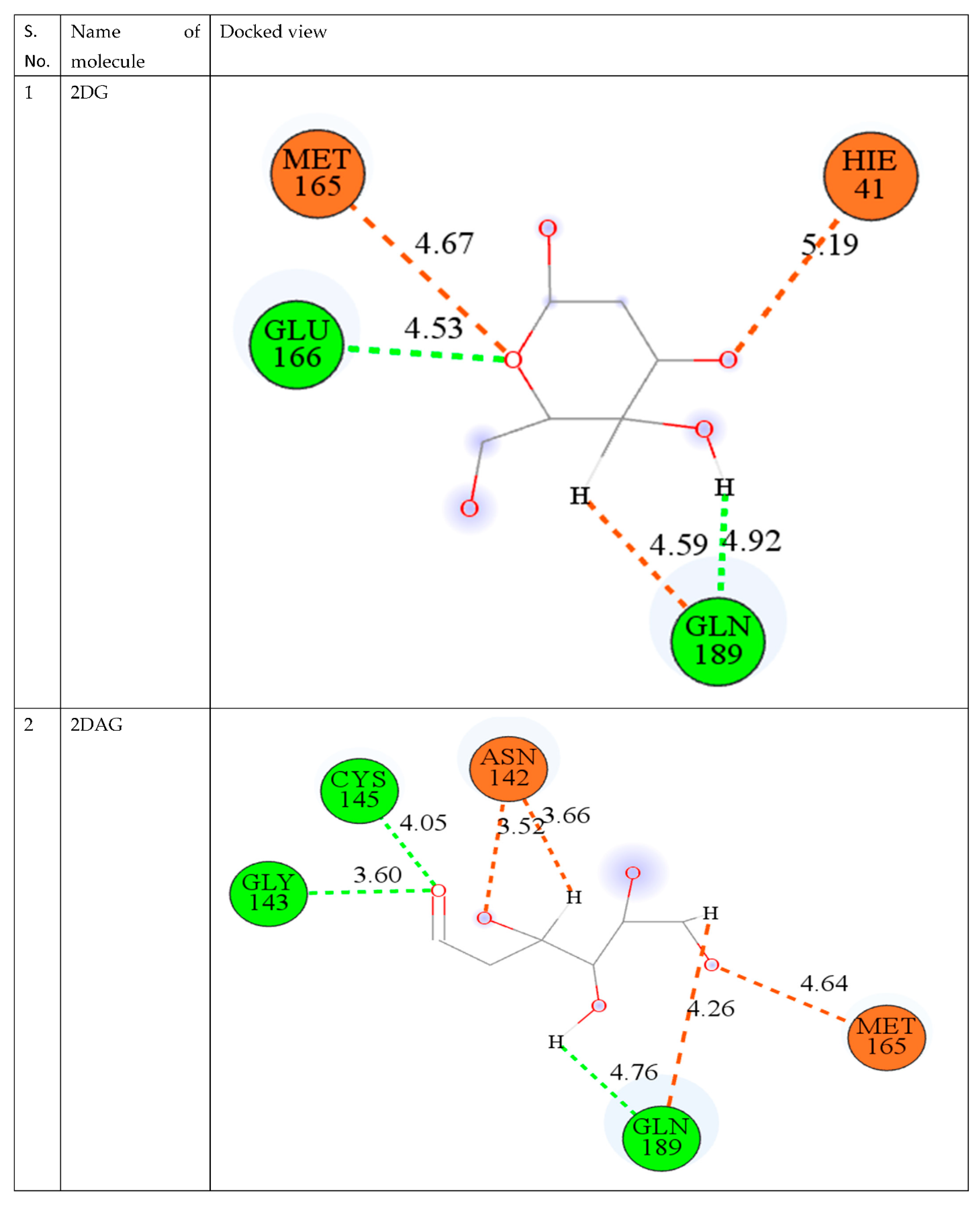
3.2. Molecular Docking
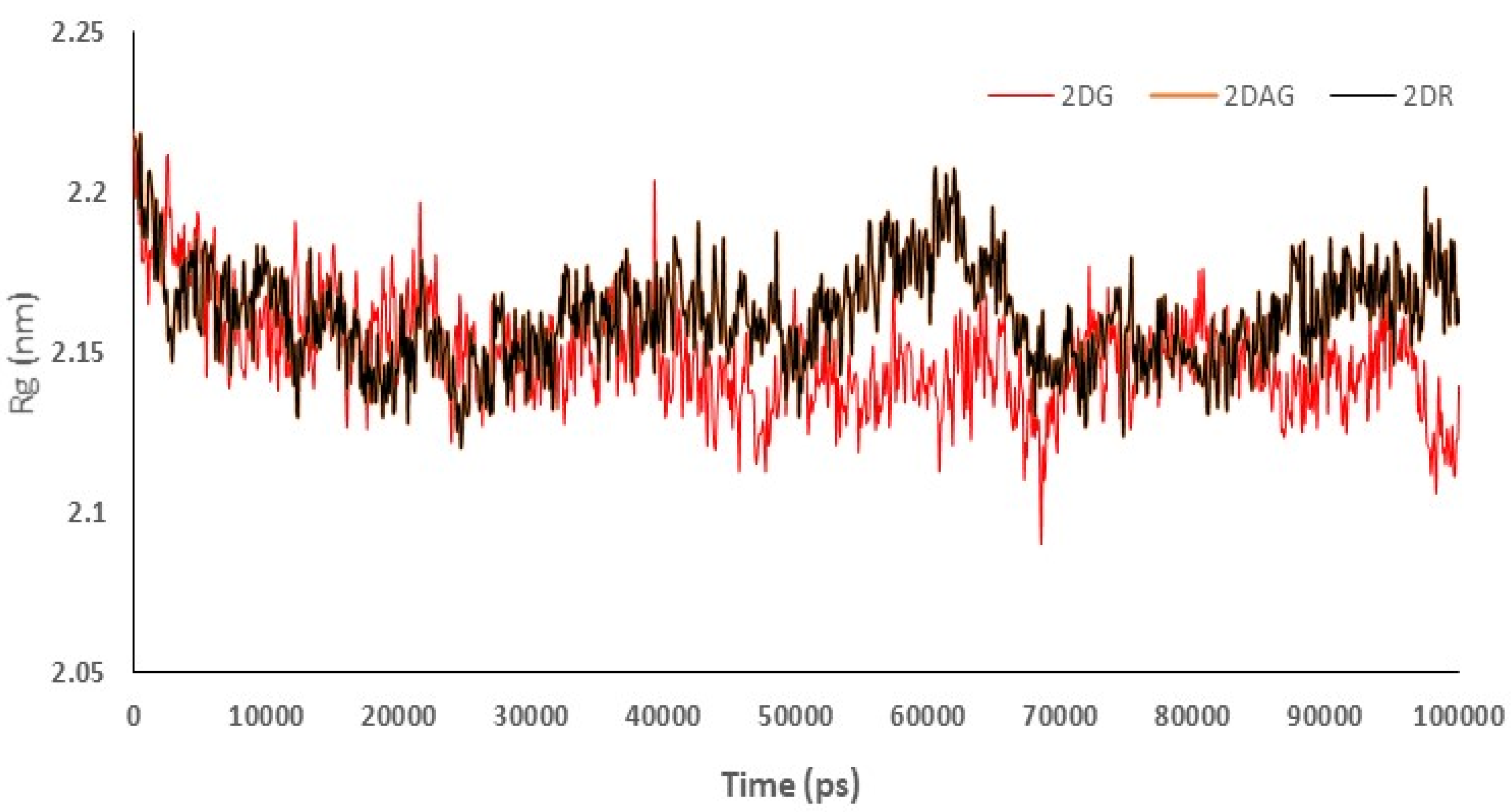
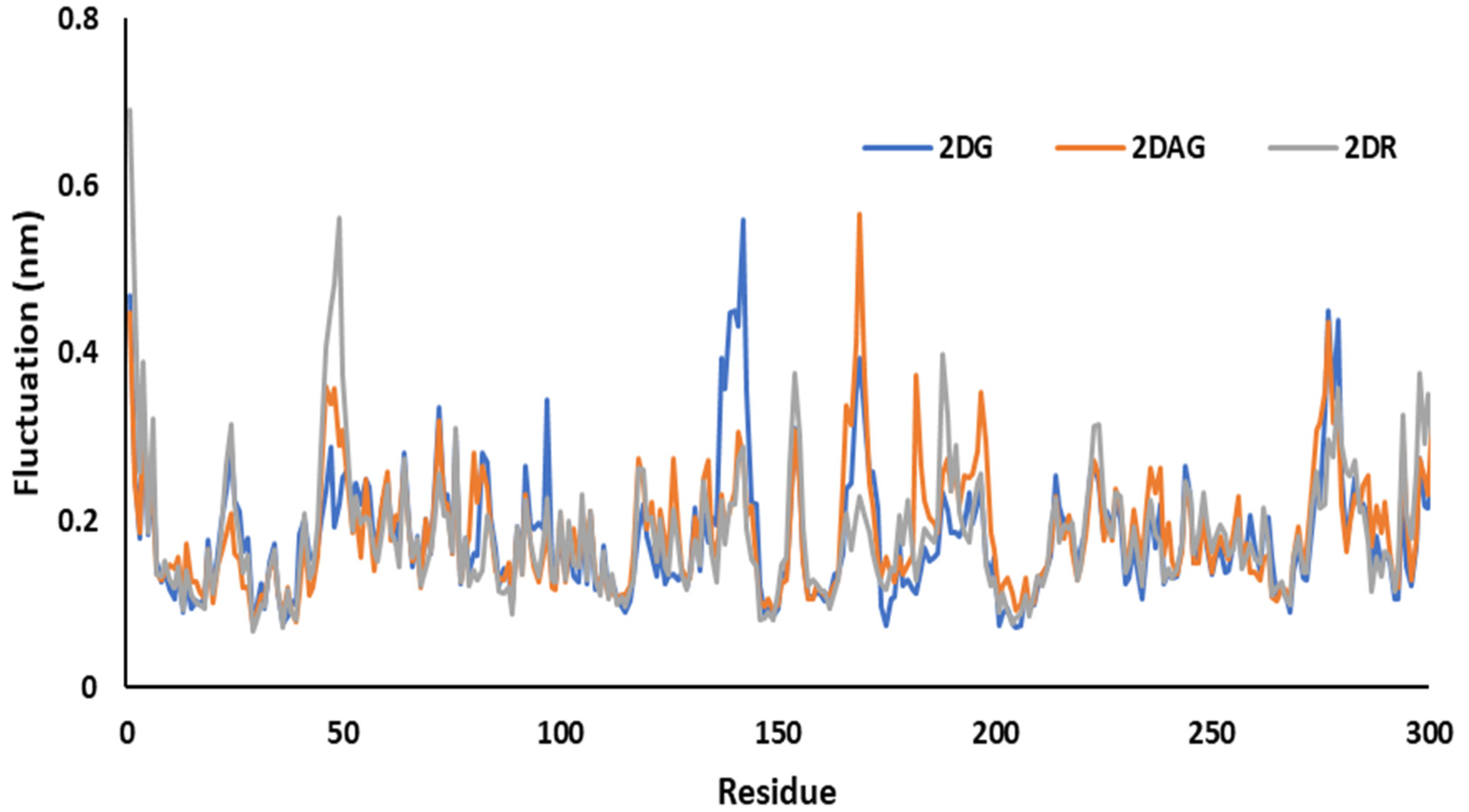
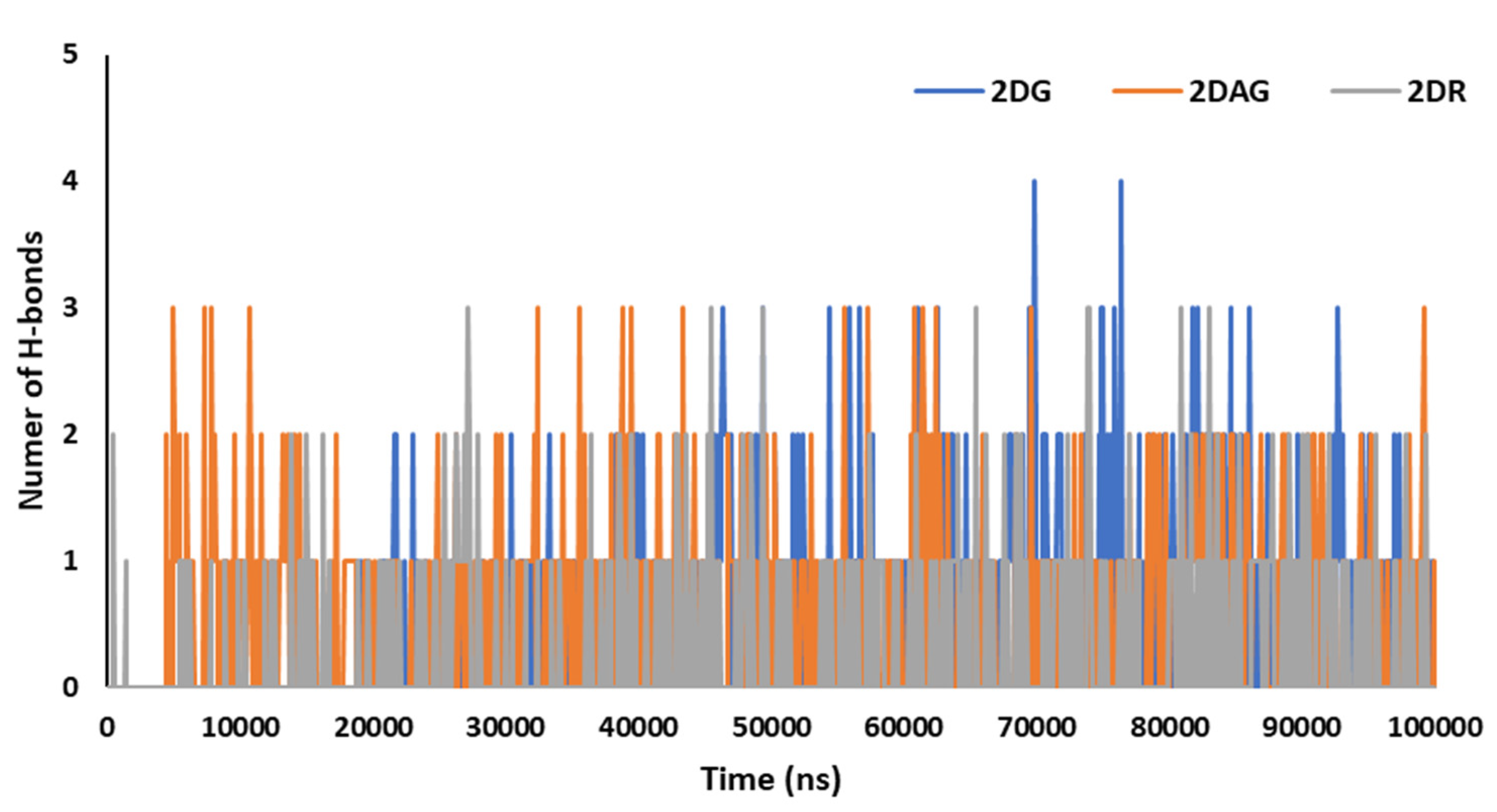
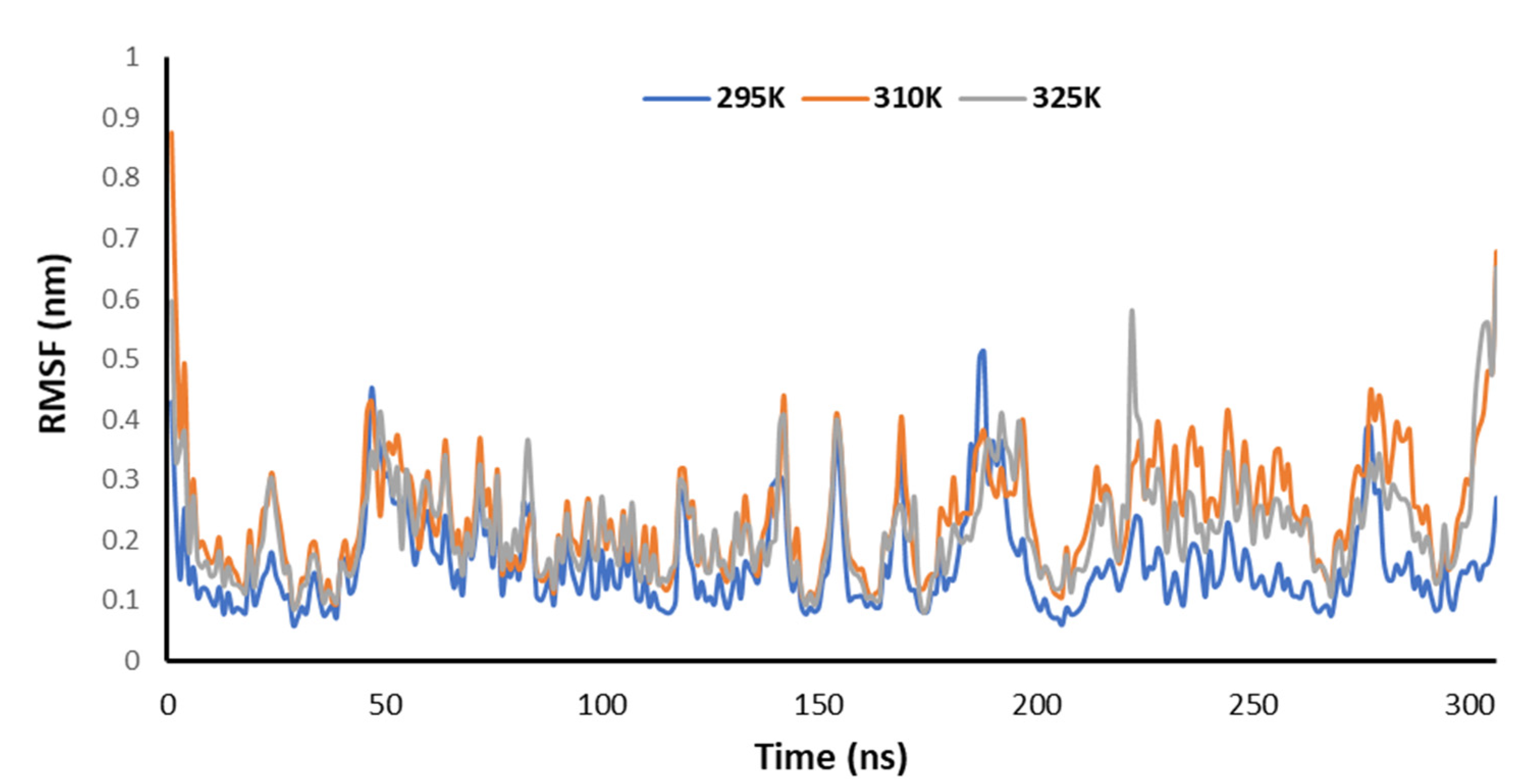
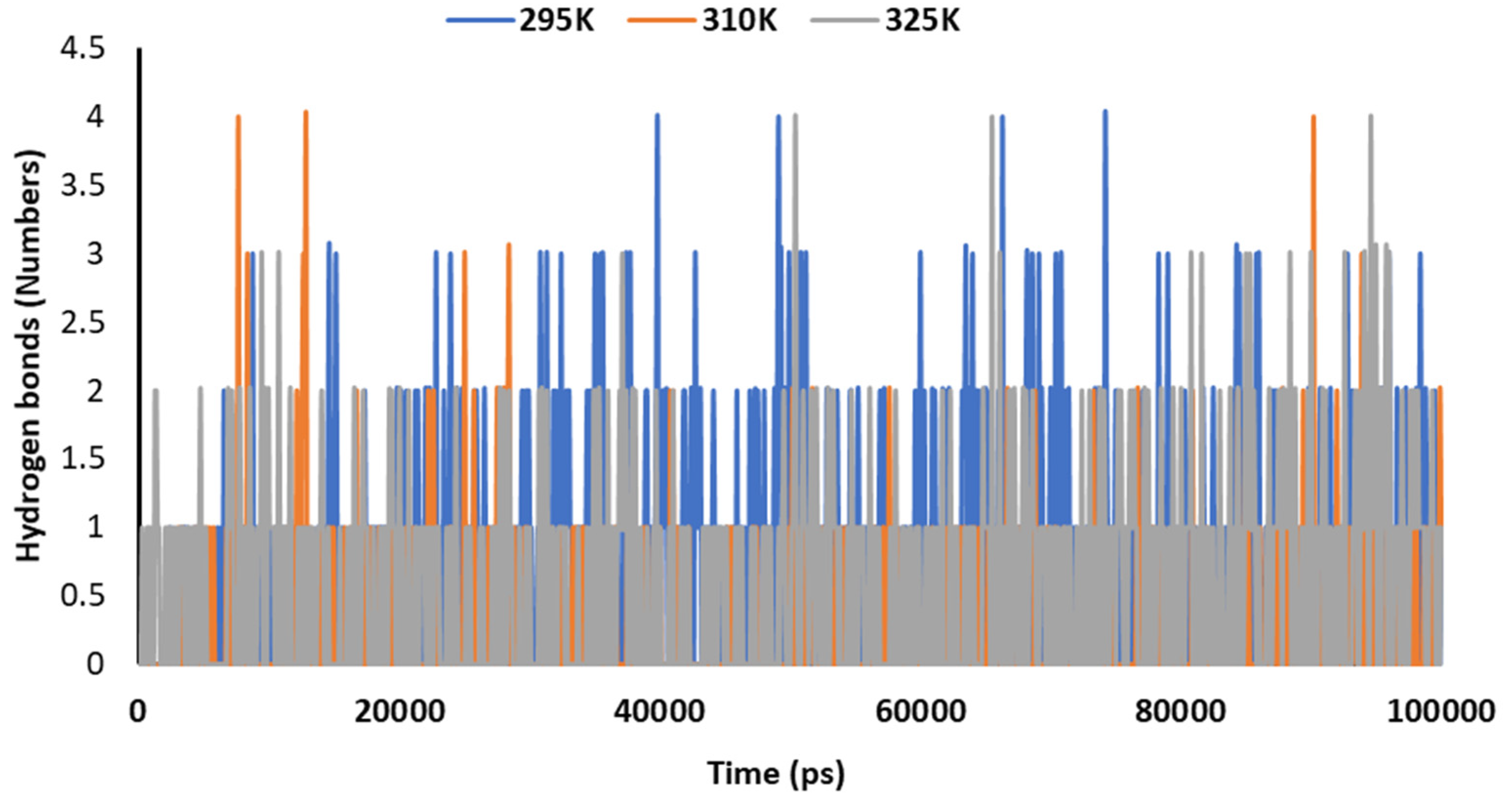
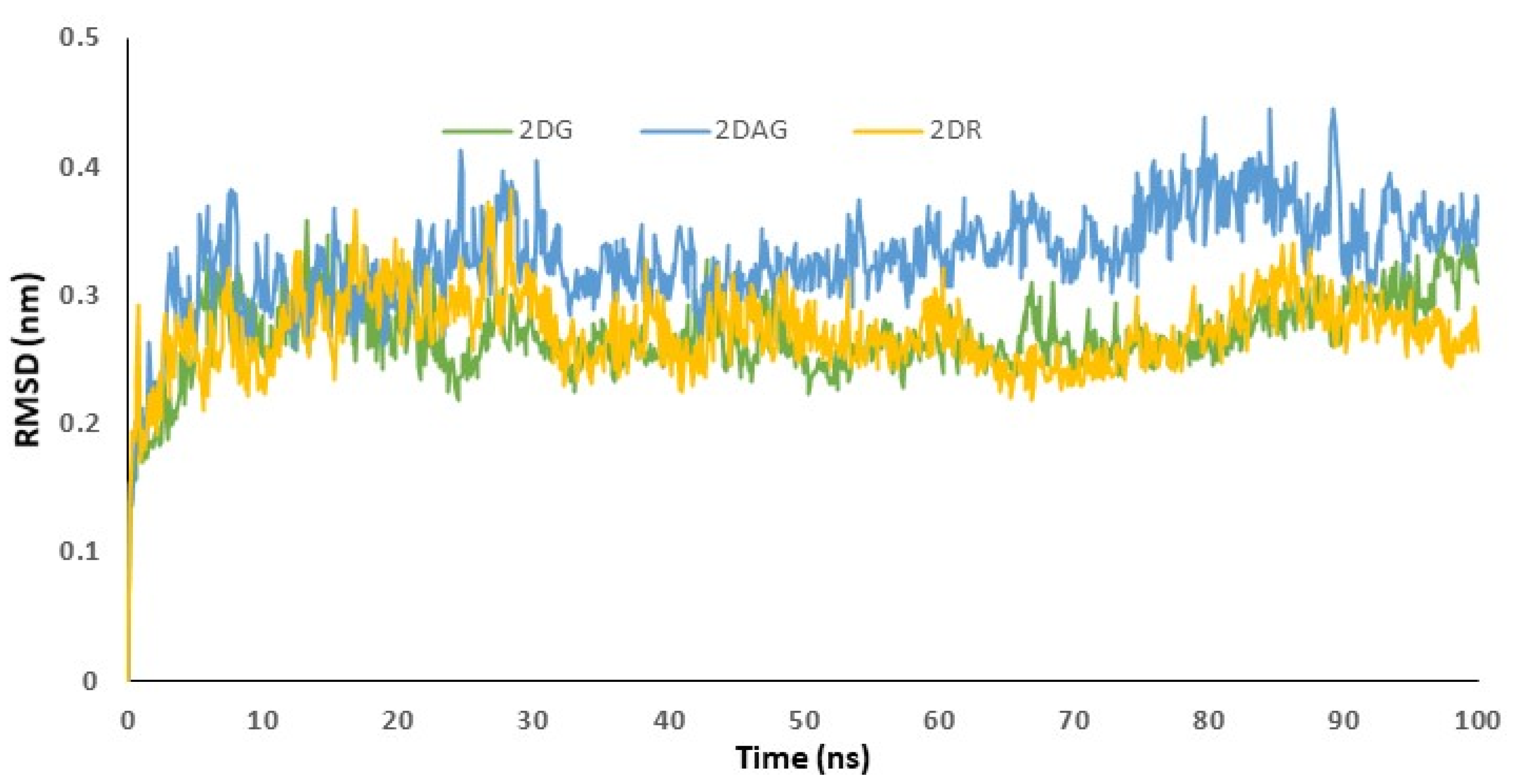
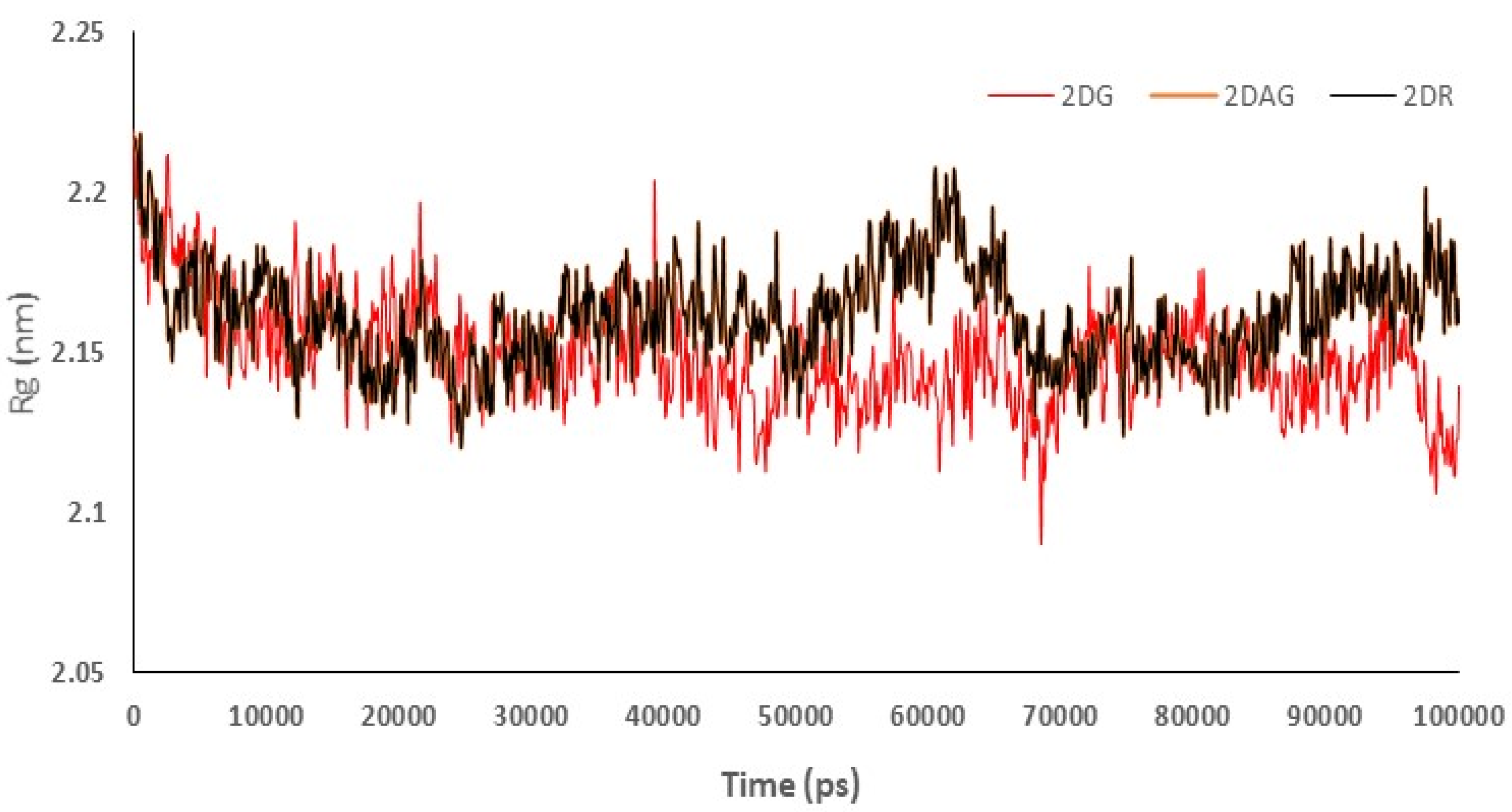
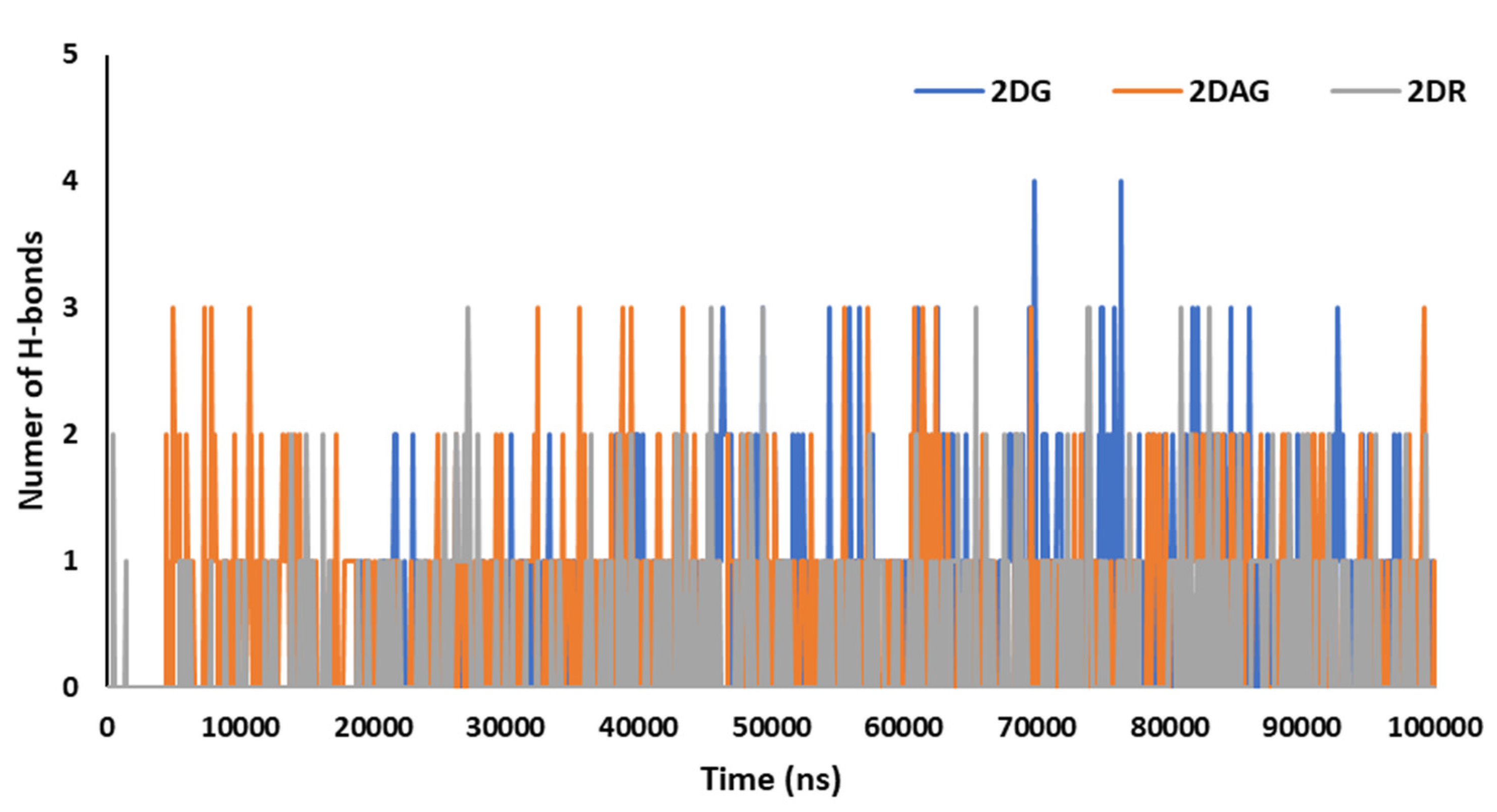
3.3. Molecular Dynamics (MD) Simulations
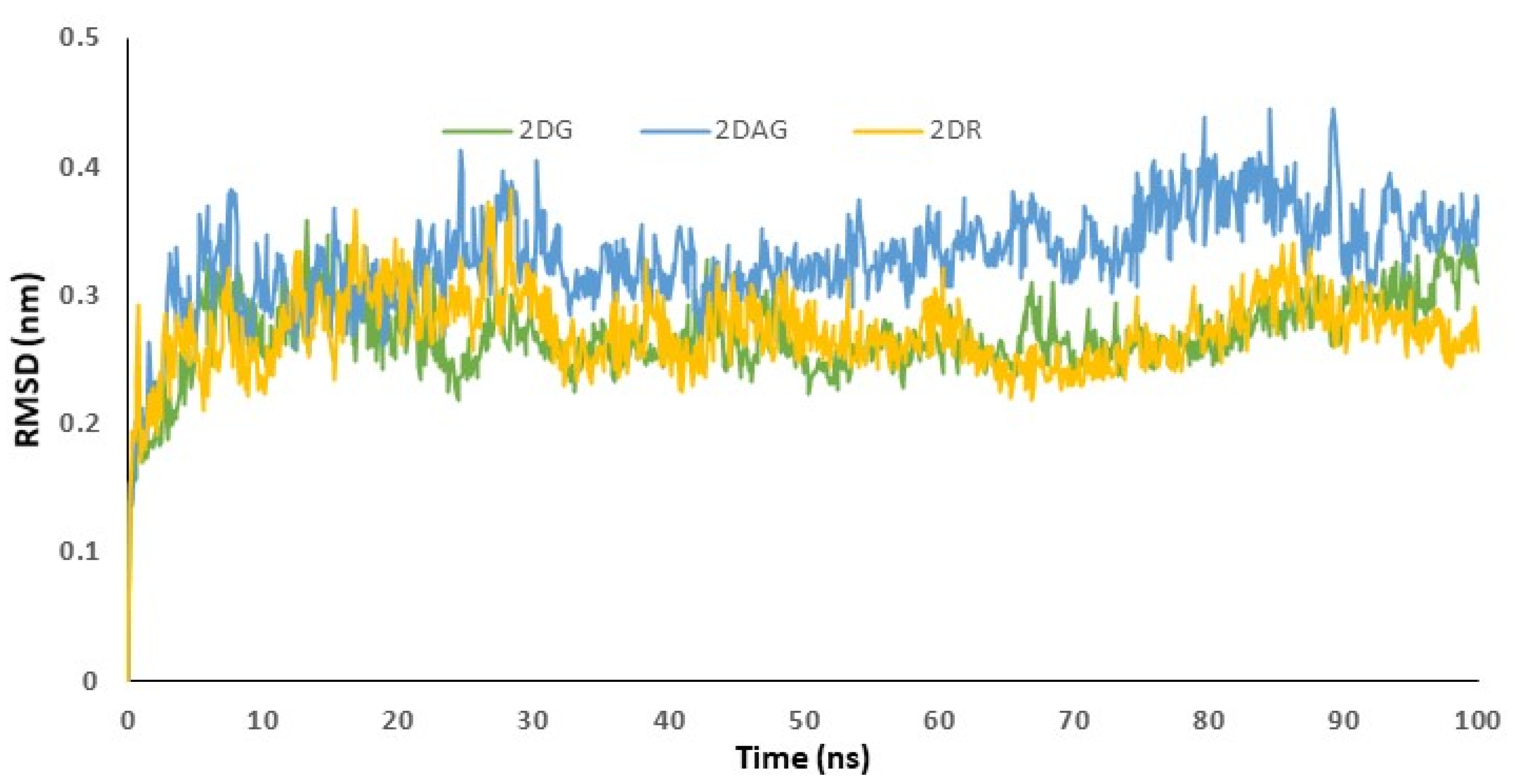
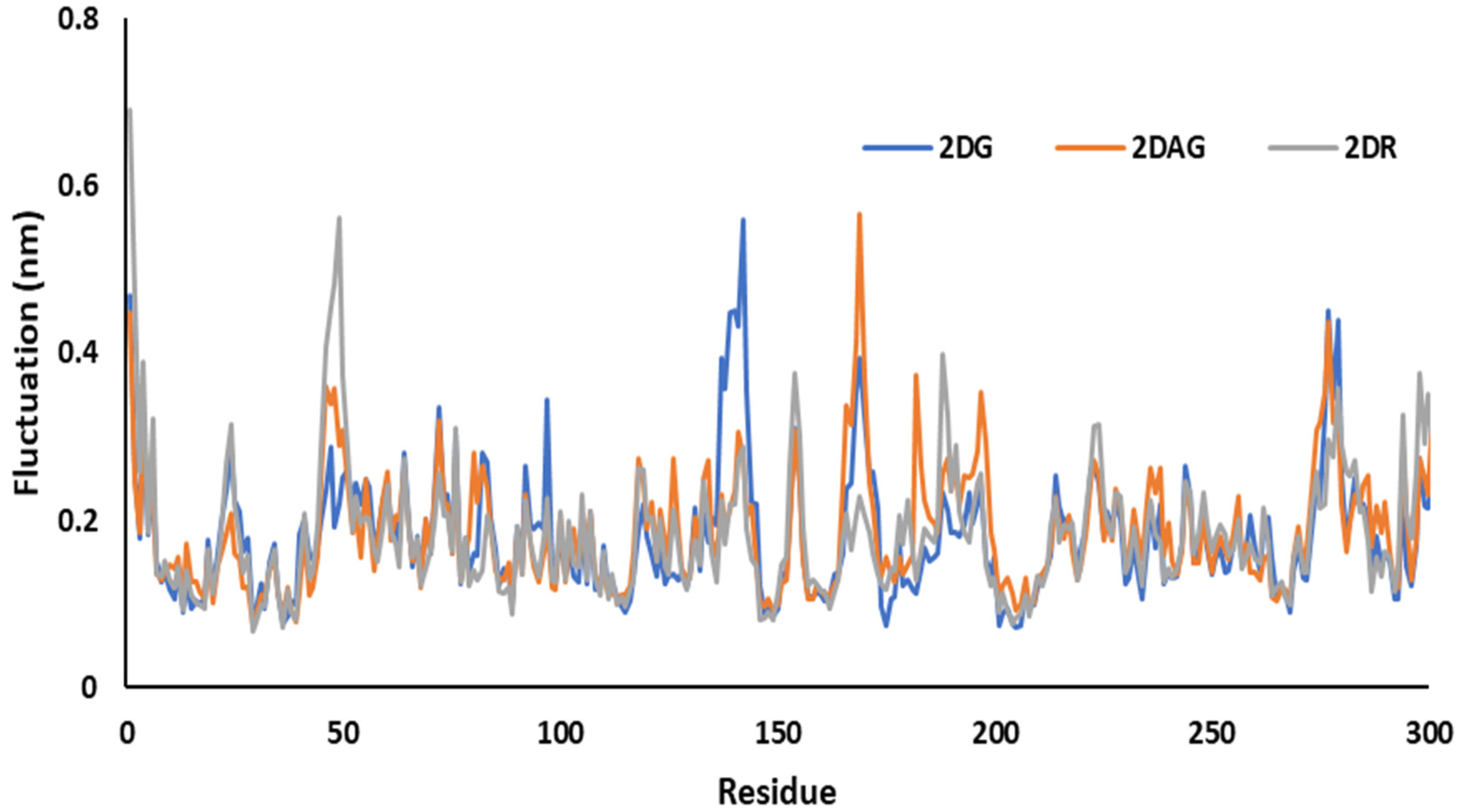
3.4. RMSD Trajectories of 2-Deoxy-D-Ribose (2DR), 2-Deoxy-Glucose (2DAG) and 2-Deoxy-D-Glucose (2DG) with the Main Protease of SARS-CoV-2
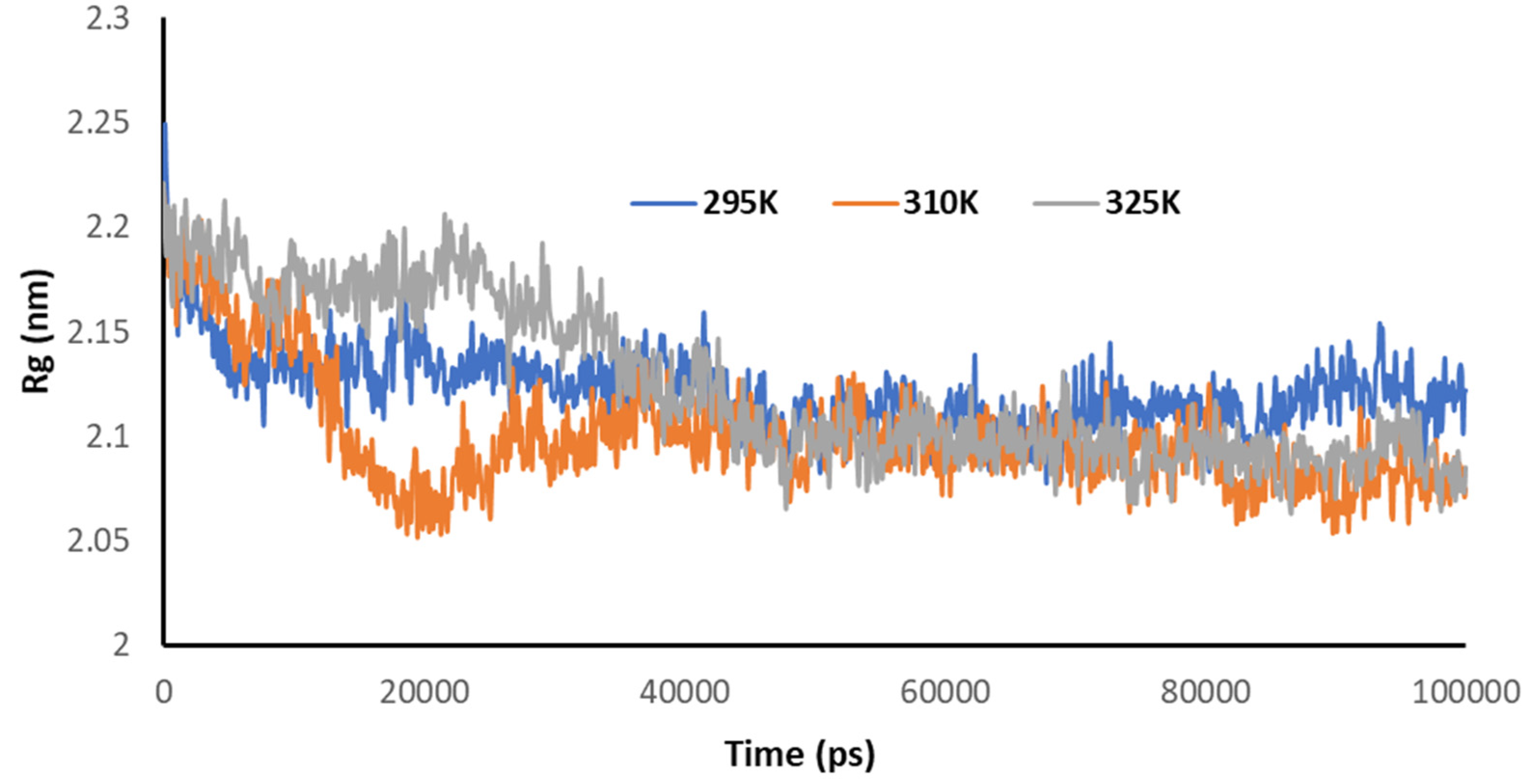
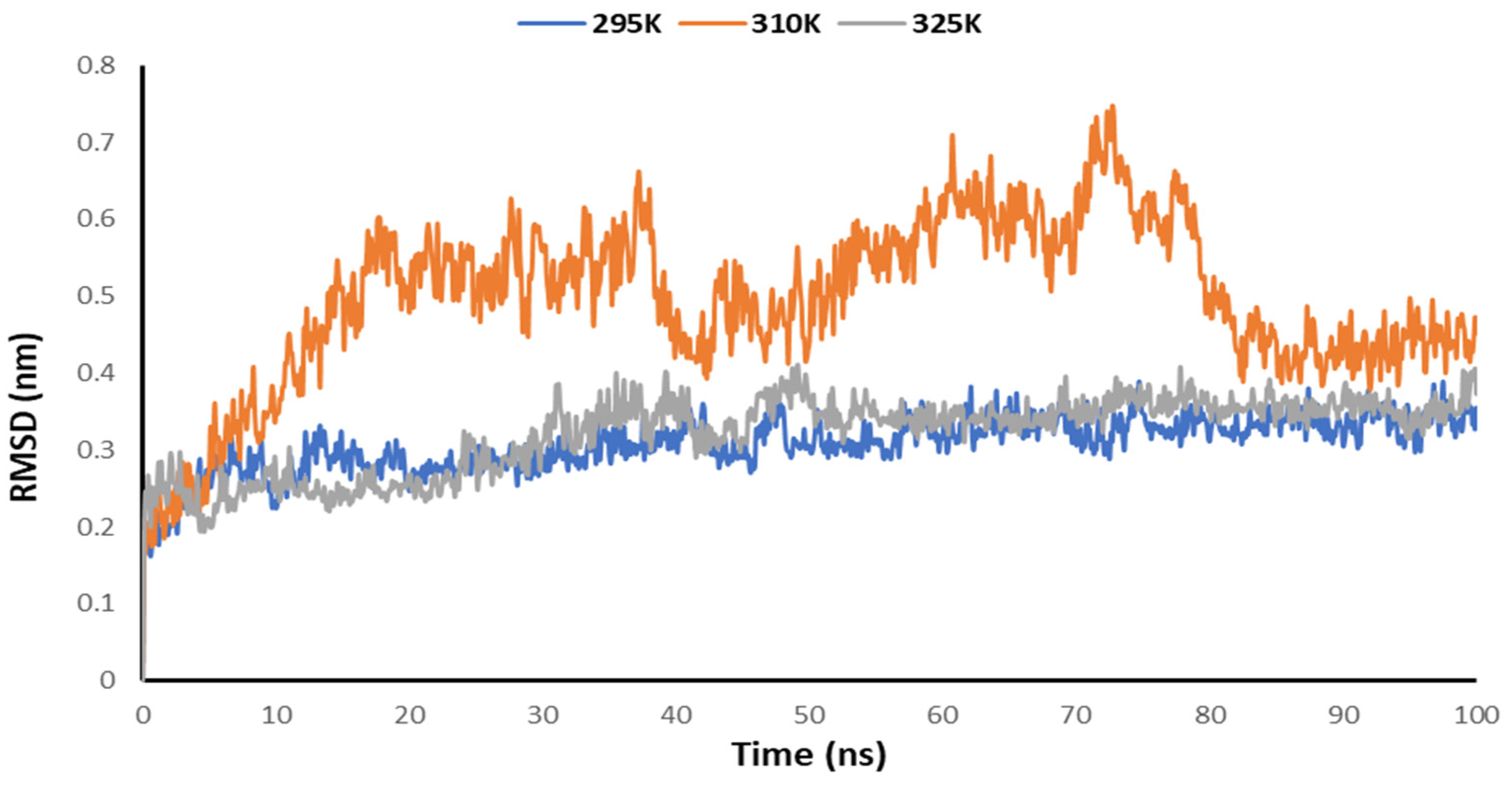
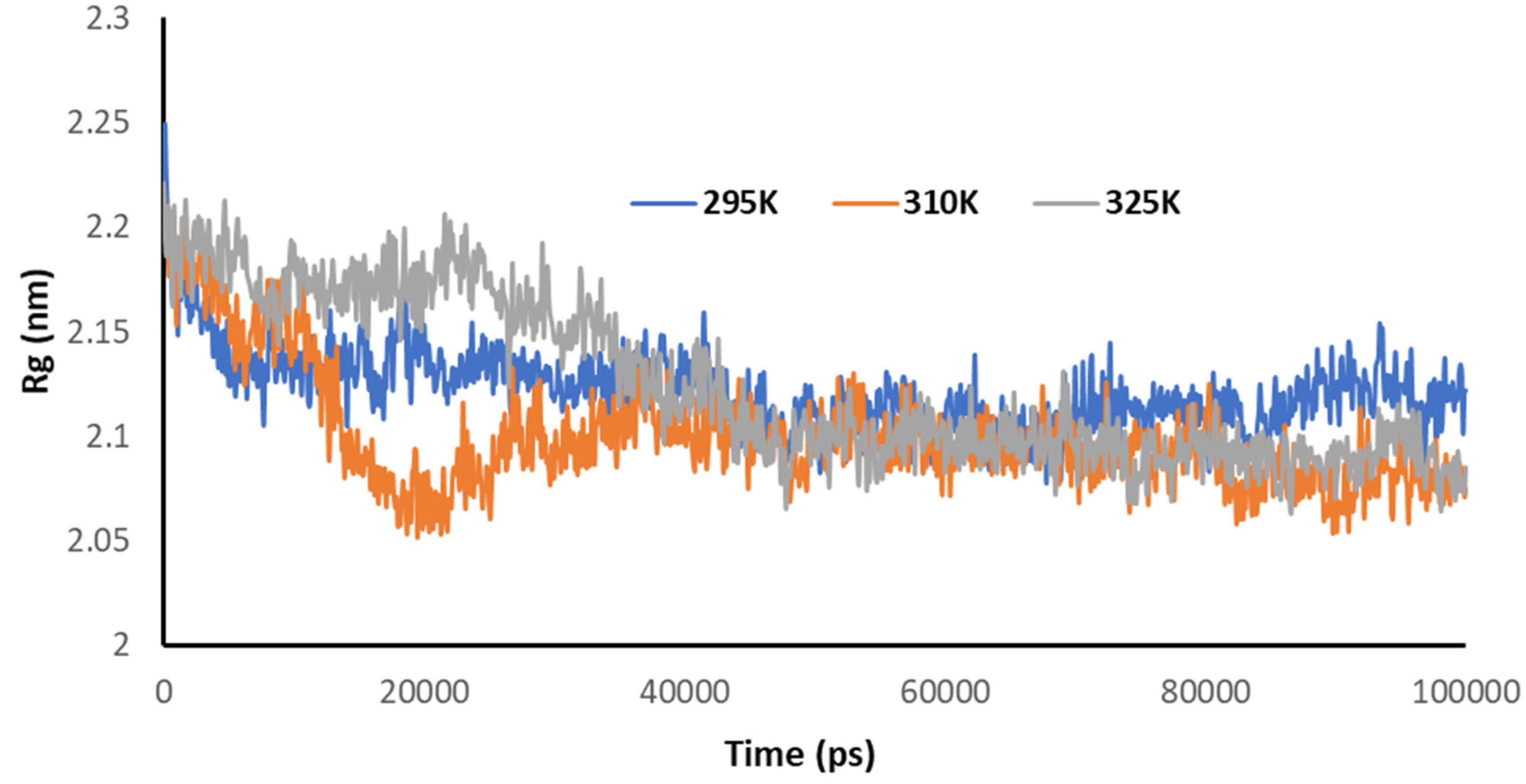
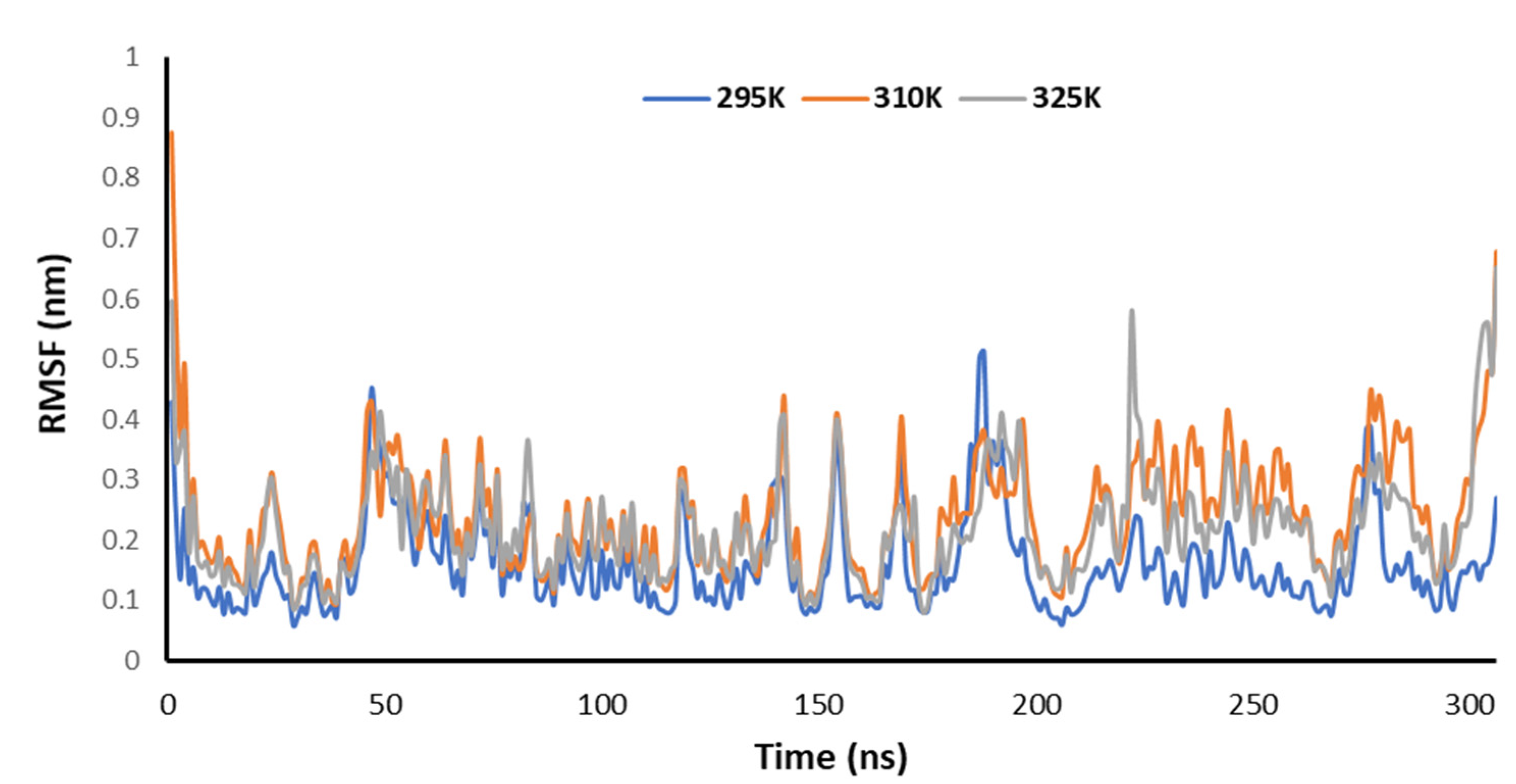
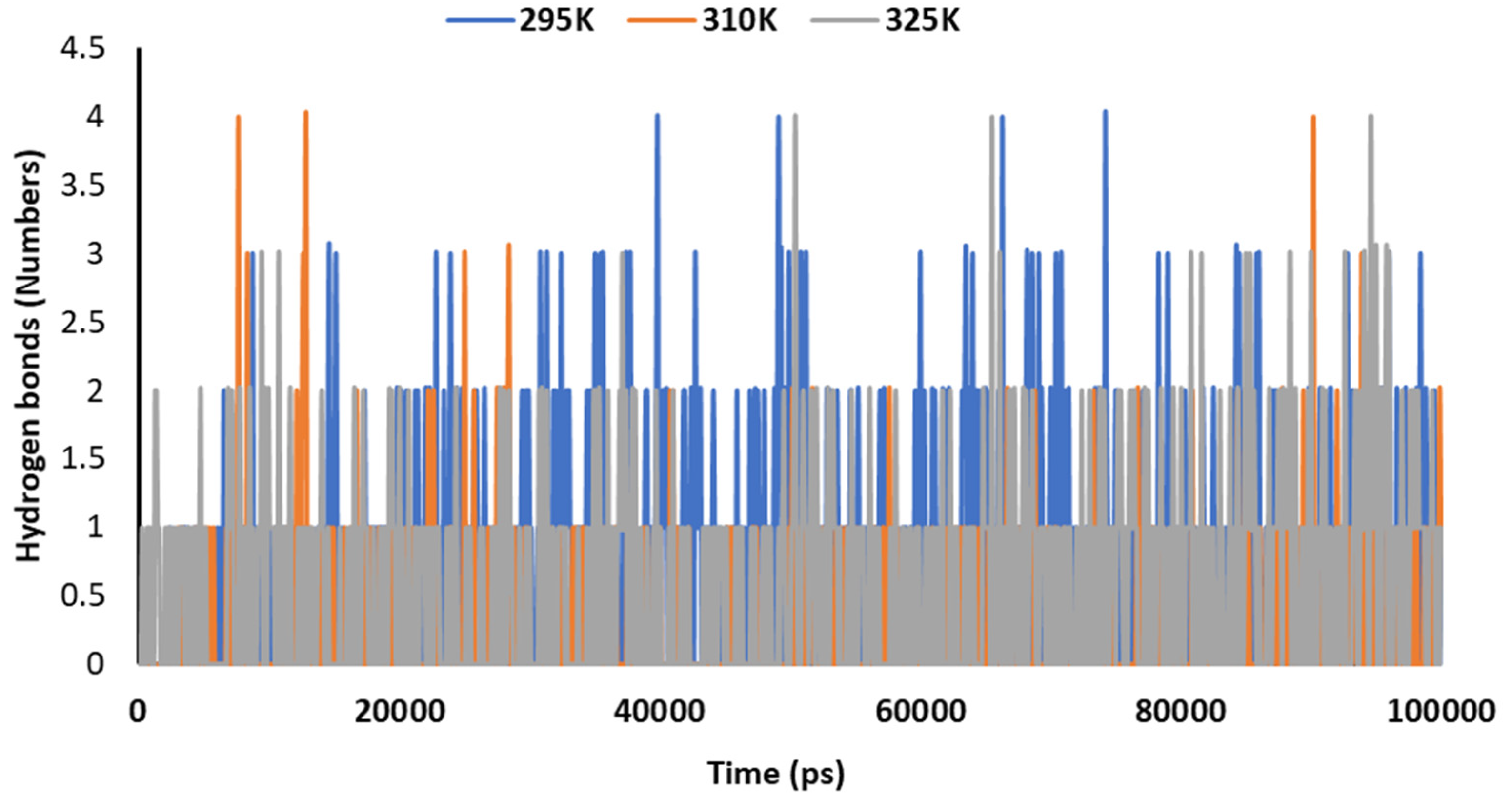
3.5. Temperature-Dependent MD Simulations for Mrpo of nCoV with 2DG
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Acknowledgments
Conflicts of Interest
References
- Singh, R.K.; Drews, M.; De la Sen, M.; Srivastava, P.K.; Trisasongko, B.H.; Kumar, M.; Pandey, M.K.; Anand, A.; Singh, S.S.; Pandey, A.K.; et al. Highlighting the compound risk of COVID-19 and environmental pollutants using geospatial technology. Sci. Rep. 2021, 11, 1–12. [Google Scholar] [CrossRef]
- Saraswat, J.; Singh, P.; Patel, R. A computational approach for the screening of potential antiviral compounds against SARS-CoV-2 protease: Ionic liquid vs herbal and natural compounds. J. Mol. Liq. 2021, 326, 115298. [Google Scholar] [CrossRef] [PubMed]
- Kumar, D.; Kumari, K.; Vishvakarma, V.K.; Jayaraj, A.; Kumar, D.; Ramappa, V.K.; Patel, R.; Kumar, V.; Dass, S.K.; Chandra, R.; et al. Promising inhibitors of main protease of novel corona virus to prevent the spread of COVID-19 using docking and molecular dynamics simulation. J. Biomol. Struct. Dyn. 2021, 39, 4671–4685. [Google Scholar] [CrossRef] [PubMed]
- Hagar, M.; Ahmed, H.A.; Aljohani, G.; Alhaddad, O.A. Investigation of some antiviral N-heterocycles as COVID 19 drug: Molecular docking and DFT calculations. Int. J. Mol. Sci. 2020, 21, 3922. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, R.; Ghosh, M.; Sahoo, S.; Padhi, S.; Misra, N.; Raina, V.; Suar, M.; Son, Y.O. Next-generation bioinformatics approaches and resources for coronavirus vaccine discovery and development—A perspective review. Vaccines 2021, 9, 812. [Google Scholar] [CrossRef]
- Panda, P.K.; Arul, M.N.; Patel, P.; Verma, S.K.; Luo, W.; Rubahn, H.G.; Mishra, Y.K.; Suar, M.; Ahuja, R. Structure-based drug designing and immunoinformatics approach for SARS-CoV-2. Sci. Adv. 2020, 6, eabb8097. [Google Scholar] [CrossRef]
- Kangabam, R.; Sahoo, S.; Ghosh, A.; Roy, R.; Silla, Y.; Misra, N.; Suar, M. Next-generation computational tools and resources for coronavirus research: From detection to vaccine discovery. Comput. Biol. Med. 2021, 128, 104158. [Google Scholar] [CrossRef]
- Gupta, A.; Kumar, S.; Kumar, R.; Choudhary, A.K.; Kumari, K.; Singh, P.; Kumar, V. COVID-19: Emergence of Infectious Diseases, Nanotechnology Aspects, Challenges, and Future Perspectives. ChemistrySelect 2020, 5, 7521–7533. [Google Scholar] [CrossRef]
- Pascarella, G.; Strumia, A.; Piliego, C.; Bruno, F.; Del Buono, R.; Costa, F.; Scarlata, S.; Agrò, F.E. COVID-19 diagnosis and management: A comprehensive review. J. Intern. Med. 2020, 288, 192–206. [Google Scholar] [CrossRef]
- Mishra, D.; Maurya, R.R.; Kumar, K.; Munjal, N.S.; Bahadur, V.; Sharma, S.; Singh, P.; Bahadur, I. Structurally modified compounds of hydroxychloroquine, remdesivir and tetrahydrocannabinol against main protease of SARS-CoV-2, a possible hope for COVID-19: Docking and molecular dynamics simulation studies. J. Mol. Liq. 2021, 335, 116185. [Google Scholar] [CrossRef]
- Peeri, N.C.; Shrestha, N.; Siddikur Rahman, M.; Zaki, R.; Tan, Z.; Bibi, S.; Baghbanzadeh, M.; Aghamohammadi, N.; Zhang, W.; Haque, U. The SARS, MERS and novel coronavirus (COVID-19) epidemics, the newest and biggest global health threats: What lessons have we learned? Int. J. Epidemiol. 2021, 49, 717–726. [Google Scholar] [CrossRef] [Green Version]
- Yavuz, S.Ş.; Ünal, S. Antiviral treatment of COVID-19. Turk. J. Med. Sci. 2020, 50, 611–619. [Google Scholar] [CrossRef] [PubMed]
- Vargas, J.R.N. The COVID-19 pandemic. Rev. Fac. Med. 2020, 68, 7–8. [Google Scholar] [CrossRef]
- Kumar, D.; Kumari, K.; Jayaraj, A.; Kumar, V.; Kumar, R.V.; Dass, S.K.; Chandra, R.; Singh, P. Understanding the binding affinity of noscapines with protease of SARS-CoV-2 for COVID-19 using MD simulations at different temperatures. J. Biomol. Struct. Dyn. 2021, 39, 2659–2672. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, L.C.; Soveri, A.; Lewandowsky, S.; Karlsson, L.; Karlsson, H.; Nolvi, S.; Karukivi, M.; Lindfelt, M.; Antfolk, J. Fearing the disease or the vaccine: The case of COVID-19. Pers. Individ. Dif. 2021, 172, 110590. [Google Scholar] [CrossRef] [PubMed]
- Bojkova, D.; Costa, R.; Reus, P.; Bechtel, M.; Jaboreck, M.; Olmer, R.; Martin, U.; Ciesek, S.; Michaelis, M.; Cinatl, J. Targeting pentose phosphate pathway for SARS-CoV-2 therapy. Metabolites 2021, 11, 699. [Google Scholar] [CrossRef]
- Halder, S.; Mehta, A.K. 2-Deoxy-D-glucose: Is this the final cure for COVID-19: Or yet another mirage? Eur. Rev. Med. Pharmacol. Sci. 2021, 25, 4448–4450. [Google Scholar] [PubMed]
- Sahra, I.B.; Laurent, K.; Giuliano, S.; Larbret, F.; Ponzio, G.; Gounon, P.; Le Marchand-Brustel, Y.; Giorgetti-Peraldi, S.; Cormont, M.; Bertolotto, C.; et al. Targeting cancer cell metabolism: The combination of metformin and 2-deoxyglucose induces p53-dependent apoptosis in prostate cancer cells. Cancer Res. 2010, 70, 2465–2475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Devor, A.; Hillman, E.M.C.; Tian, P.; Waeber, C.; Teng, I.C.; Ruvinskaya, L.; Shalinsky, M.H.; Zhu, H.; Haslinger, R.H.; Narayanan, S.N.; et al. Stimulus-induced changes in blood flow and 2-deoxyglucose uptake dissociate in ipsilateral somatosensory cortex. J. Neurosci. 2008, 28, 14347–14357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zagorodna, O.; Martin, S.M.; Rutkowski, D.T.; Kuwana, T.; Spitz, D.R.; Knudson, C.M. 2-Deoxyglucose-induced toxicity is regulated by Bcl-2 family members and is enhanced by antagonizing Bcl-2 in lymphoma cell lines. Oncogene 2012, 31, 2738–2749. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.Q.; Yung, K.L.K.; Chung, S.K.; Chung, S.M.S. Aldo-keto reductases-mediated cytotoxicity of 2-deoxyglucose: A novel anticancer mechanism. Cancer Sci. 2018, 109, 1970–1980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, D.; Fei, Q.; Li, J.; Zhang, C.; Sun, Y.; Zhu, C.; Wang, F.; Sun, Y. 2-Deoxyglucose reverses the promoting effect of insulin on colorectal cancer cells in vitro. PLoS ONE 2016, 11, e0151115. [Google Scholar] [CrossRef]
- Khan, T.; He, Y.; Kryza, T.; Harrington, B.S.; Gunter, J.H.; Sullivan, M.A.; Cuda, T.; Rogers, R.; Davies, C.M.; Broomfield, A.; et al. Disruption of glycogen utilization markedly improves the efficacy of carboplatin against preclinical models of clear cell ovarian carcinoma. Cancers 2020, 12, 869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishino, K.; Kudo, M.; Peng, W.X.; Kure, S.; Kawahara, K.; Teduka, K.; Kawamoto, Y.; Kitamura, T.; Fujii, T.; Yamamoto, T.; et al. 2-Deoxy-D-glucose increases GFAT1 phosphorylation resulting in endoplasmic reticulum-related apoptosis via disruption of protein N-glycosylation in pancreatic cancer cells. Biochem. Biophys. Res. Commun. 2018, 501, 668–673. [Google Scholar] [CrossRef] [PubMed]
- Amin Hussen, N.H. Docking study of naringin binding with COVID-19 main protease enzyme. Iraqi J. Pharm. Sci. 2021, 29, 231–238. [Google Scholar] [CrossRef]
- Singh, P.; Kumar, D.; Vishvakarma, V.K.; Yadav, P.; Jayaraj, A.; Kumari, K. Computational approach to study the synthesis of noscapine and potential of stereoisomers against nsP3 protease of CHIKV. Heliyon 2019, 5, e02795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, M.B.; Kumar, A.; Jain, P.; Singh, P.; Kumari, K. An insight of novel eutectic mixture between thiazolidine-2,4-dione and zinc chloride: Temperature-dependent density functional theory approach. J. Phys. Org. Chem. 2021. [Google Scholar] [CrossRef]
- Gupta, A.; Sharma, P.; Jayaram, B. ParDOCK: An All Atom Energy Based Monte Carlo Docking Protocol for Protein-Ligand Complexes. Protein Pept. Lett. 2007, 14, 632–646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bekker, H.; Berendsen, H.J.C.; Dijkstra, E.J.; Achterop, S.; Vondrumen, R.; Vanderspoel, D.; Sijbers, A.; Keegstra, H.; Renardus, M.K.R. Gromacs—A Parallel Computer for Molecular-Dynamics Simulations; DeGroot, R.A., Nadrchal, J., Eds.; World Scientific Publishing: Singapore, 1993; pp. 252–256. [Google Scholar]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindah, E. Gromacs: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Paz, J.O.; Batchelor, W.D.; Pedersen, P. WebGro: A web-based soybean management decision support system. Agron. J. 2004, 96, 1771–1779. [Google Scholar] [CrossRef]
- Brown, T. ChemDraw. Sci. Teach. 2014, 81, 67. [Google Scholar]
- Goddard, T.D.; Huang, C.C.; Meng, E.C.; Pettersen, E.F.; Couch, G.S.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Meeting modern challenges in visualization and analysis. Protein Sci. 2018, 27, 14–25. [Google Scholar] [CrossRef] [PubMed]
- Butt, S.S.; Badshah, Y.; Shabbir, M.; Rafiq, M. Molecular Docking Using Chimera and Autodock Vina Software for Nonbioinformaticians. JMIR Bioinform. Biotechnol. 2020, 1, e14232. [Google Scholar] [CrossRef]
- Kumar, V.V.; Indra, B.; Kamlesh, K.; Prashant, S. Thiazolidinones: Potential Human Novel Coronavirus (SARS-CoV-2) Protease Inhibitors Against COVID-19. chemRxiv 2020. Available online: https://chemrxiv.org/engage/chemrxiv/article-details/60c74fb3bdbb8987e4a39ded (accessed on 28 December 2021). [CrossRef]
- Vishvakarma, V.K.; Kumari, K.; Patel, R.; Dixit, V.S.; Singh, P.; Mehrotra, G.K.; Chandra, R.; Chakrawarty, A.K. Theoretical model to investigate the alkyl chain and anion dependent interactions of gemini surfactant with bovine serum albumin. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2015, 143, 319–323. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Zhu, Q.; Huang, C.; Yang, M.; Li, J.; Chen, Y.; Yang, B.; Zhao, X. Comparative cytotoxicity of halogenated aromatic DBPs and implications of the corresponding developed QSAR model to toxicity mechanisms of those DBPs: Binding interactions between aromatic DBPs and catalase play an important role. Water Res. 2020, 170, 115283. [Google Scholar] [CrossRef]
- Vishvakarma, V.K.; Nand, B.; Kumar, V.; Kumari, K.; Bahadur, I.; Singh, P. Xanthene based hybrid analogues to inhibit protease of novel corona Virus: Molecular docking and ADMET studies. Comput. Toxicol. 2020, 16, 100140. [Google Scholar] [CrossRef]
- Meena, M.K.; Kumar, D.; Kumari, K.; Kaushik, N.K.; Kumar, R.V.; Bahadur, I.; Vodwal, L.; Singh, P. Promising inhibitors of nsp2 of CHIKV using molecular docking and temperature-dependent molecular dynamics simulations. J. Biomol. Struct. Dyn. 2020, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Kumari, K.; Kumar, A.; Bahadur, I.; Singh, P. Investigate the interaction of testosterone/progesterone with ionic liquids on varying the anion to combat COVID-19: Density functional theory calculations and molecular docking approach. J. Phys. Org. Chem. 2021, 34, e4273. [Google Scholar] [CrossRef]
- Kumar, R.V.; Srivastava, D.; Singh, V.; Kumar, U.; Vishvakarma, V.K.; Singh, P.; Kumar, D.; Kumar, R. Characterization, biological evaluation and molecular docking of mulberry fruit pectin. Sci. Rep. 2020, 10, 21789. [Google Scholar] [CrossRef] [PubMed]
- Kumar, D.; Kumari, K.; Chandra, R.; Jain, P.; Vodwal, L.; Gambhir, G.; Singh, P. A review targeting the infection by CHIKV using computational and experimental approaches. J. Biomol. Struct. Dyn. 2021, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Vishvakarma, V.K.; Chandra, R.; Singh, P. An experimental and theoretical approach to understand Fever, DENF & its cure. Infect. Disord. Drug Targets 2021, 21, 495–513. [Google Scholar] [CrossRef] [PubMed]
- Sinha, S.K.; Prasad, S.K.; Islam, M.A.; Gurav, S.S.; Patil, R.B.; AlFaris, N.A.; Aldayel, T.S.; AlKehayez, N.M.; Wabaidur, S.M.; Shakya, A. Identification of bioactive compounds from Glycyrrhiza glabra as possible inhibitor of SARS-CoV-2 spike glycoprotein and non-structural protein-15: A pharmacoinformatics study. J. Biomol. Struct. Dyn. 2021, 39, 4686–4700. [Google Scholar] [CrossRef] [PubMed]
- Kumar, D.; Singh, P.; Jayaraj, A.; Kumar, V.; Kumari, K.; Patel, R. A Theoretical Model to Study the Interaction of Erythro-Noscapines with nsP3 protease of Chikungunya Virus. ChemistrySelect 2019, 4, 4892–4900. [Google Scholar] [CrossRef]
- Sharma, S.; Deep, S. In-silico drug repurposing for targeting SARS-CoV-2 main protease (Mpro). J. Biomol. Struct. Dyn. 2020, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Lindahl, E.; Bjelkmar, P.; Larsson, P.; Cuendet, M.A.; Hess, B. Implementation of the charmm force field in GROMACS: Analysis of protein stability effects from correction maps, virtual interaction sites, and water models. J. Chem. Theory Comput. 2010, 6, 459–466. [Google Scholar] [CrossRef]
- Lindorff-Larsen, K.; Piana, S.; Palmo, K.; Maragakis, P.; Klepeis, J.L.; Dror, R.O.; Shaw, D.E. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins Struct. Funct. Bioinform. 2010, 78, 1950–1958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schüttelkopf, A.W.; Van Aalten, D.M.F. PRODRG: A tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr. Sect. D Biol. Crystallogr. 2004, 60, 1355–1363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oostenbrink, C.; Villa, A.; Mark, A.E.; Van Gunsteren, W.F. A biomolecular force field based on the free enthalpy of hydration and solvation: The GROMOS force-field parameter sets 53A5 and 53A6. J. Comput. Chem. 2004, 25, 1656–1676. [Google Scholar] [CrossRef]
- Vishvakarma, V.K.; Singh, M.B.; Jain, P.; Kumari, K.; Singh, P. Hunting the main protease of SARS-CoV-2 by plitidepsin: Molecular docking and temperature-dependent molecular dynamics simulations. Amino Acids. 2021, 1–9. [Google Scholar] [CrossRef]
- Vishvakarma, V.K.; Pal, S.; Singh, P.; Bahadur, I. Interactions between main protease of SARS-CoV-2 and testosterone or progesterone using computational approach. J. Mol. Struct. 2021, 131965. [Google Scholar] [CrossRef]
- Kumar, D.; Kumari, K.; Jayaraj, A.; Singh, P. Development of a theoretical model for the inhibition of nsP3 protease of Chikungunya virus using pyranooxazoles. J. Biomol. Struct. Dyn. 2020, 38, 3018–3034. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16; Rev. C.01; Gaussian, Inc.: Pittsburgh, PA, USA, 2016. [Google Scholar]
- Kumar, A.; Kumari, K.; Bahadur, I.; Singh, P. Temperature dependent DFT studies to understand the physiochemical interaction of lithium chloride cluster ions in presence of syringic acid. J. Chem. Thermodyn. 2021, 152, 106277. [Google Scholar] [CrossRef]
- Kumar, A.; Kumar, D.; Kumar, R.; Singh, P.; Chandra, R.; Kumari, K. DFT and docking studies of designed conjugates of noscapines & repurposing drugs: Promising inhibitors of main protease of SARS-CoV-2 and falcipan-2. J. Biomol. Struct. Dyn. 2020, 1–21. [Google Scholar] [CrossRef]
- Vishvakarma, V.K.; Singh, P.; Kumar, V.; Kumari, K.; Patel, R.; Chandra, R. Pyrrolothiazolones as Potential Inhibitors for the nsP2B-nsP3 Protease of Dengue Virus and Their Mechanism of Synthesis. ChemistrySelect 2019, 4, 9410–9419. [Google Scholar] [CrossRef]
- Singh, H.; Singh, A.; Banipal, T.S.; Singh, P.; Bahadur, I. Temperature and concentration dependent physicochemical interactions of L-ascorbic acid in aqueous LiCl solution: Experimental and theoretical study. Colloids Surf. A Physicochem. Eng. Asp. 2021, 623, 126672. [Google Scholar] [CrossRef]
- Miranda, F.S.; Ronconi, C.M.; Sousa, M.O.B.; Silveira, G.Q.; Vargas, M.D. 6-Aminocoumarin-naphthoquinone conjugates: Design, synthesis, photophysical and electrochemical properties and DFT calculations. J. Braz. Chem. Soc. 2014, 25, 133–142. [Google Scholar] [CrossRef]
- Tai, C.K.; Chen, Y.J.; Chang, H.W.; Yeh, P.L.; Wang, B.C. DFT and TD-DFT investigations of metal-free dye sensitizers for solar cells: Effects of electron donors and π-conjugated linker. Comput. Theor. Chem. 2011, 971, 42–50. [Google Scholar] [CrossRef]
- Langhoff, S.R.; Bauschlicher, C.W. A Theoretical Study of the Electric Dipole Moment Function of SiO; Elsevier: Amsterdam, The Netherlands, 1993. [Google Scholar]
- Hagar, M.; Ahmed, H.A.; Alhaddadd, O.A. DFT calculations and mesophase study of coumarin esters and its azoesters. Crystals 2018, 8, 359. [Google Scholar] [CrossRef] [Green Version]
- Wungu, T.D.K.; Marsha, S.E. Density Functional Theory (DFT) Study of Molecularly Imprinted Polymer (MIP) Methacrylic Acid (MAA) with D-Glucose. IOP Conf. Ser. Mater. Sci. Eng. 2017, 214, 012004. [Google Scholar] [CrossRef] [Green Version]
- Singh, V.K.; Chaurasia, H.; Mishra, R.; Srivastava, R.; Naaz, F.; Kumar, P.; Singh, R.K. Docking, ADMET prediction, DFT analysis, synthesis, cytotoxicity, antibacterial screening and QSAR analysis of diarylpyrimidine derivatives. J. Mol. Struct. 2022, 1247, 131400. [Google Scholar] [CrossRef]
- Kumar, A.; Kumari, K.; Singh, S.; Bahdur, I.; Singh, P. Noscapine anticancer drug designed with ionic liquids to enhance solubility: DFT and ADME approach. J. Mol. Liq. 2021, 325, 115159. [Google Scholar] [CrossRef]
- Vishvakarma, V.K.; Shukla, N.; Kumari, K.; Patel, R.; Singh, P. A model to study the inhibition of nsP2B-nsP3 protease of dengue virus with imidazole, oxazole, triazole thiadiazole, and thiazolidine based scaffolds. Heliyon 2019, 5, e02124. [Google Scholar] [CrossRef] [Green Version]
- Schreiner, W.; Karch, R.; Knapp, B.; Ilieva, N. Relaxation Estimation of RMSD in Molecular Dynamics Immunosimulations. Comput. Math. Methods Med. 2012, 2012, 173521. [Google Scholar] [CrossRef]
- Kumar, D.; Meena, M.K.; Kumari, K.; Patel, R.; Jayaraj, A.; Singh, P. In-silico prediction of novel drug-target complex of nsp3 of CHIKV through molecular dynamic simulation. Heliyon 2020, 6, e04720. [Google Scholar] [CrossRef]
- Kumar, D.; Meena, M.K.; Kumari, K.; Kumar, R.V.; Bahadur, I.; Jain, P.; Singh, P. Exploring the effect of temperature on inhibition of non-structural protease 3 of Chikungunya virus using molecular dynamics simulations and thermodynamics parameters. J. Mol. Liq. 2021, 335, 116164. [Google Scholar] [CrossRef]
- Prasanth, D.S.N.B.K.; Murahari, M.; Chandramohan, V.; Panda, S.P.; Atmakuri, L.R.; Guntupalli, C. In silico identification of potential inhibitors from Cinnamon against main protease and spike glycoprotein of SARS CoV-2. J. Biomol. Struct. Dyn. 2020, 39, 1–15. [Google Scholar] [CrossRef]
- Shree, P.; Mishra, P.; Selvaraj, C.; Singh, S.K.; Chaube, R.; Garg, N.; Tripathi, Y.B. Targeting COVID-19 (SARS-CoV-2) main protease through active phytochemicals of ayurvedic medicinal plants–Withania somnifera (Ashwagandha), Tinospora cordifolia (Giloy) and Ocimum sanctum (Tulsi)–a molecular docking study. J. Biomol. Struct. Dyn. 2022, 40, 190–203. [Google Scholar] [CrossRef]
- Bello, M.; Martínez-Muñoz, A.; Balbuena-Rebolledo, I. Identification of saquinavir as a potent inhibitor of dimeric SARS-CoV2 main protease through MM/GBSA. J. Mol. Model. 2020, 26, 340. [Google Scholar] [CrossRef]
- Lemkul, J.A.; Allen, W.J.; Bevan, D.R. Practical considerations for building GROMOS-compatible small-molecule topologies. J. Chem. Inf. Model. 2010, 50, 2221–2235. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Solvent | Sum of Electronic and Zero-Point Energies | Sum of Electronic and Thermal Energies | Sum of Electronic and Thermal Enthalpies | Sum of Electronic and Thermal Free Energies | Optimization Energy | Dipole Moment | |
|---|---|---|---|---|---|---|---|
| 2DG | Default | −611.93 | −611.92 | −611.91 | −611.97 | −612.12 | 3.4 |
| Water | −611.95 | −611.94 | −611.94 | −611.98 | −612.14 | 4.7 | |
| 2DAG | Default | −611.93 | −611.92 | −611.91 | −611.97 | −612.12 | 8.17 |
| Water | −611.95 | −611.94 | −611.94 | −611.99 | −612.14 | 10.04 | |
| 2DR | Default | −497.40 | −497.39 | −497.39 | −497.44 | −497.56 | 2.35 |
| Water | −497.42 | −497.41 | −497.45 | −497.45 | −497.58 | 3.1 |
| S. No. | CMPD | Binding Energy (kcal/mol) |
|---|---|---|
| 1. | 2DG | −2.40 |
| 2. | 2DR | −2.22 |
| 3. | 2DAG | −2.88 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Raman, A.P.S.; Kumari, K.; Jain, P.; Vishvakarma, V.K.; Kumar, A.; Kaushik, N.; Choi, E.H.; Kaushik, N.K.; Singh, P. In Silico Evaluation of Binding of 2-Deoxy-D-Glucose with Mpro of nCoV to Combat COVID-19. Pharmaceutics 2022, 14, 135. https://doi.org/10.3390/pharmaceutics14010135
Raman APS, Kumari K, Jain P, Vishvakarma VK, Kumar A, Kaushik N, Choi EH, Kaushik NK, Singh P. In Silico Evaluation of Binding of 2-Deoxy-D-Glucose with Mpro of nCoV to Combat COVID-19. Pharmaceutics. 2022; 14(1):135. https://doi.org/10.3390/pharmaceutics14010135
Chicago/Turabian StyleRaman, Anirudh Pratap Singh, Kamlesh Kumari, Pallavi Jain, Vijay Kumar Vishvakarma, Ajay Kumar, Neha Kaushik, Eun Ha Choi, Nagendra Kumar Kaushik, and Prashant Singh. 2022. "In Silico Evaluation of Binding of 2-Deoxy-D-Glucose with Mpro of nCoV to Combat COVID-19" Pharmaceutics 14, no. 1: 135. https://doi.org/10.3390/pharmaceutics14010135
APA StyleRaman, A. P. S., Kumari, K., Jain, P., Vishvakarma, V. K., Kumar, A., Kaushik, N., Choi, E. H., Kaushik, N. K., & Singh, P. (2022). In Silico Evaluation of Binding of 2-Deoxy-D-Glucose with Mpro of nCoV to Combat COVID-19. Pharmaceutics, 14(1), 135. https://doi.org/10.3390/pharmaceutics14010135