
Celecoxib-Loaded Solid Lipid Nanoparticles for Colon Delivery: Formulation Optimization and In Vitro Assessment of Anti-Cancer Activity


 ,
, 
Abstract
1. Introduction
2. Materials and Methods
2.1. Preparation of Solid Lipid Nanoparticles
2.2. Analytical Method
2.3. Evaluation of the Prepared SLN
2.3.1. Particle Size and Polydispersity Evaluation
2.3.2. Measurement of Zeta Potential
2.3.3. Measurement of Drug Entrapment and Drug Loading
2.3.4. Differential Scanning Calorimetry
2.3.5. Fourier Transform Infrared Spectroscopy
2.3.6. Particle Morphology
2.4. In Vitro Release Profile Study
2.5. Release Kinetic Analysis
2.6. Cell Lines
2.7. In Vitro Cytotoxicity Studies
2.8. Statistical Analysis
3. Results
3.1. Particle Size and Zeta Potential
3.2. Drug Loading and Entrapment Efficiency
3.3. Differential Scanning Calorimetry
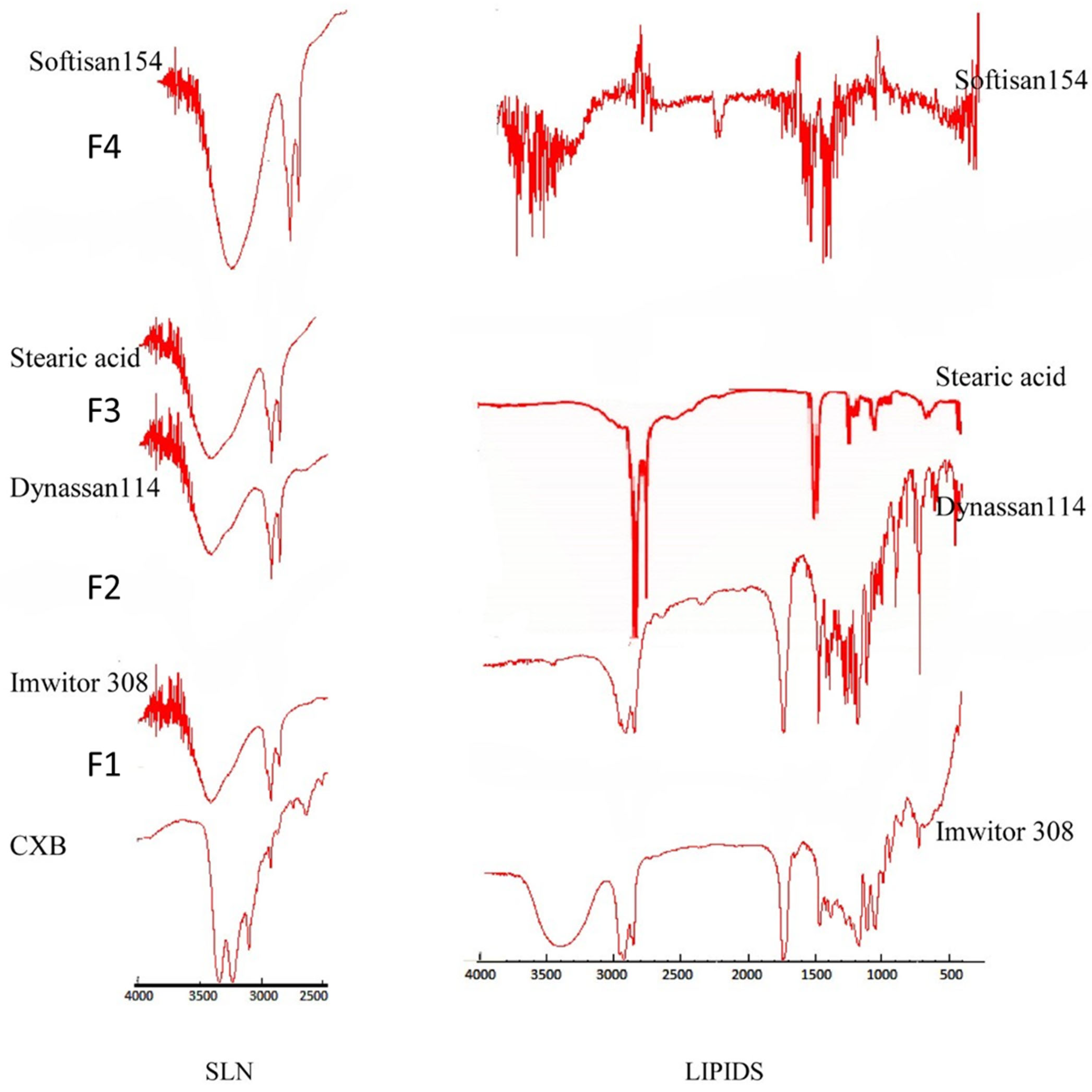
3.4. Fourier Transform Infrared Spectroscopy
3.5. Particle Morphology
3.6. In Vitro Release
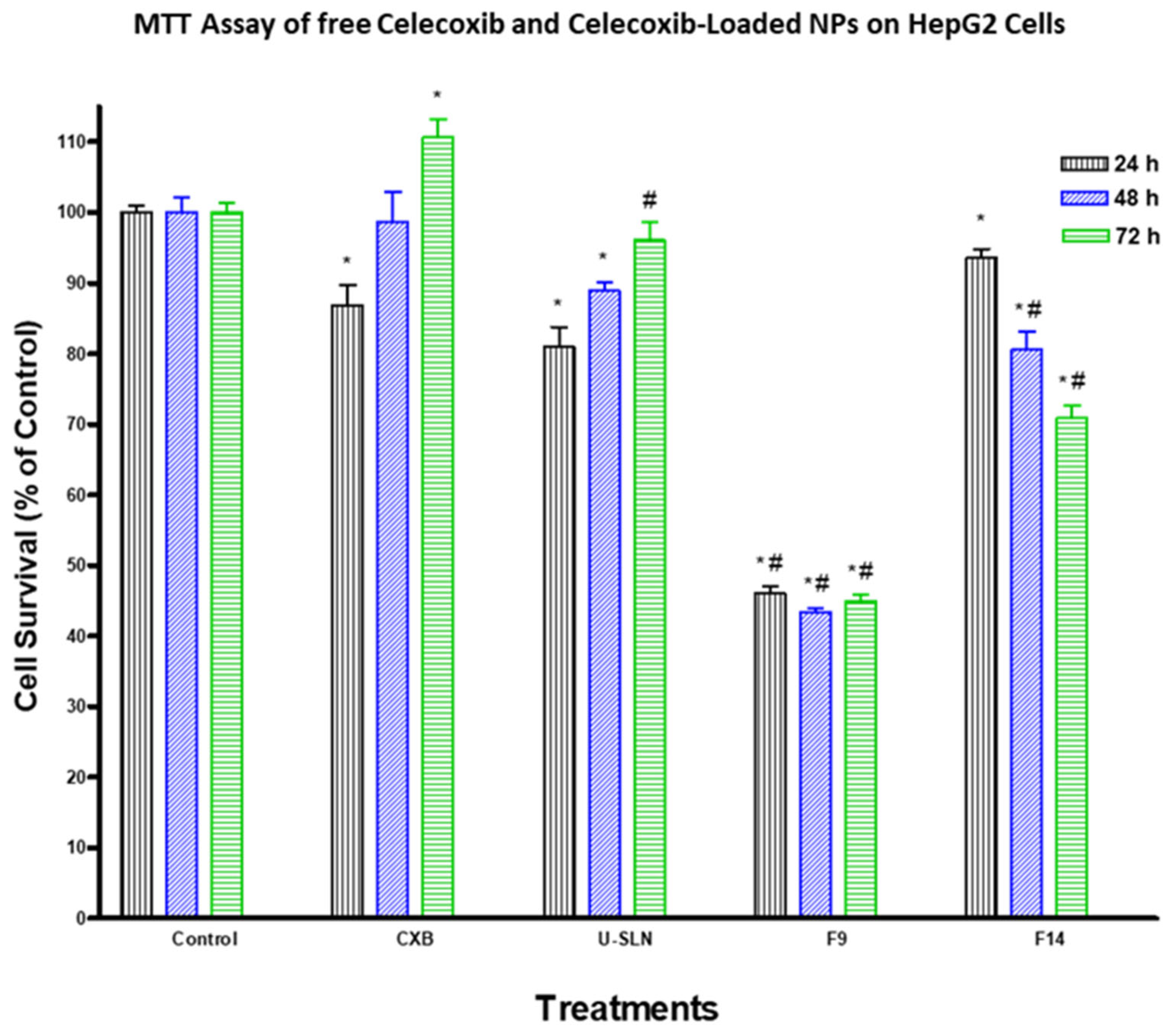
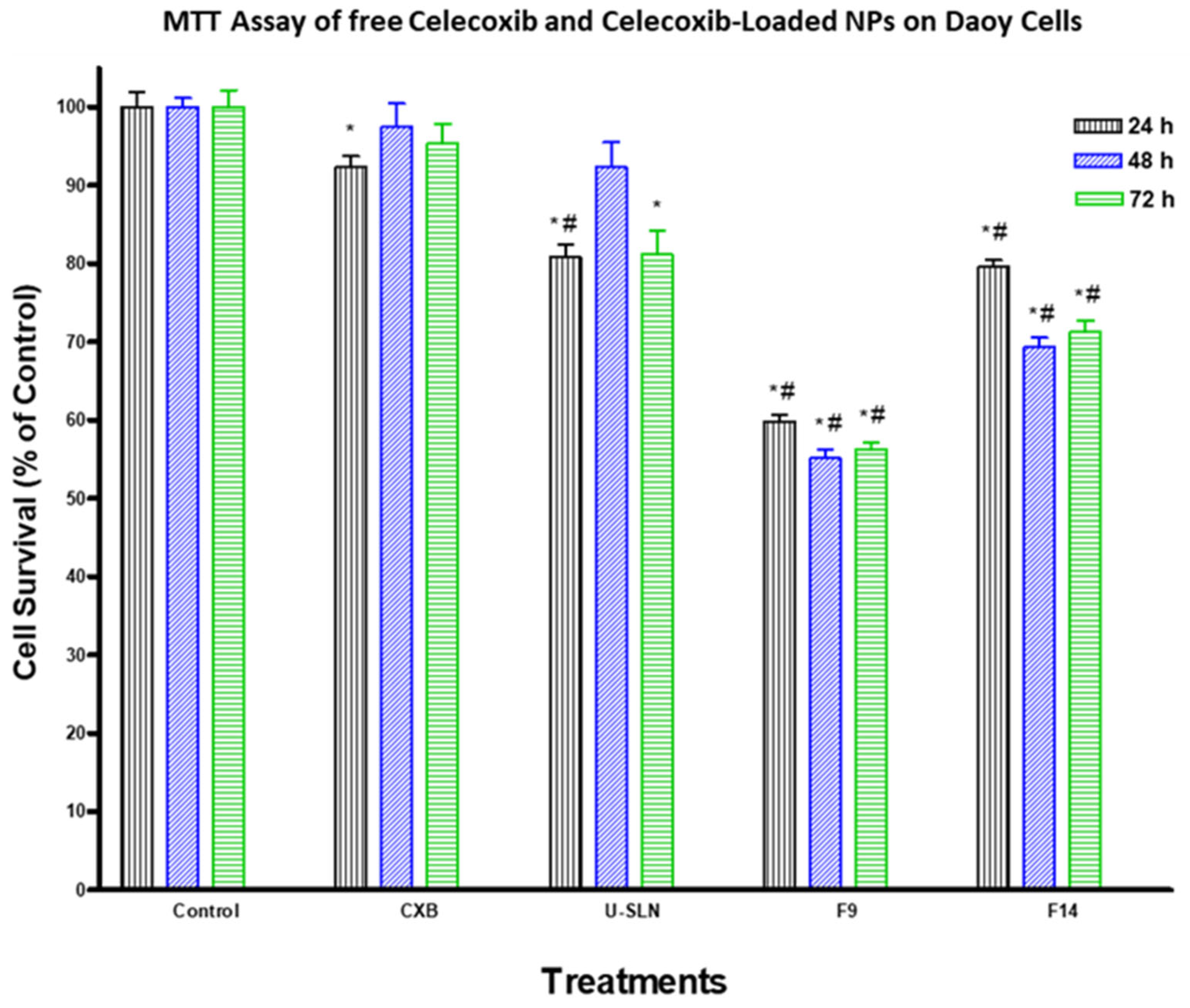
3.7. In Vitro Cytotoxicity
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Paulson, S.; Vaughn, M.; Jessen, S.; Lawal, Y.; Gresk, C.; Yan, B.; Maziasz, T.; Cook, C.; Karim, A. Pharmacokinetics of Celecoxib after oral administration in dogs and humans: Effet of food and site of absorption. J. Pharmacol Exp. Ther. 2001, 297, 638–654. [Google Scholar]
- Fischer, S.M.; Lo, H.H.; Godron, G.B.; Seibert, K.; Kelloff, G.; Lubet, R.A.; Conti, C. Chemotherapy activity of celecoxib, a specific cyclooxygenase-2 inhibitor, and indomethacin against ultraviolet light-induced skin carcinogenesis. Mol. Carcinogen. 1999, 25, 231–240. [Google Scholar] [CrossRef]
- Harris, R.E.; Alshafie, G.A.; Abou-Issa, H.; Seibert, K. Chemoprevention of breast cancer in rats by celecoxib, a cyclooxygenase-2 inhibitor. Cancer Res. 2000, 60, 2101–2103. [Google Scholar] [PubMed]
- Steinbach, G.; Lynch, P.M.; Philips, R.K.S. The effect of celecoxib, a cyclooxygenase-2 inhibitor, in famililal adenomatous polyposis. N. Engl. J. Med. 2000, 342, 1946–1952. [Google Scholar] [CrossRef] [PubMed]
- Amin, A.R.; Attur, M.; Patel, R.N. Superinduction of cyclooxygenase-2 activity in human osteoarthritis-affected cartilage. Influence of nitric oxide. J. Clin. Investig. 1997, 99, 1231–1237. [Google Scholar] [CrossRef]
- Qian, M.; Qian, D.; Jing, H.; Li, Y.; Ma, C.; Zhou, Y. Combined cetuximab and celecoxib treatment exhibits a synergistic anticancer effect on human oral squamous cell carcinoma in vitro and in vivo. Oncol. Rep. 2014, 32, 1681–1688. [Google Scholar] [CrossRef]
- Valverde, A.; Peñarando, J.; Cañas, A.; López-Sánchez, L.M.; Conde, F.; Guil-Luna, S.; Hernández, V.; Villar, C.; Morales-Estévez, C.; de la Haba-Rodríguez, J.; et al. The addition of celecoxib improves the antitumor effect of cetuximab in colorectal cancer: Role of EGFR-RAS-FOXM1-β-catenin signaling axis. Oncotarget 2017, 8, 21754–21769. [Google Scholar] [CrossRef]
- Tive, L. Celecoxib clinical profile. Rhemuatology 2000, 39, 21–28. [Google Scholar] [CrossRef]
- Subramanian, N.; Ghosal, S.; Moulik, S. Topical delivery of celecoxib using microemulsion. Acta Pol. Pharm. 2004, 52, 263–267. [Google Scholar]
- Garti, N.; Avrahami, M.; Aserin, A. Improved solubilization of Celecoxib in U-type nonionic microemulsions and their strauctural transitions with progressive aqueous dilution. J. Colloid Interface Sci. 2006, 299, 352–365. [Google Scholar] [CrossRef]
- Homar, M.; Ubrich, N.; El Ghazouani, F.; Kristl, J.; Kerc, J.; Maincent, P. Influence of polymers on the bioavailability of microencapsulated celecoxib. J. Microencapsule 2007, 24, 621–633. [Google Scholar] [CrossRef] [PubMed]
- Margulis, K.; Neofytou, E.A.; Beygui, R.E.; Zare, R.N. Celecoxib Nanoparticles for Therapeutic Angiogenesis. ACS Nano 2015, 9, 9416–9426. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, M.M.; Abd-Elgawad, A.H.; Soliman, O.A.; Jablonski, M.M. Stability and Ocular Pharmacokinetics of Celecoxib-Loaded Nanoparticles Topical Ophthalmic Formulations. J. Pharm. Sci. 2016, 105, 3691–3701. [Google Scholar] [CrossRef] [PubMed]
- Bhatt, V.; Shete, G.; Bansal, A.K. Mechanism of generation of drug nanocrystals in celecoxib: Mannitol nanocrystalline solid dispersion. Int. J. Pharm. 2015, 495, 132–139. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Gong, M.Q.; Liu, B.Y.; Zhuo, R.X.; Cheng, S.X. Co-delivery of multiple drug resistance inhibitors by polymer/inorganic hybrid nanoparticles to effectively reverse cancer drug resistance. Colloidal Surf. B Biointerfaces 2017, 149, 250–259. [Google Scholar] [CrossRef] [PubMed]
- Gowada, R.; Kardos, G.; Sharma, A.; Singh, S.; Robertson, G.P. Nanoparticle-Based Celecoxib and Plumbagin for the Synergistic Treatment of Melanoma. Mol. Cancer Ther. 2017, 16, 440–452. [Google Scholar] [CrossRef]
- Crivelli, B.; Bari, E.; Perteghella, S.; Catenacci, L.; Mocchi, M.; Farago, S.; Tripodo, G.; Prina-Mello, A.; Torre, M.L. Silk fibroin nanoparticles for celecoxib and curcumin delivery: ROS-scavenging and anti-inflammatory activities in an in vitro model of osteoarthritis. Eur. J. Pharm. Biopharm. 2019, 137. [Google Scholar] [CrossRef]
- Gugulothu, D.; Kulkarni, A.; Patravale, V.; Dandekar, P. pH-sensitive nanoparticles of curcumin-celecoxib combination: Evaluating drug synergy in ulcerative colitis model. J. Pharm. Sci. 2014, 137, 687–696. [Google Scholar] [CrossRef]
- Kim, T.H.; Jeong, Y.L.; Jin, S.G.; Pei, J.; Jung, T.Y.; Moon, K.S.; Kim, I.Y.; Kang, S.S.; Jung, S. Preparation of polylactide-co-glycolide nanoparticles incorporating celecoxib and their antitumor activity against brain tumor cells. Int. J. Nanomed. 2011, 6, 2621–2631. [Google Scholar] [CrossRef][Green Version]
- Shi, L.; Xu, L.; Wu, C.; Xue, B.; Jin, X.; Yang, J.; Zhu, X. Celecoxib-Induced Self-Assembly of Smart Albumin-Doxorubicin Conjugate for Enhanced Cancer Therapy. ACS Appl. Mater. Interfaces 2018, 10, 8555–8565. [Google Scholar] [CrossRef]
- Zhang, B.; Jin, K.; Jiang, T.; Want, L.; Shen, S.; Luo, Z.; Tuo, Y.; Liu, X.; Hu, Y.; Pang, Z. Celecoxib normalizes the tumor microenvironment and enhances small nanotherapeutics delivery to A549 tumors in nude mice. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef]
- Emami, J.; Pourmashhadi, A.; Sadeghi, H.; Varshosaz, J.; Hamishehkar, H. Formulation and optimization of celecoxib-loaded PLGA nanoparticles by the Taguchi design and their in vitro cytotoxicity for lung cancer therapy. Pharm. Dev. Technol. 2015, 20, 791–800. [Google Scholar] [CrossRef]
- Yassin, A.E.B.; Albekairy, A.; Alkatheri, A.; Sharma, R.K. Anticancer-loaded solid lipid nanoparticles: High potential advancement in chemotherapy. Dig. J. Nanomater. Biostructures 2013, 8, 905–916. [Google Scholar]
- Arduino, I.; Liu, Z.; Rahikkala, A.; Figueiredo, P.; Correia, A.; Cutrignelli, A.; Denora, N.; Santos, H.A. Preparation of cetyl palmitate-based PEGylated solid lipid nanoparticles by microfluidic technique. Acta Biomater. 2021, 121, 566–578. [Google Scholar] [CrossRef]
- Mehnert, W.; Mader, K. Solid lipid nanoparticles Production, characterization and applications. Adv. Drug Deliv. Rev. 2001, 47, 165–196. [Google Scholar] [CrossRef]
- Schöler, N.; Hahn, H.; Müller, R.H.; Liesenfeld, O. Effect of lipid matrix and size of solid lipid nanoparticles (SLN) on the viability and cytokine production of macrophages. Int. J. Pharm. 2002, 231, 167–176. [Google Scholar] [CrossRef]
- Schöler, N.; Olbrich, C.; Tabatt, K.; Müller, R.H.; Hahn, H.; Liesenfeld, O. Surfactant, but not the size of solid lipid nanoparticles (SLN) influences viability and cytokine production of macrophages. Int. J. Pharm. 2001, 221, 57–67. [Google Scholar] [CrossRef]
- Albekery, M.A.; Alharbi, K.T.; Alarifi, S.; Ahmad, D.; Omer, M.E.; Massadeh, S.; Yassin, A.E. Optimization of a nanostructured lipid carriers’ system for enhancing biopharmaceutical properties of valsartan. Dig. J. Nanomater. Biostructures 2017, 12, 381–389. [Google Scholar]
- Peltier, S.; Oger, J.M.; Lagarce, F.; Couet, W.; Benoît, J.P. Enhanced Oral Paclitaxel Bioavailability After Administration of Paclitaxel-Loaded Lipid Nanocapsules. Pharm. Res. 2006, 23, 1243–1250. [Google Scholar] [CrossRef] [PubMed]
- Serpe, L.; Guido, M.; Canaparo, R.; Muntoni, E.; Cavalli, R.; Panzanelli, P.; Pepa, C.; Carlo, D.; Bargoni, A.; Mauro, M.; et al. Interacellular Accumulation and Cytotoxicity of Doxorubicin with Different Pharmaceutical Formulations in Human Cancer Cell Lines. J. Nanosci. Nanotechnol. 2006, 6, 3062–3069. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, C.; Mehnert, W. Freeze-drying of drug-free and drug-loaded solid lipid nanoparticles (SLN). Int. J. Pharm. 1997, 157, 171–179. [Google Scholar] [CrossRef]
- Jalalizadeh, H.; Amini, M.; Ziaee, V.; Safa, A.; Farsam, H.; Shafiee, A. Determiniation of celecoxib in human plasma by high-performance liquid chromatography. J. Pharm. Biomed. Analysis 2004, 35, 665–670. [Google Scholar] [CrossRef]
- Yassin, A.E.B.; Anwer, M.K.; Mowafy, H.A.; El-Bagory, I.M.; Bayomi, M.A.; Alsarra, I.A. Optimization of 5-fluorouracil solid-lipid nanoparticles: A preliminary study to treat colon cancer. Int. J. Med. Sci. 2010, 7, 398–408. [Google Scholar] [CrossRef]
- Pathak, P.; Nagarsenker, M. Formulation and Evaluation of Lidocaine Lipid Nanosystems for Dermal Delivery. AAPS PharmaSciTech 2009, 10, 2393–2401. [Google Scholar] [CrossRef]
- Yamaoka, K.; Nakagawa, T.; Uno, T. Application of Akaike’s Information Criterion (AIC) in the Evaluation of Linear Pharmacokinetic Equations. J. Pharmacokinet. Biopharm. 1978, 6, 165–175. [Google Scholar] [CrossRef] [PubMed]
- Hussain, R.F.; Nouri, A.M.E.; Oliver, R.T.D. A new approach for measurement of cytotoxicity using colorimetric assay. J. Immunol. Methods 1993, 160, 89–96. [Google Scholar] [CrossRef]
- Badran, M.M.; Alomrani, A.H.; Harisa, G.I.; Ashour, A.E.; Kumar, A.; Yassin, A.E. Novel docetaxel chitosan-coated PLGA/PCL nanoparticles with magnified cytotoxicity and bioavailability. Biomed. Pharmacother. 2018, 106, 1461–1468. [Google Scholar] [CrossRef]
- Elumalai, P.; Gunadharini, D.N.; Senthilkumar, K. Induction of apoptosis in human breast cancer cells by nimbolide through extrinsic and intrinsic pathway. Toxicol. Lett. 2012, 215, 131–142. [Google Scholar] [CrossRef] [PubMed]
- Mulhen, A.Z. Feste Lipid-Nanopartikl mit prolongierter Wirkstoffiberation: Herstellung, Langzeitstabilitat, Charakterisierung, Freisetzungsverhalten und-Mechanismen. Ph.D. Thesis, Freie Universitat, Berlin, Germany, 1996. [Google Scholar]
- Zhao, Y.; Wang, C.; Chow, A.H.L.; Ren, K.; Gong, T.; Zhang, Z.; Zheng, Y. Self-nanoemulsifying drug delivery system for oral delivery of Zedoary essential oil: Formulation and bioavailability studies. Int. J. Pharm. 2010, 383, 170–177. [Google Scholar] [CrossRef] [PubMed]
- Asasutjarit, R.; Lorenzen, S.L.; Sirivichaykul, S.; Ruxrungtham, K.; Ruktanonchai, U.; Ritthidej, G.C. Effect of solid lipid nanoparticles formulation compositions on particle size and zeta potential and potential for in-vitro pHIS-HIV-hugag transfection. Pharm. Res. 2007, 6, 1098–1107. [Google Scholar] [CrossRef]
- Vivek, K.; Reddy, H.; Murthy, R.S.R. Investigations of the effect of the lipid matrix on drug entrapment, in vitro release and physical stability of Olanzapine-Loaded Solid Lipid Nanoparticles. AAPS PharmaSciTech 2007, 83, 16–24. [Google Scholar] [CrossRef]
- Patil, H.; Feng, X.; Ye, X.; Majumdar, S.; Repka, M.A. Continuous production of fenofibrate solid lipid nanoparticles by hot-melt extrusion technology: A systematic study based on a quality by design approach. AAPS J. 2015, 17, 194–205. [Google Scholar] [CrossRef]
- Bummer, P.M. Physical chemical considerations of lipid-based oral drug delivery- solid lipid nanoparticles. Crit. Rev. Ther. Drug Carrier Syst. 2004, 21, 1–20. [Google Scholar] [CrossRef]
- Bommareddy, G.S.; Paker-Leggs, S.; Saripella, K.K.; Neau, S.H. Extruded and spheronized beads containing Carbopol® 974P to deliver nonelectrolytes and salts of weakly basic drugs. Int. J. Pharm. 2006, 321, 62–71. [Google Scholar] [CrossRef]
- Fillery-Travis, A.J.; Foster, L.H.; Robins, M.M. Stability of emulsions stabilised by two physiological surfactants: L-alpha-phosphatidylcholine and sodium taurocholate. Biophys. Chem. 1995, 54, 253–260. [Google Scholar] [CrossRef]
- Müller, R.H.; Mäder, K.; Gohla, S. Solid lipid nanoparticles (SLN) for controlled drug delivery–A review of the state of the art. Eur. J. Pharm. Biopharm. 2000, 50, 161–177. [Google Scholar] [CrossRef]
- Abdelbary, G.; Fahmy, R.H. Diazepam-loaded Solid Lipid Nanoparticle: Design and Characterization. AAPS PharmaSciTech 2009, 10, 211–219. [Google Scholar] [CrossRef] [PubMed]
- Bhaskar, K.; Anbu, J.; Ravichandiran, V.; Venkateswarlu, V.; Rao, Y.M. Lipid nanoparticles for transdermal for transdermal delivery of flurbiprofen: Formulation, in vitro, ex vivo and in vivo studies. Lipids Health Dis. 2009, 8, 1476–1511. [Google Scholar] [CrossRef] [PubMed]
- Illing, A.; Unruh, T. Investigations on the flow behavior of dispersions of solid triglycerides nanoparticles. Int. J. Pharm. 2004, 284, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Unruh, T.; Bunjes, H.; Westesen, K. Observation of size dependent melting in lipid nanoparticles. J. Phys. Chem. B. 1999, 47, 10373–10377. [Google Scholar] [CrossRef]
- Unruh, T.; Bunjes, H.; Westesen, K.; Koch, M.H.J. Investigations on the melting behavior of triglyceride nanoparticles. Colloid Polym. Sci. 2001, 279, 398–403. [Google Scholar] [CrossRef]
- Bunjes, H.; Koch, M.H.J.; Westesen, K. Effects of surfactants on the crystallization and polymorphism of lipid nanoparticles. Prog. Colloid Polym. Sci. 2002, 121, 7–10. [Google Scholar] [CrossRef]
- Bunjes, H.; Koch, M.H.J.; Westesen, K. Effect of Particle Size on Colloidal Solid Triglycerides. Langmuir 2000, 12, 5234–5241. [Google Scholar] [CrossRef]
- Schubert, M.A.; Schicke, B.C.; Müller-Goymann, C.C. Thermal analysis of the crystallization and melting behavior of lipid matrices and lipid nanoparticles containing high amounts of lecithin. Int. J. Pharm. 2005, 298, 242–254. [Google Scholar] [CrossRef] [PubMed]
- Mühlen, A.Z.; Schwarz, C.; Mehnert, W. Solid lipid nanoparticles (SLN) for controlled drug delivery–Drug release and release mechanism. Eur. J. Pharm. Biopharm. 1998, 45, 149–155. [Google Scholar] [CrossRef]
- Cavalli, R.; Aquilano, D.; Carlooti, M.E.; Gasco, M.R. Study by X-ray powder diffraction and differential scanning calorimetry of two model drugs, phenothiazine and nifedipine, incorporated into lipid nanoparticles. Eur. J. Pharm. Biopharm. 1995, 41, 329–333. [Google Scholar]
- Cavalli, R.; Caputo, O.; Marengo, E.; Pattarino, F.; Gasco, M.R. The effect of the components of microemulsions on both size and crystalline structure of solid lipid nanoparticles (SLN) containing a series of model molecules. Pharmazie 1998, 53, 392–396. [Google Scholar]
- Ivanova, B.; Kolev, T. Structural elucidation of organic compounds. In Linearly Polarized IR Spectroscop, Theory and Applications for Structural Analysis; CRC Press, Ed.; Taylor & Francis Group, LLC: Boca Raton, FL, USA, 2012; pp. 73–104. [Google Scholar]
- Barakat, N.S.; Al-Suwayeh, S.A.; Taha, E.I.; Yassin, A.E. A new pressure-controlled colon delivery capsule for chronotherapeutic treatment of nocturnal asthma. J. Drug Target. 2011, 19, 365–372. [Google Scholar] [CrossRef]
- Iscan, Y.; Hekimoglu, S.; Sargon, M.F.; Hincal, A.A. DEET-loaded solid lipid particles for skin delivery: In vitro release and skin permeation characteristics in different vehicles. J. Microencapsul. 2006, 23, 315–327. [Google Scholar] [CrossRef]
- Muhlen, A.Z.; Mehner, W. Drug release and release mechanism of prednisolone loaded solid lipid nanoparticles. Pharmazie 1998, 53, 552–555. [Google Scholar]
- Shwartz, C.; Freitas, C.; Mehnert, W.; Muller, R.H. Sterilization and physical stability of drug- free and etomidate loaded solid lipid nanoparticles. Proc. Int. Symp. Cont. Rel. Bioact. Mater. 1995, 22, 766–767. [Google Scholar]
- Ibrahim, W.M.; AlOmrani, A.H.; Yassin, A.E. Novel sulpiride-loaded solid lipid nanoparticles with enhanced intestinal permeability. Int. J. Nanomed. 2014, 9, 129–144. [Google Scholar] [CrossRef]
- Alarifi, S.; Massadeh, S.; Al-Agamy, M.; Al Aamery, M.; Albekairy, A.; Yassin, A.E.Y. Enhancement of ciprofloxacin activity by incorporating it in solid lipid nanoparticles. Trop. J. Pharm. Res. 2020, 19, 909–918. [Google Scholar] [CrossRef]
- Kiss, E.L.; Berkó, S.; Gácsi, A.; Kovács, A.; Katona, G.; Soós, J.; Csányi, E.; Gróf, I.; Harazin, A.; Deli, M.A.; et al. Design and Optimization of Nanostructured Lipid Carrier Containing Dexamethasone for Ophthalmic Use. Pharmaceutics 2019, 14, 679. [Google Scholar] [CrossRef] [PubMed]
- Silva, A.C.; Lopes, C.M.; Fonseca, J.; Soares, M.E.; Santos, D.; Souto, E.B.; Ferreira, D. Risperidone Release from Solid Lipid Nanoparticles (SLN): Validated HPLC Method and Modelling Kinetic Profile. Curr. Pharm. Anal. 2012, 8, 307–316. [Google Scholar] [CrossRef]
- Kushwaha, S.K.S.; Rai, A.K.; Parveen, H. Development & Pharmaceutical Characterization of Isoniazid Loaded Solid Lipid Nanoparticle Drug Delivery Approach. Curr. Drug Ther. 2019, 14, 228–238. [Google Scholar] [CrossRef]
- Kishore, N.; Raja, M.D.; Dhanalekshmi, U.M.; Saranya, B.; Reddy, P.N. Formulation and evaluation of NSAID-Loaded Tristearin Solid Lipid Nanoparticles. Int. J. Med. Res. 2011, 4, 217–223. [Google Scholar]
- Wang, H.; Ke, F.; Zheng, J. Hedgehog-glioma-associated oncogene homolog-1 signaling in colon cancer cells and its role in the celecoxib-mediated anti-cancer effect. Oncol. Lett. 2014, 8, 2203–2208. [Google Scholar] [CrossRef][Green Version]
- Limasale, Y.D.; Tezcaner, A.; Özen, C.; Keskin, D.; Banerjee, S. Epidermal growth factor receptor-targeted immunoliposomes for delivery of celecoxib to cancer cells. Int. J. Pharm. 2015, 479, 364–373. [Google Scholar] [CrossRef]
- Erdoğ, A.; Limasale, Y.D.; Keskin, D.; Tezcaner, A.; Banerjee, S. In vitro characterization of a liposomal formulation of celecoxib containing 1,2-distearoyl-sn-glycero-3-phosphocholine, cholesterol, and polyethylene glycol and its functional effects against colorectal cancer cell lines. J. Pharm. Sci. 2013, 102, 3666–3677. [Google Scholar] [CrossRef]
- Liu, N.B.; Peng, T.; Pan, C.; Yao, Y.Y.; Shen, B.; Leng, J. Overexpression of cyclooxygenase-2 in human HepG2, Bel-7402 and SMMC-7721 hepatoma cell lines and mechanism of cyclooxygenase-2 selective inhibitor celecoxib-induced cell growth inhibition and apoptosis. World J. Gastroenterol. 2005, 11, 6281–6287. [Google Scholar] [CrossRef] [PubMed]
- Chlapek, P.; Neradil, J.; Redova, M.; Zitterbart, K.; Sterba, J.; Veselska, R. The ATRA-induced differentiation of medulloblastoma cells is enhanced with LOX/COX inhibitors: An analysis of gene expression. Cancer Cell Int. 2014, 14, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; Yao, J.; Zhu, X.; Qi, Y. Nanomedicines: Redefining traditional medicine. Biomed. Pharmacother. 2021, 134, 111103. [Google Scholar] [CrossRef] [PubMed]
- Ali Khan, A.; Mudassir, J.; Mohtar, N.; Darwis, Y. Advanced drug delivery to the lymphatic system: Lipid-based nanoformulations. Int. J. Nanomed. 2013, 8, 2733–2744. [Google Scholar] [CrossRef]
- Paliwal, R.; Rai, S.; Vaidya, B.; Khatri, K.; Goyal, A.K.; Mishra, N.; Mehta, A.; Vyas, S.P. Effect of lipid core material on characteristics of solid lipid nanoparticles designed for oral lymphatic delivery. Nanomedicine 2009, 5, 184–191. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Formula | Lipid | Surfactant/Co-Surfactant | |||||
|---|---|---|---|---|---|---|---|
| Stearic Acid | Dynasan 114 | Imwitor 308 | Softisan 154 | Tween 80 | Cremophor EL | Sodium Deoxycholate | |
| F1 | -- | -- | 450 mg | -- | 45 mg | -- | 22.5 mg |
| F2 | -- | 450 mg | -- | -- | 45 mg | -- | 22.5 mg |
| F3 | 450 mg | -- | -- | -- | 45 mg | -- | 22.5 mg |
| F4 | -- | -- | -- | 450 mg | 45 mg | -- | 22.5 mg |
| F5 | -- | -- | 450 mg | -- | 90 mg | -- | 22.5 mg |
| F6 | -- | -- | -- | 450 mg | 90 mg | -- | 22.5 mg |
| F7 | -- | 450 mg | -- | -- | 90 mg | -- | 22.5 mg |
| F8 | 450 mg | -- | -- | -- | 90 mg | -- | 22.5 mg |
| F9 | -- | 225 mg | 225 mg | -- | 90 mg | -- | 22.5 mg |
| F10 | 225 mg | 225 mg | -- | -- | 90 mg | -- | 22.5 mg |
| F11 | 225 mg | -- | 225 mg | -- | 90 mg | -- | 22.5 mg |
| F12 | -- | 225 mg | -- | 225 mg | 90 mg | -- | 22.5 mg |
| F13 | -- | 225 mg | 225 mg | -- | 45 mg | -- | 22.5 mg |
| F14 | -- | 225 mg | 225 mg | -- | -- | 90 mg | 22.5 mg |
| F15 | 225 mg | -- | 225 mg | -- | 45 mg | -- | 22.5 mg |
| F16 | 225 mg | -- | 225 mg | -- | -- | 90 mg | 22.5 mg |
| Formulation | %EE | %DL |
|---|---|---|
| F1 | 90.9 ± 1.12 | 9.17 ± 0.12 |
| F2 | 90.5 ± 0.62 | 9.14 ± 0.06 |
| F3 | 95.0 ± 0.72 | 9.55 ± 0.07 |
| F4 | 93.7 ± 1.63 | 9.42 ± 0.62 |
| F5 | 88.7 ± 3.10 | 8.97 ± 0.32 |
| F6 | 93.0 ± 0.93 | 9.36 ± 0.39 |
| F7 | 93.3 ± 2.09 | 9.38 ± 0.21 |
| F8 | 92.0 ± 4.2 | 9.27 ± 0.16 |
| F9 | 86.8 ± 1.59 | 8.97 ± 0.16 |
| F10 | 87.4 ± 0.98 | 8.85 ± 0.10 |
| F11 | 92.2 ± 1.82 | 9.29 ± 0.18 |
| F12 | 94.1 ± 2.67 | 9.46 ± 0.28 |
| F13 | 96.6 ± 1.19 | 9.96 ± 0.12 |
| F14 | 95.5 ± 2.29 | 9.59 ± 0.23 |
| F15 | 96.1 ± 3.38 | 9.65 ± 0.35 |
| F16 | 95.4 ± 2.04 | 9.58 ± 0.21 |
| Lipid | Composition | Peak Temp °C | Δ H j/g | SLN Peak Temp °C | SLN Δ H j/g |
|---|---|---|---|---|---|
| Imwitor 308 | Mono-glyceryl ester with caprylic acid, 8-C fatty acid | 41.19 | −234.93 | 32.65 | −27.58 |
| Dynasan 114 | Tri-glyceryl ester with myristic acid, 14-C fatty acid | 56.79 | −44.07 | 69.27 | −45.26 |
| Softisan 154 | Tri-glyceryl ester of a blend of 16-C and 18-C saturated fatty acid | 58.59 | −234.96 | 54.8 | −40.97 |
| Stearic acid | Fatty acid containing 18-C saturated hydrocarbon chain | 74.12 | −238.03 | 57.31 | −93.28 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alajami, H.N.; Fouad, E.A.; Ashour, A.E.; Kumar, A.; Yassin, A.E.B. Celecoxib-Loaded Solid Lipid Nanoparticles for Colon Delivery: Formulation Optimization and In Vitro Assessment of Anti-Cancer Activity. Pharmaceutics 2022, 14, 131. https://doi.org/10.3390/pharmaceutics14010131
Alajami HN, Fouad EA, Ashour AE, Kumar A, Yassin AEB. Celecoxib-Loaded Solid Lipid Nanoparticles for Colon Delivery: Formulation Optimization and In Vitro Assessment of Anti-Cancer Activity. Pharmaceutics. 2022; 14(1):131. https://doi.org/10.3390/pharmaceutics14010131
Chicago/Turabian StyleAlajami, Hamdan N., Ehab A. Fouad, Abdelkader E. Ashour, Ashok Kumar, and Alaa Eldeen B. Yassin. 2022. "Celecoxib-Loaded Solid Lipid Nanoparticles for Colon Delivery: Formulation Optimization and In Vitro Assessment of Anti-Cancer Activity" Pharmaceutics 14, no. 1: 131. https://doi.org/10.3390/pharmaceutics14010131
APA StyleAlajami, H. N., Fouad, E. A., Ashour, A. E., Kumar, A., & Yassin, A. E. B. (2022). Celecoxib-Loaded Solid Lipid Nanoparticles for Colon Delivery: Formulation Optimization and In Vitro Assessment of Anti-Cancer Activity. Pharmaceutics, 14(1), 131. https://doi.org/10.3390/pharmaceutics14010131