
Prediction of Drug–Drug Interaction Potential of Tegoprazan Using Physiologically Based Pharmacokinetic Modeling and Simulation

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Tegoprazan PBPK Model Development
2.1.1. Absorption
2.1.2. Distribution
2.1.3. Elimination
2.2. Tegoprazan PBPK Model Refinement and Verification
2.3. Prediction of a DDI Potential
3. Results
3.1. PK Predictions of Tegoprazan
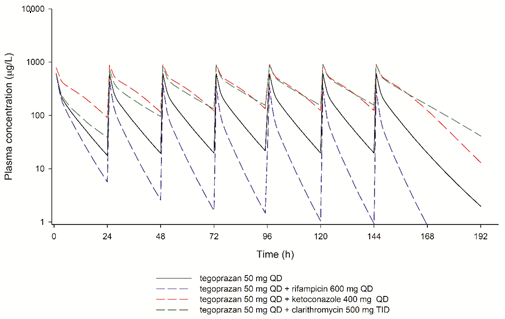
3.2. Performance of the PBPK Model in Predicting DDI
3.3. DDI Potential of Tegoprazan
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Takahashi, N.; Take, Y. Tegoprazan, a Novel Potassium-Competitive Acid Blocker to Control Gastric Acid Secretion and Motility. J. Pharmacol. Exp. Ther. 2017, 364, 275–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, S.; Choi, H.Y.; Kim, Y.H.; Nam, J.Y.; Kim, B.; Song, G.S.; Lim, H.-S.; Bae, K.-S. Randomised clinical trial: Safety, tolerability, pharmacokinetics, and pharmacodynamics of single and multiple oral doses of tegoprazan (CJ-12420), a novel potassium-competitive acid blocker, in healthy male subjects. Aliment. Pharmacol. Ther. 2019, 50, 751–759. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.G.; Yoo, H.; Lee, J.W.; Song, G.S.; Lee, S.; Kim, M.-G. Comparison of pharmacokinetic characteristics of two Tegoprazan (CJ-12420) formulations in healthy male subjects. Transl. Clin. Pharmacol. 2019, 27, 80–85. [Google Scholar] [CrossRef] [Green Version]
- Ghim, J.; Chin, M.C.; Jung, J.; Lee, J.; Kim, S.; Kim, B.; Song, G.S.; Ms, Y.C.; Shin, J. Pharmacokinetics and Pharmacodynamics of Tegoprazan Coadministered With Amoxicillin and Clarithromycin in Healthy Subjects. J. Clin. Pharmacol. 2020, 61, 913–922. [Google Scholar] [CrossRef] [PubMed]
- Antunes, C.; Aleem, A.; Curtis, S.A. Gastroesophageal Reflux Disease; [Updated 18 July 2021]; StatPearls Publishing: Treasure Island, FL, USA, 2021. Available online: https://www.ncbi.nlm.nih.gov/books/NBK441938/ (accessed on 10 August 2021).
- Roberts-Thomson, I.C. Rise and fall of peptic ulceration: A disease of civilization? J. Gastroenterol. Hepatol. 2018, 33, 1321–1326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuepfer, L.; Niederalt, C.; Wendl, T.; Schlender, J.; Willmann, S.; Lippert, J.; Block, M.; Eissing, T.; Teutonico, D. Applied Concepts in PBPK Modeling: How to Build a PBPK/PD Model. CPT: Pharmacomet. Syst. Pharmacol. 2016, 5, 516–531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shebley, M.; Sandhu, P.; Riedmaier, A.E.; Jamei, M.; Narayanan, R.; Patel, A.; Peters, S.A.; Reddy, V.P.; Zheng, M.; De Zwart, L.; et al. Physiologically Based Pharmacokinetic Model Qualification and Reporting Procedures for Regulatory Submissions: A Consortium Perspective. Clin. Pharmacol. Ther. 2018, 104, 88–110. [Google Scholar] [CrossRef]
- Zhang, X.; Yang, Y.; Grimstein, M.; Fan, J.; Grillo, J.A.; Huang, S.; Zhu, H.; Wang, Y. Application of PBPK Modeling and Simulation for Regulatory Decision Making and Its Impact on US Prescribing Information: An Update on the 2018-2019 Submissions to the US FDA’s Office of Clinical Pharmacology. J. Clin. Pharmacol. 2020, 60, S160–S178. [Google Scholar] [CrossRef]
- Huang, S.-M.; Rowland, M. The Role of Physiologically Based Pharmacokinetic Modeling in Regulatory Review. Clin. Pharmacol. Ther. 2012, 91, 542–549. [Google Scholar] [CrossRef]
- Luzon, E.; Blake, K.; Cole, S.; Nordmark, A.; Versantvoort, C.; Gil Berglund, E. Physiologically based pharmacokinetic modeling in regulatory decision-making at the European Medicines Agency. Clin. Pharmacol. Ther. 2016, 102, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Yoon, D.Y.; Sunwoo, J.; Shin, N.; Kim, A.R.; Kim, B.T.; Song, G.S.; Jang, I.; Lee, S. Effect of meal timing on pharmacokinetics and pharmacodynamics of tegoprazan in healthy male volunteers. Clin. Transl. Sci. 2020, 14, 934–941. [Google Scholar] [CrossRef] [PubMed]
- Jamei, M.; Turner, D.; Yang, J.; Neuhoff, S.; Polak, S.; Rostami-Hodjegan, A.; Tucker, G. Population-Based Mechanistic Prediction of Oral Drug Absorption. AAPS J. 2009, 11, 225–237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodgers, T.; Rowland, M. Mechanistic Approaches to Volume of Distribution Predictions: Understanding the Processes. Pharm. Res. 2007, 24, 918–933. [Google Scholar] [CrossRef] [PubMed]
- Houston, J.B. Utility of in vitro drug metabolism data in predicting in vivo metabolic clearance. Biochem. Pharmacol. 1994, 47, 1469–1479. [Google Scholar] [CrossRef]
- Abduljalil, K.; Cain, T.; Humphries, H.; Rostami-Hodjegan, A. Deciding on Success Criteria for Predictability of Pharmacokinetic Parameters from In Vitro Studies: An Analysis Based on In Vivo Observations. Drug Metab. Dispos. 2014, 42, 1478–1484. [Google Scholar] [CrossRef] [PubMed]
- Tsamandouras, N.; Rostami-Hodjegan, A.; Aarons, L. Combining the ‘bottom up’ and ‘top down’ approaches in pharmacokinetic modelling: Fitting PBPK models to observed clinical data. Br. J. Clin. Pharmacol. 2014, 79, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Niwa, T.; Morimoto, M.; Hirai, T.; Hata, T.; Hayashi, M.; Imagawa, Y. Effect of penicillin-based antibiotics, amoxicillin, ampicillin, and piperacillin, on drug-metabolizing activities of human hepatic cytochromes P450. J. Toxicol. Sci. 2016, 41, 143–146. [Google Scholar] [CrossRef] [Green Version]
- Rodvold, K.A. Clinical Pharmacokinetics of Clarithromycin. Clin. Pharmacokinet. 1999, 37, 385–398. [Google Scholar] [CrossRef]
- Oo, C.; Chen, Y.-C. The need for multiple doses of 400 mg ketoconazole as a precipitant inhibitor of a CYP3A substrate in an in vivo drug-drug interaction study. J. Clin. Pharmacol. 2009, 49, 368–369. [Google Scholar] [CrossRef]
- Ke, A.B.; Zamek-Gliszczynski, M.J.; Higgins, J.W.; Hall, S.D. Itraconazole and Clarithromycin as Ketoconazole Alternatives for Clinical CYP3A Inhibition Studies. Clin. Pharmacol. Ther. 2014, 95, 473–476. [Google Scholar] [CrossRef]
- Baneyx, G.; Parrott, N.; Meille, C.; Iliadis, A.; Lavé, T. Physiologically based pharmacokinetic modeling of CYP3A4 induction by rifampicin in human: Influence of time between substrate and inducer administration. Eur. J. Pharm. Sci. 2014, 56, 1–15. [Google Scholar] [CrossRef]
- Han, S.; Choi, H.Y.; Kim, Y.H.; Nam, J.Y.; Kim, B.; Song, G.S.; Lim, H.-S.; Bae, K.-S. Effect of Food on the Pharmacokinetics and Pharmacodynamics of a Single Oral Dose of Tegoprazan. Clin. Ther. 2021. [Google Scholar] [CrossRef]
- Wagner, C.; Pan, Y.; Hsu, V.; Grillo, J.A.; Zhang, L.; Reynolds, K.S.; Sinha, V.; Zhao, P. Predicting the Effect of Cytochrome P450 Inhibitors on Substrate Drugs: Analysis of Physiologically Based Pharmacokinetic Modeling Submissions to the US Food and Drug Administration. Clin. Pharmacokinet. 2014, 54, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Wagner, C.; Pan, Y.; Hsu, V.; Sinha, V.; Zhao, P. Predicting the Effect of CYP3A Inducers on the Pharmacokinetics of Substrate Drugs Using Physiologically Based Pharmacokinetic (PBPK) Modeling: An Analysis of PBPK Submissions to the US FDA. Clin. Pharmacokinet. 2015, 55, 475–483. [Google Scholar] [CrossRef] [PubMed]
- Sunwoo, J.; Oh, J.; Moon, S.J.; Ji, S.C.; Lee, S.H.; Yu, K.-S.; Kim, H.S.; Lee, A.; Jang, I.-J. Safety, tolerability, pharmacodynamics and pharmacokinetics of DWP14012, a novel potassium-competitive acid blocker, in healthy male subjects. Aliment. Pharmacol. Ther. 2018, 48, 206–218. [Google Scholar] [CrossRef] [Green Version]
- US Food and Drug Administration. Clinical Drug Interaction Studies. Available online: https://www.fda.gov/media/134581/download (accessed on 2 March 2021).
- Van Duijn, B.; Ypey, D.L.; De Goede, J.; Verveen, A.A.; Hekkens, W. A model study of the regulation of gastric acid secretion. Am. J. Physiol. Liver Physiol. 1989, 257, G157–G168. [Google Scholar] [CrossRef] [PubMed]
- Tompkins, L.M.; Wallace, A.D. Mechanisms of cytochrome P450 induction. J. Biochem. Mol. Toxicol. 2007, 21, 176–181. [Google Scholar] [CrossRef]
- Marsousi, N.; Desmeules, J.A.; Rudaz, S.; Daali, Y. Prediction of drug-drug interactions using physiologically-based pharmacokinetic models of CYP450 modulators included in Simcyp software. Biopharm. Drug Dispos. 2017, 39, 3–17. [Google Scholar] [CrossRef]
- Funakoshi, R.; Tomoda, Y.; Kudo, T.; Furihata, K.; Kusuhara, H.; Ito, K. Effects of proton pump inhibitors, esomeprazole and vonoprazan, on the disposition of proguanil, a CYP2C19 substrate, in healthy volunteers. Br. J. Clin. Pharmacol. 2019, 85, 1454–1463. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters and Models | Value | Source | |
|---|---|---|---|
| Physiochemical properties | MW | 387.38 | Experimental data |
| Log P | 3 | Experimental data | |
| pKa | Ampholyte | Experimental data | |
| pKa 1: 5.2 | |||
| pKa 2: 12 | |||
| B/P | 0.868 | Experimental data | |
| fu | 0.124 | Experimental data | |
| Absorption | ADAM model | Data | Data |
| fuGut | 0.008 | Predicted using method 2 (Rodgers and Rowland 2007) | |
| Peff,man | 12.397 | Predicted using PAMPA permeability data | |
| PAMPA | 68.4 | Experimental data | |
| Distribution | Minimal PBPK model + SAC | ||
| Vss | 1.128 | Predicted using method 2 (Rodgers and Rowland 2007) | |
| Q | 24.4 | Estimated | |
| VSAC | 0.66 | Estimated | |
| Kp scalar | 0.33 | Estimated | |
| Elimination | CYP1A2 CLint | 2.5 | Experimental data |
| CYP2C9 CLint | 2.6 | Experimental data | |
| CYP2C19 CLint | 3.6 | Experimental data | |
| CYP2D6 CLint | 2 | Experimental data | |
| CYP3A4 CLint | 30.34 | Estimated | |
| CLR | 1.31 | Experimental data | |
| Treatment | Dose (mg) | n | Tmax (h) * | Cmax (μg/L) | AUCinf or AUCτ (μg∙h/L) ** | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Pred. | Obs. | Pred. | Obs. | Pred. | Obs. | Ratio (Pred./Obs.) | Pred. | Obs. | Ratio (Pred./Obs.) | ||
| Single oral dose | 25 | 100 | 12 | 0.95 | 0.75 | 310.4 | 335.6 | 0.92 | 1479.4 | 1340.0 | 1.03 |
| (0.50–1.62) | (0.50–3.00) | ||||||||||
| 50 | 100 | 24 | 0.95 | 1.00 | 620.6 | 759.1 | 0.82 | 2958.6 | 2903.0 | 1.02 | |
| (0.50–1.62) | (0.50–2.00) | ||||||||||
| 100 | 100 | 12 | 0.95 | 1.00 | 1241.2 | 1434.5 | 0.87 | 5916.6 | 5998.1 | 0.99 | |
| (0.50–1.62) | (0.50–1.00) | ||||||||||
| Multiple oral doses † | 50 | 100 | 6 | 0.94 | 1.00 | 638.9 | 842.8 | 0.76 | 2969.5 | 2954.9 | 1.00 |
| (0.51–1.59) | (0.50–1.03) | ||||||||||
| 100 | 100 | 6 | 0.95 | 1.25 | 1277.6 | 1149.7 | 1.11 | 5929.4 | 4768.4 | 1.24 | |
| (0.50–1.58) | (0.50–3.00) | ||||||||||
| Treatment | n | Tmax (h) * | Cmax (μg/L) | AUCτ (μg∙h/L) | Fold Increase | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Pred. | Obs. | Pred. | Obs. | Pred. | Obs. | Ratio (Pred./Obs.) | Pred. | Obs. | Ratio (Pred./Obs.) | Pred. CmaxR | Obs. CmaxR | Pred. AUCR | Obs. AUCR | |
| T 200 mg QD † | 100 | 24 | 0.95 | 1.00 | 2554.8 | 1868.6 | 1.37 | 11,838.9 | 10,817.6 | 1.09 | ||||
| (0.50–1.58) | (0.50–4.00) | |||||||||||||
| T 200 mg QD + C 500 mg BID † | 100 | 24 | 1.04 | 1.50 | 3491.4 | 3096.0 | 1.13 | 28,881.4 | 27,796.4 | 1.04 | 1.37 | 1.66 | 2.44 | 2.57 |
| (0.55–1.62) | (1.00–4.00) | |||||||||||||
| T 100 mg BID †† | 100 | 20 | 0.95 | 1.30 | 1411.3 | 1018.4 | 1.39 | 5921.6 | 5955.9 | 0.99 | ||||
| (0.51–1.55) | (0.50–6.00) | |||||||||||||
| T 100 mg BID + C 500 mg BID + A 1000 mg BID ††† | 100 | 20 | 1.03 | 2.50 | 2268.2 | 2285.6 | 0.99 | 14,897.5 | 16,045.0 | 0.93 | 1.61 | 2.24 | 2.52 | 2.69 |
| (0.55–1.55) | (1.00–3.00) | |||||||||||||
| Perpetrator | Predicted Cmax (μg/L) | Predicted AUCτ (μg∙h/L) | Predicted Fold Increase | |
|---|---|---|---|---|
| CmaxR | AUCR | |||
| Clarithromycin 250 mg BID | 768.7 | 4896.3 | 1.20 | 1.63 |
| Clarithromycin 500 mg BID | 887.8 | 7455.8 | 1.40 | 2.57 |
| Clarithromycin 500 mg TID | 933.5 | 8356.4 | 1.47 | 2.96 |
| Ketoconazole 200 mg QD | 905.8 | 7633.2 | 1.44 | 2.84 |
| Ketoconazole 400 mg QD | 936.2 | 8382.8 | 1.49 | 3.14 |
| Rifampicin 450 mg QD | 367.8 | 931.7 | 0.57 | 0.31 |
| Rifampicin 600 mg QD | 353.7 | 873.5 | 0.55 | 0.29 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yoon, D.Y.; Lee, S.; Jang, I.-J.; Kim, M.; Lee, H.; Kim, S.; Kim, B.; Song, G.S.; Rhee, S.-j. Prediction of Drug–Drug Interaction Potential of Tegoprazan Using Physiologically Based Pharmacokinetic Modeling and Simulation. Pharmaceutics 2021, 13, 1489. https://doi.org/10.3390/pharmaceutics13091489
Yoon DY, Lee S, Jang I-J, Kim M, Lee H, Kim S, Kim B, Song GS, Rhee S-j. Prediction of Drug–Drug Interaction Potential of Tegoprazan Using Physiologically Based Pharmacokinetic Modeling and Simulation. Pharmaceutics. 2021; 13(9):1489. https://doi.org/10.3390/pharmaceutics13091489
Chicago/Turabian StyleYoon, Deok Yong, SeungHwan Lee, In-Jin Jang, Myeongjoong Kim, Heechan Lee, Seokuee Kim, Bongtae Kim, Geun Seog Song, and Su-jin Rhee. 2021. "Prediction of Drug–Drug Interaction Potential of Tegoprazan Using Physiologically Based Pharmacokinetic Modeling and Simulation" Pharmaceutics 13, no. 9: 1489. https://doi.org/10.3390/pharmaceutics13091489
APA StyleYoon, D. Y., Lee, S., Jang, I.-J., Kim, M., Lee, H., Kim, S., Kim, B., Song, G. S., & Rhee, S.-j. (2021). Prediction of Drug–Drug Interaction Potential of Tegoprazan Using Physiologically Based Pharmacokinetic Modeling and Simulation. Pharmaceutics, 13(9), 1489. https://doi.org/10.3390/pharmaceutics13091489