
Cationic Liposomes as Vectors for Nucleic Acid and Hydrophobic Drug Therapeutics

 , ,
, , 
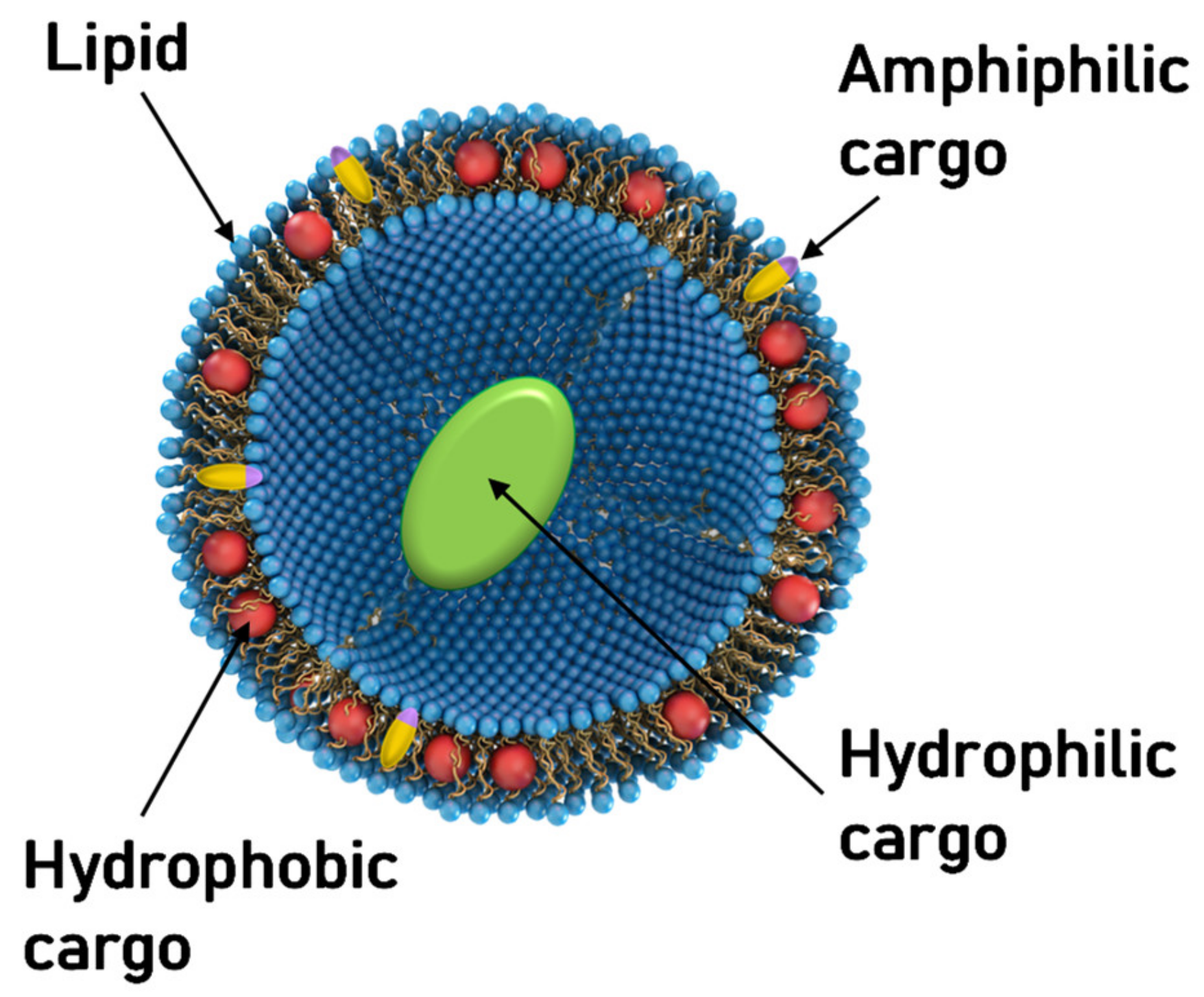{kind=link}
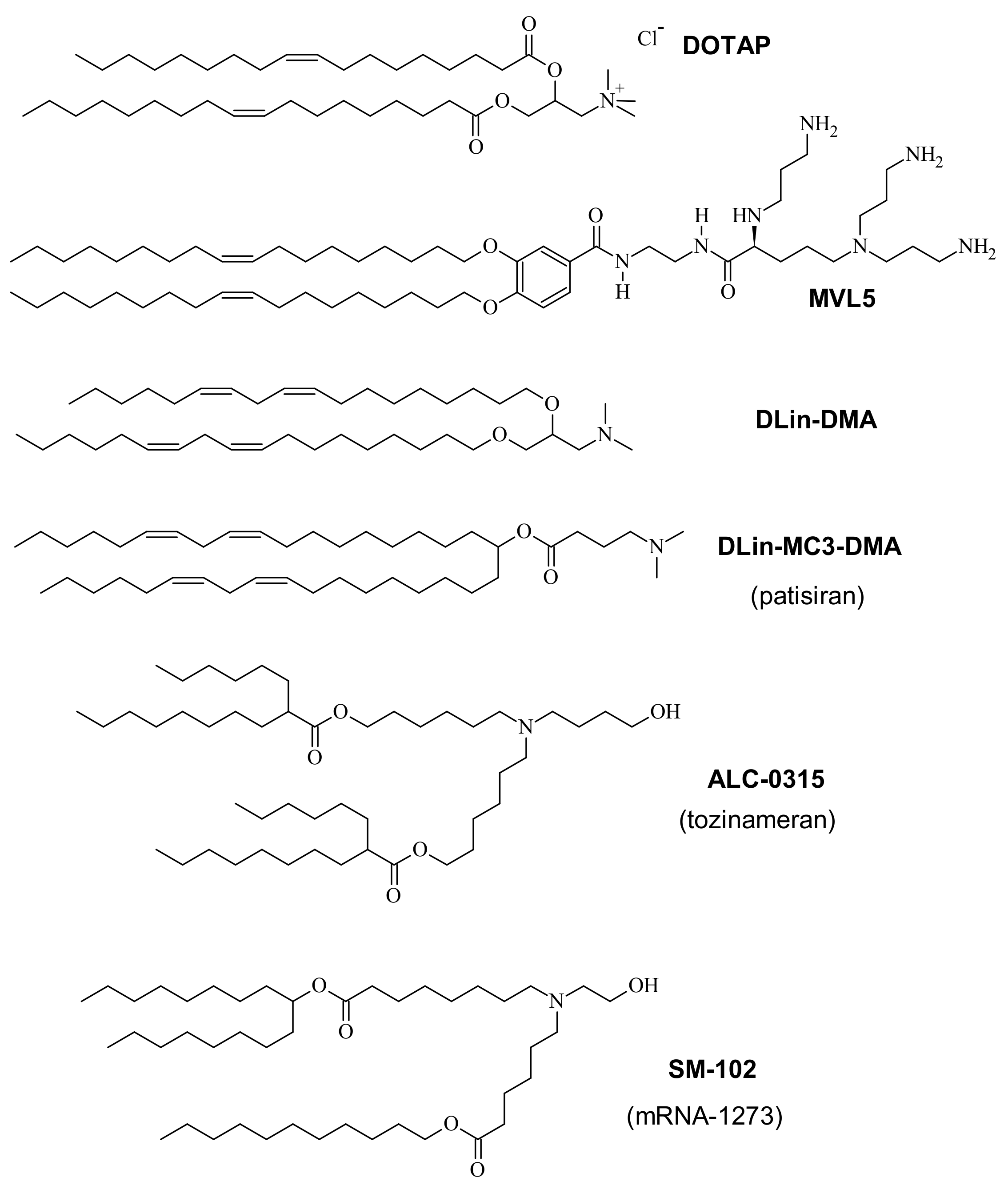{kind=link}
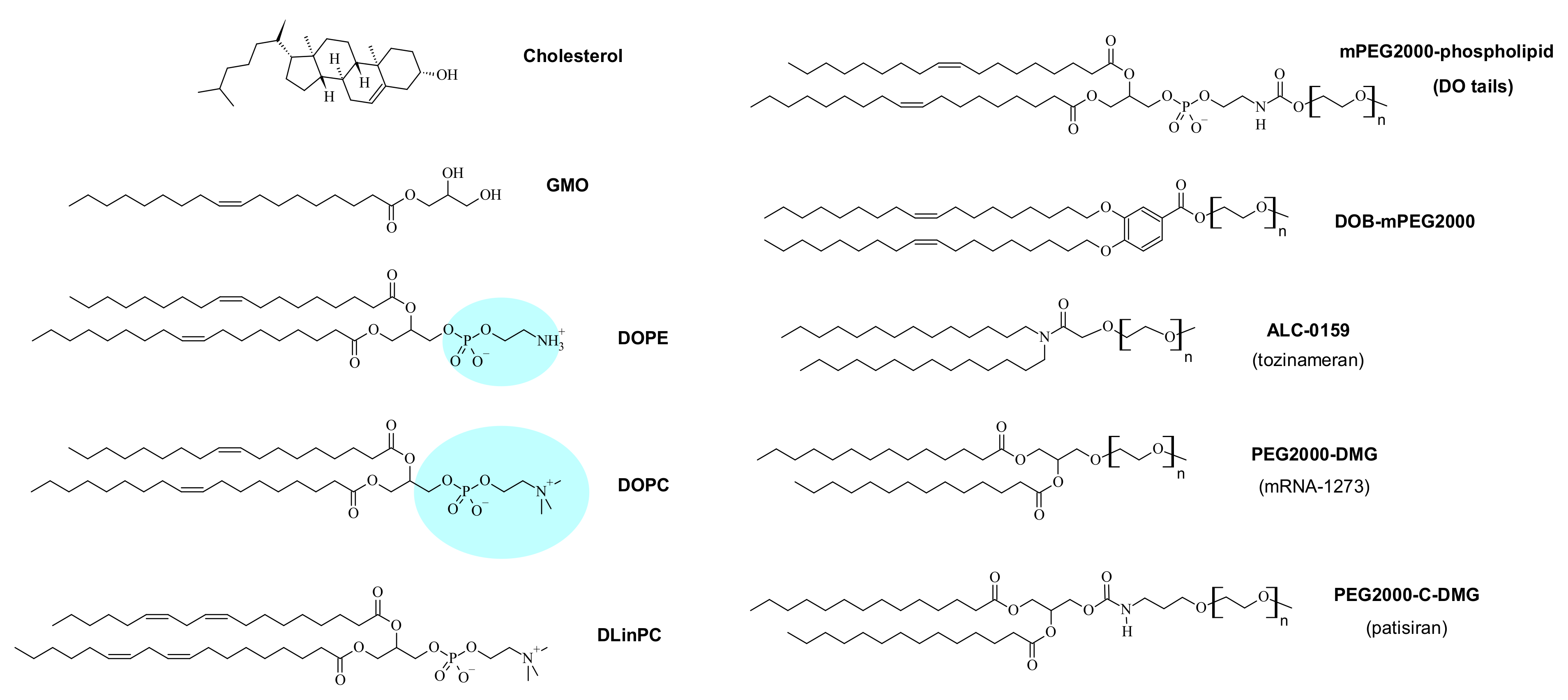{kind=link}
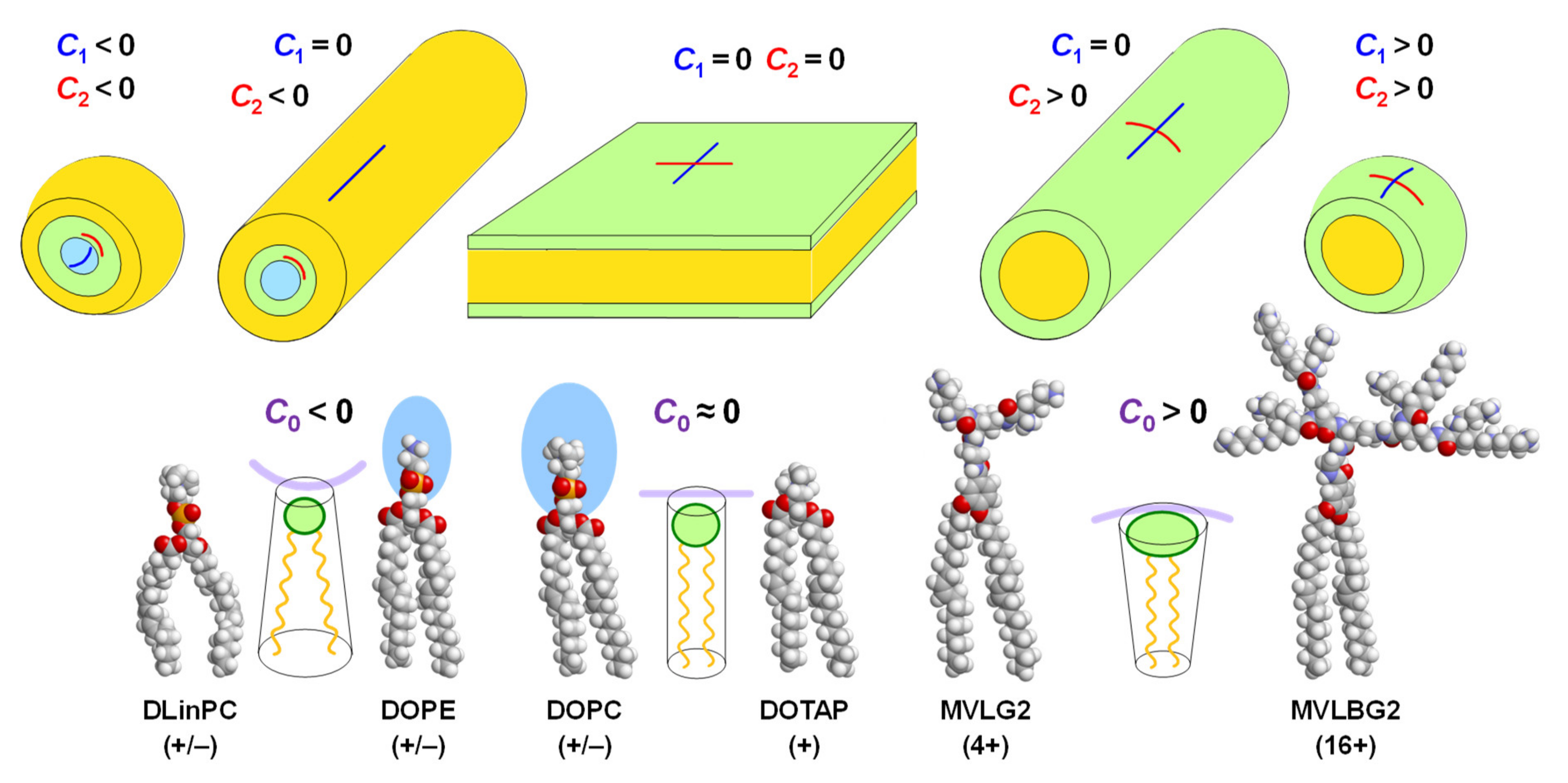{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
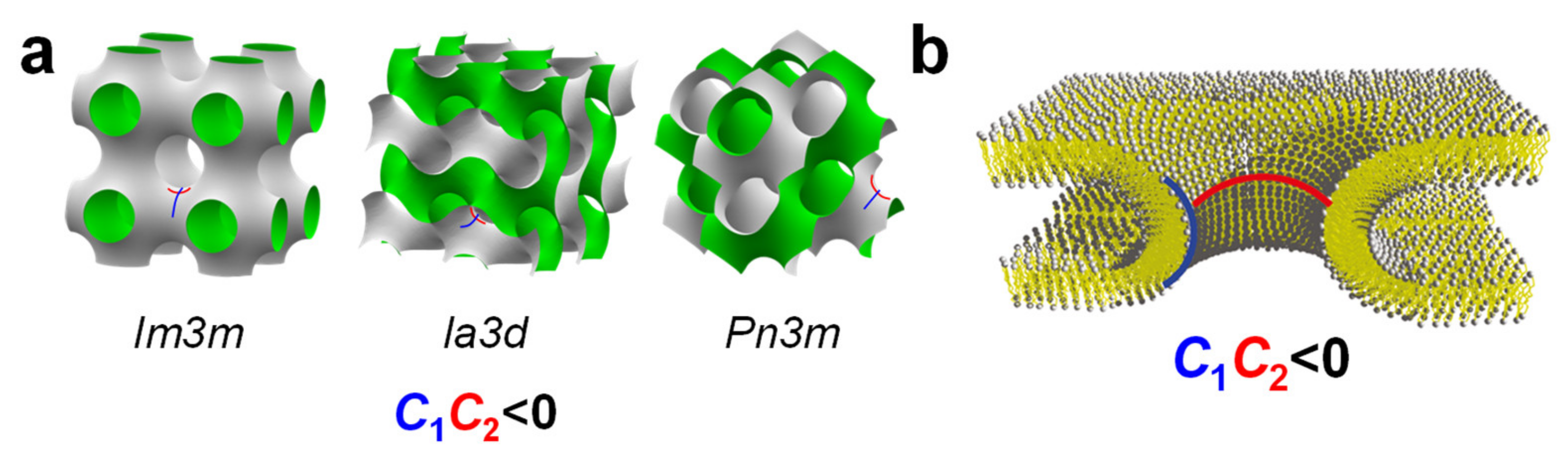
2. Lipid Shape and Membrane Curvature Elastic Energy Determine Their Self-Assembled Structures
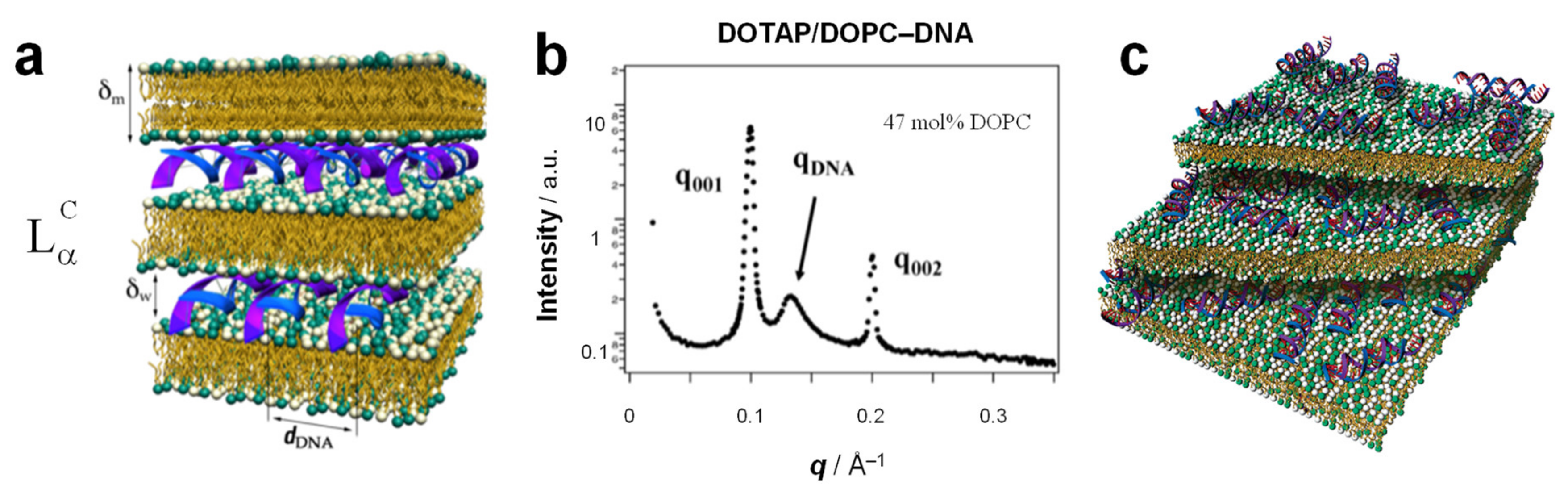
3. The Lamellar LαC Phase of Cationic Liposome–DNA Complexes
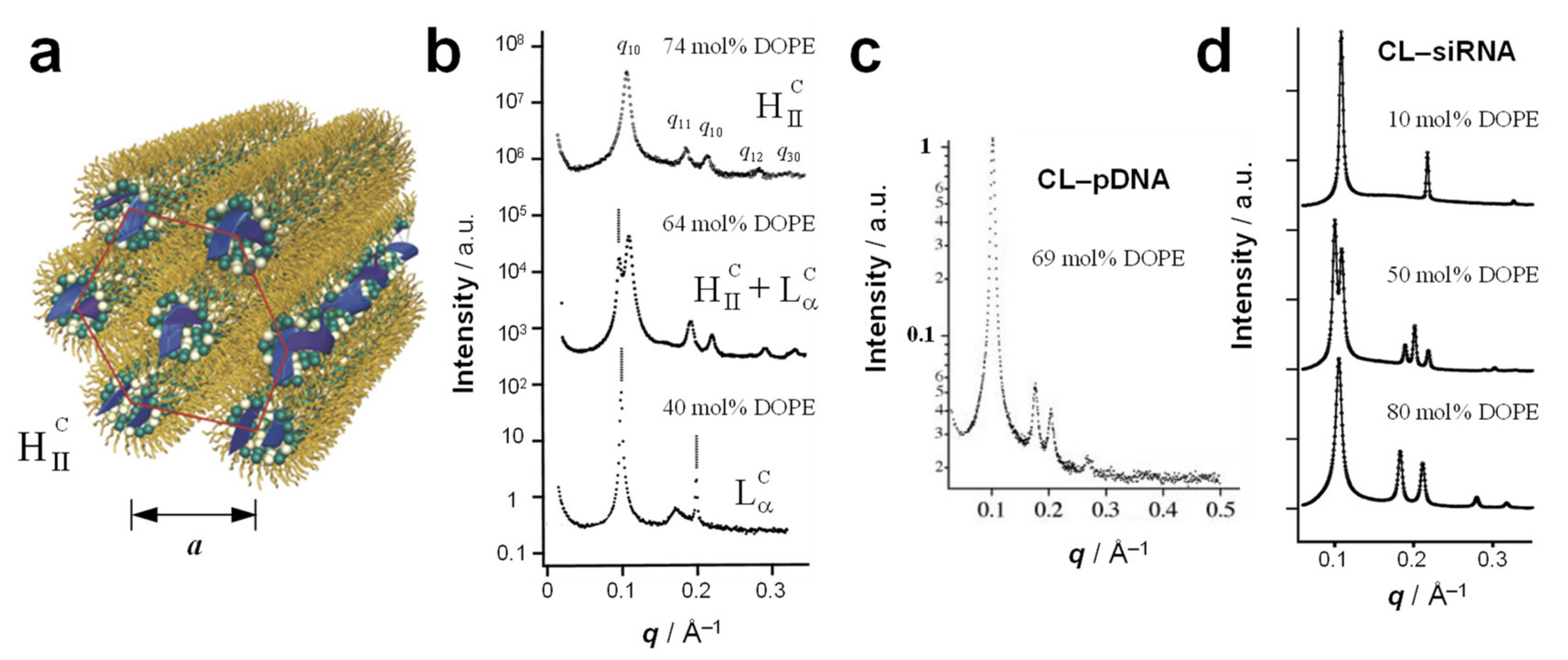
4. The Inverse Hexagonal (HIIC) Phase
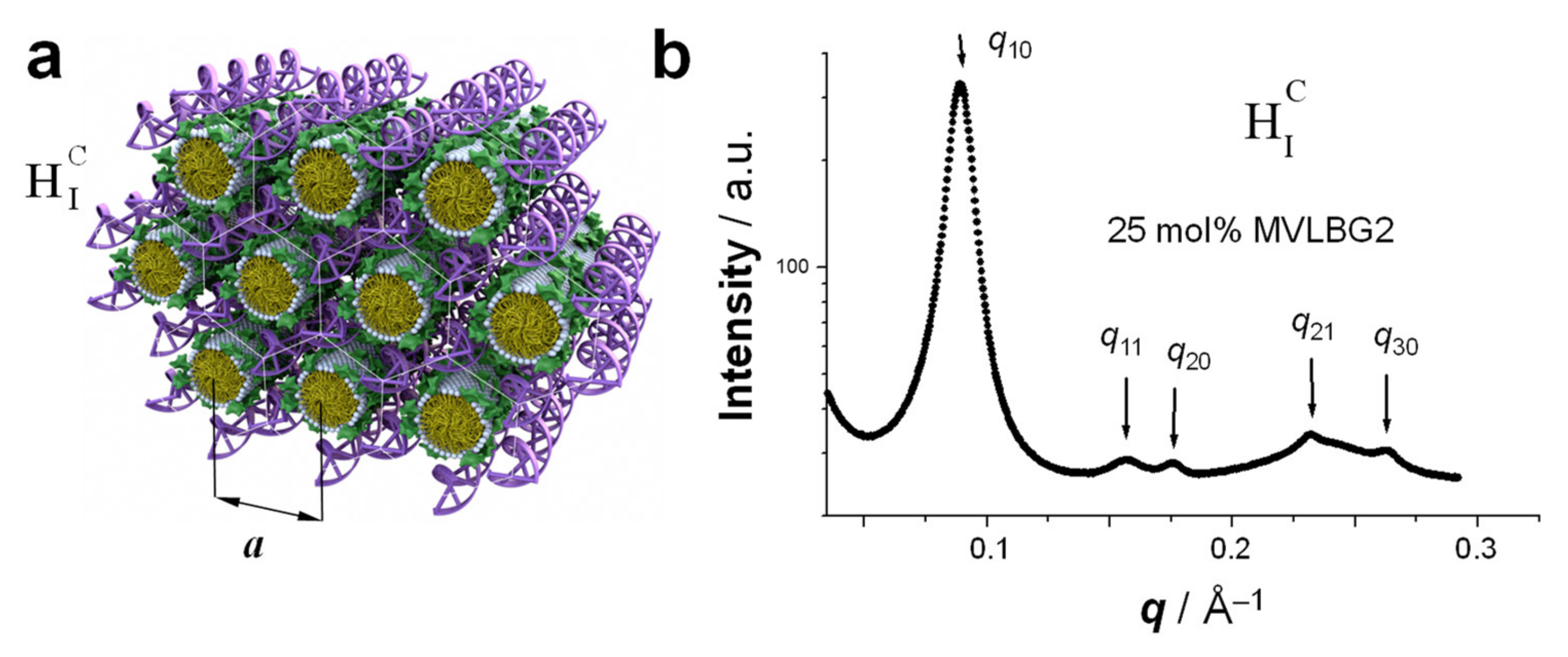
5. Hexagonally Ordered Cylindrical Micelles Embedded in a DNA Honeycomb Lattice: The HIC Phase
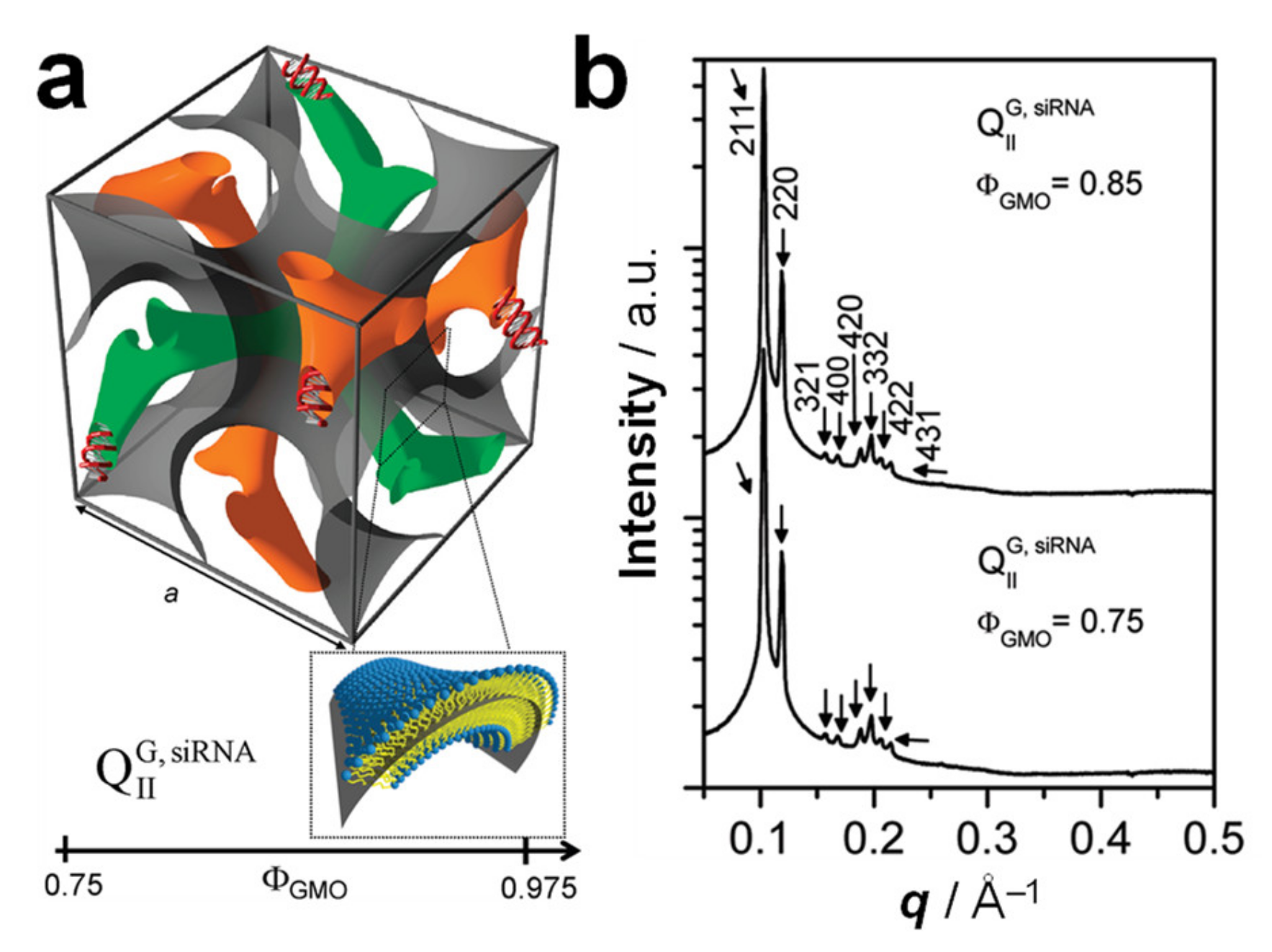
6. Cubic Lipid Phases with Embedded Nucleic Acid
7. Transfection Efficiency and the Structure of CL–DNA Complexes
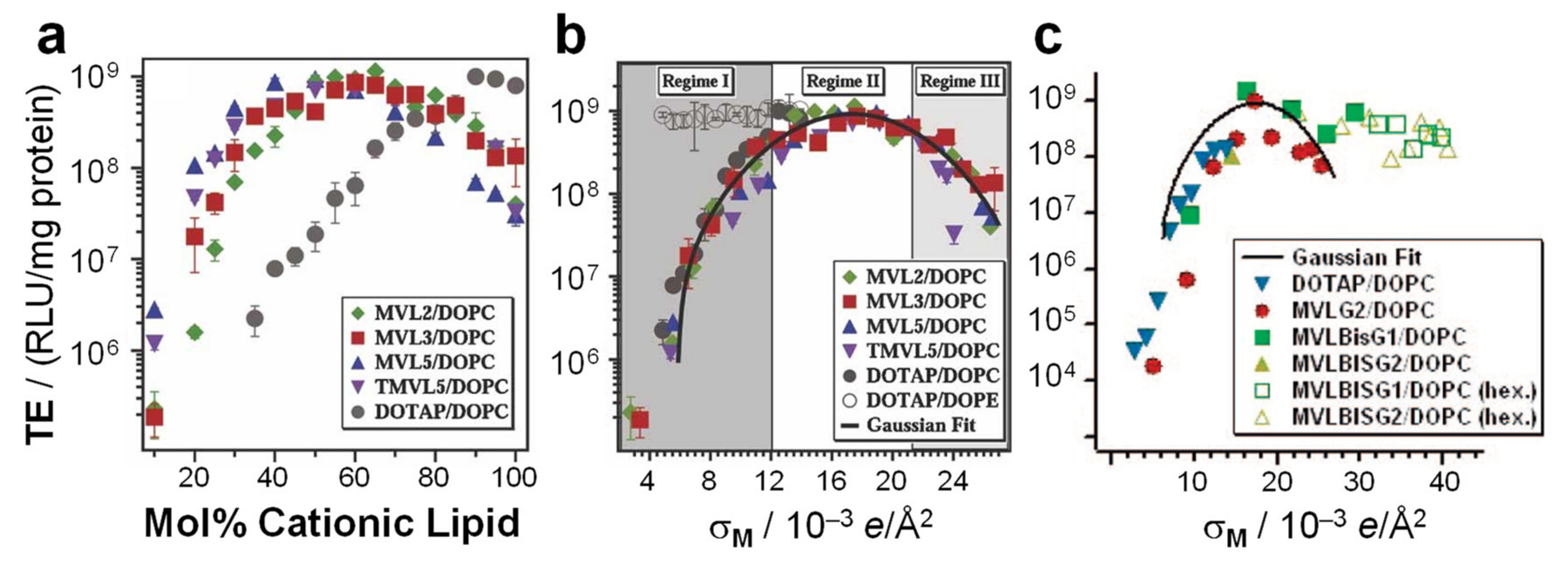
7.1. The Early Rise of DOPE and Its Relation to Complex Structure
7.2. Membrane Charge Density as a Universal Parameter for Transfection by Lamellar CL–DNA Complexes
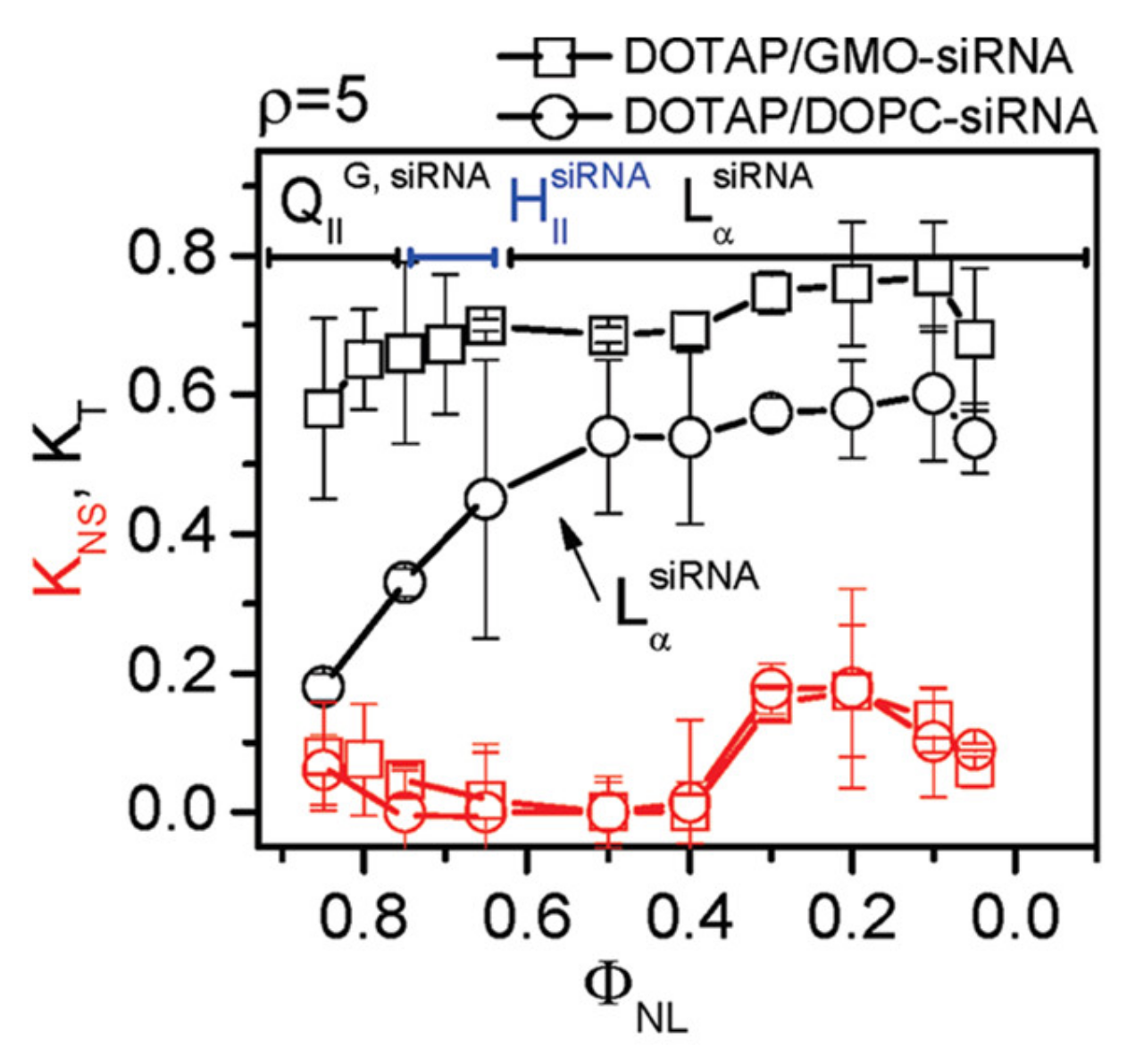
7.3. Highly Efficient Gene Silencing with Cubic CL–siRNA Complexes
8. From In Vitro to In Vivo
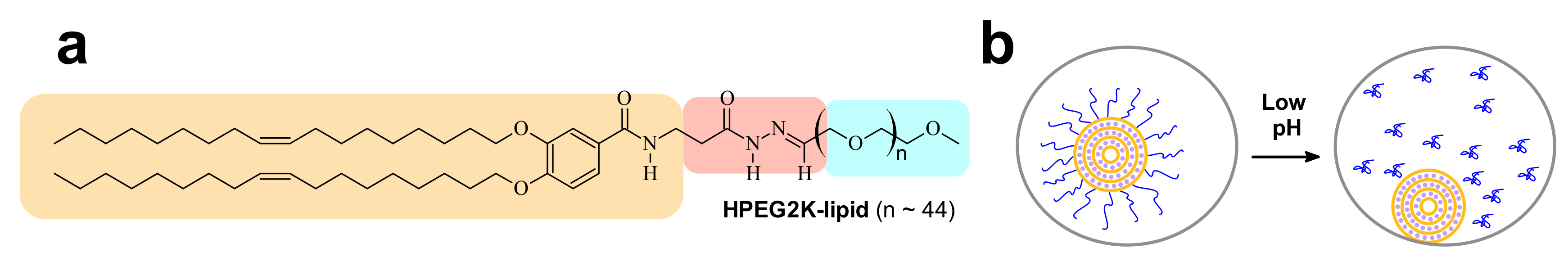
8.1. Low pH-Induced dePEGylation
8.2. Affinity Targeting of PEGylated CL–DNA Complexes
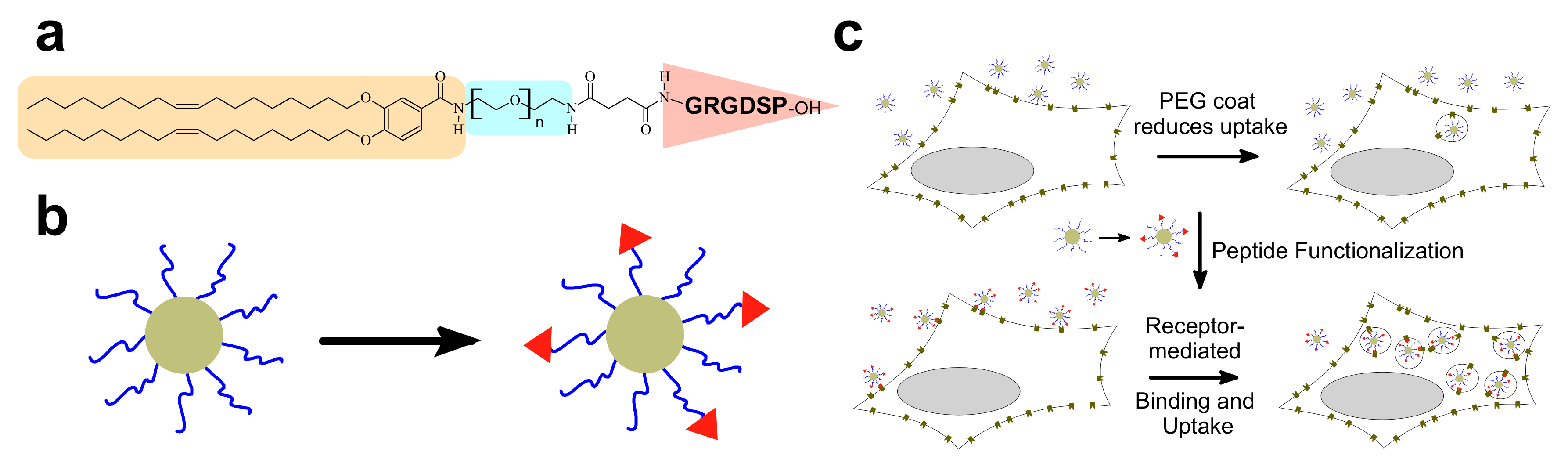
8.3. Organ- and Disease-Specific Targeting Peptides
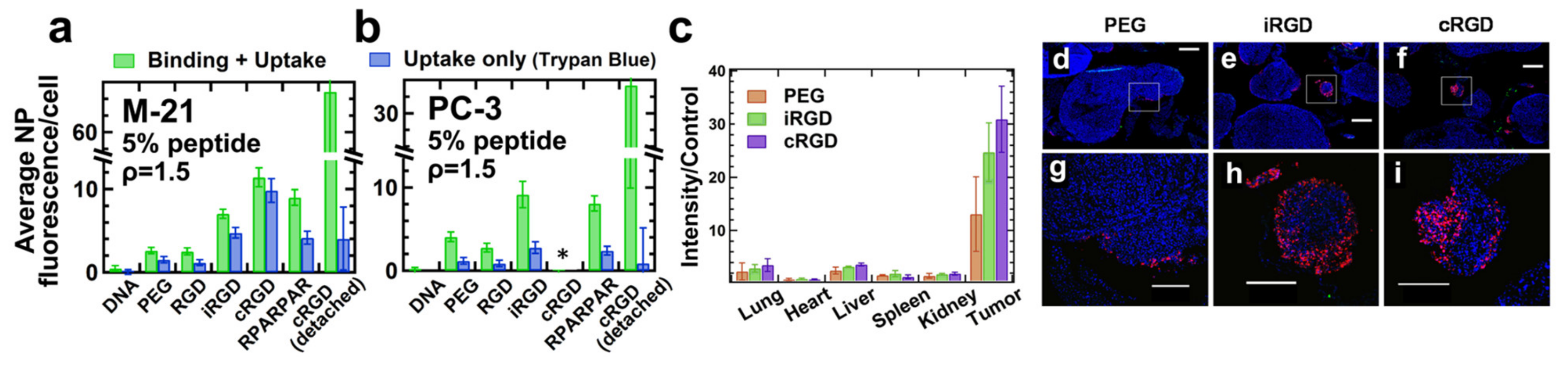
8.4. Peptide Ligands Promote Tumor Targeting and Penetration of CL–DNA NPs In Vivo
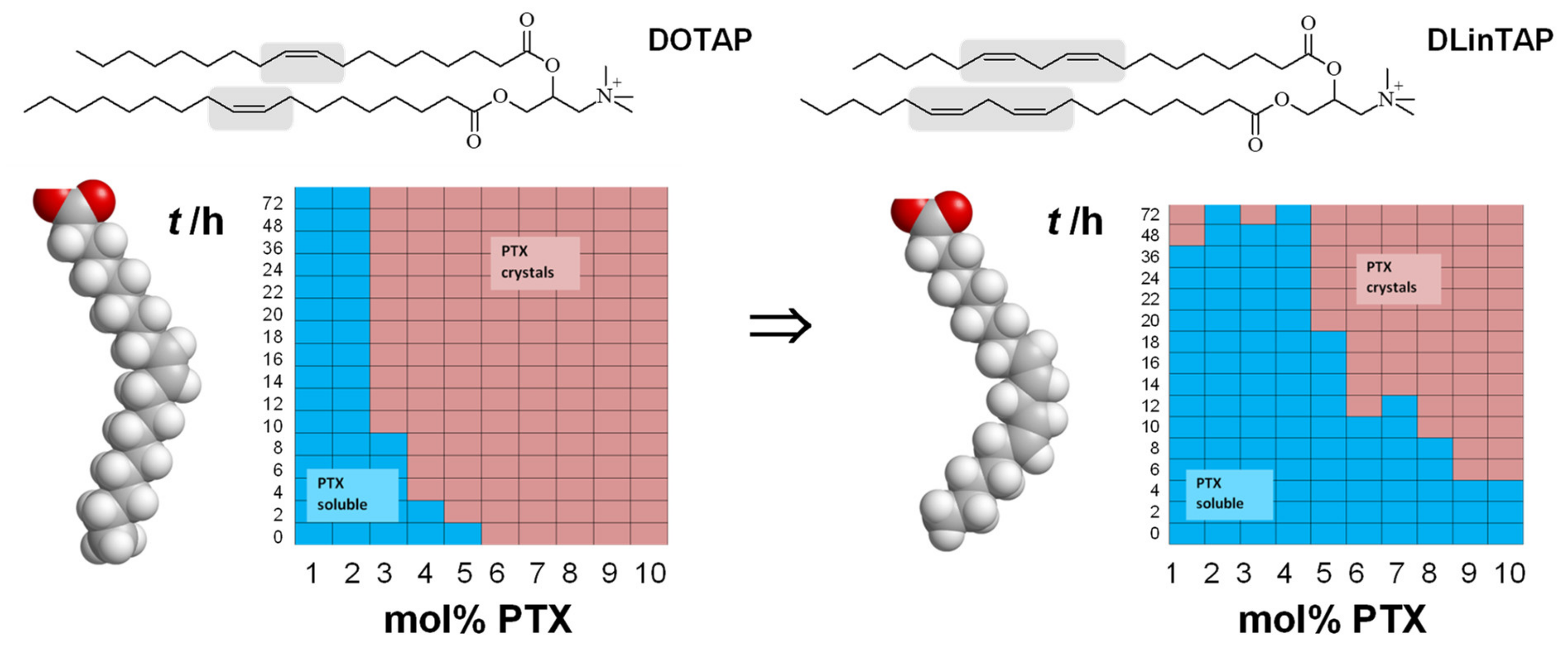
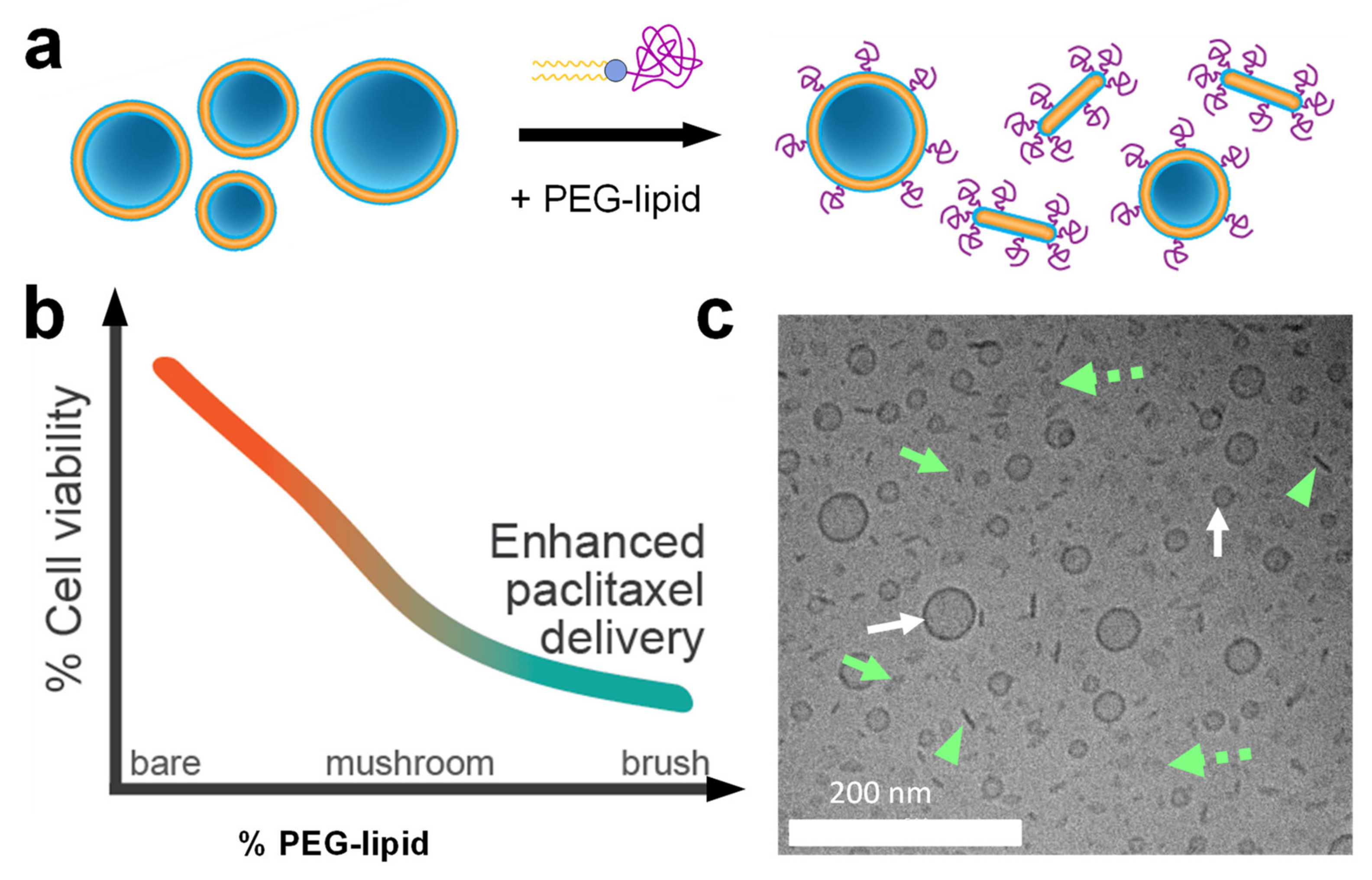
9. Cationic Liposomes for The Delivery of Hydrophobic Drugs
10. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Israelachvili, J.N. Intermolecular and Surface Forces, 3rd ed.; Elsevier: Amsterdam, The Netherlands, 2011. [Google Scholar] [CrossRef]
- Bangham, A.D.; Horne, R.W. Negative staining of phospholipids and their structural modification by surface-active agents as observed in the electron microscope. J. Mol. Biol. 1964, 8, 660–668. [Google Scholar] [CrossRef]
- Bangham, A. Model for biological membranes. New Sci. 1971, 49, 63–64. [Google Scholar]
- Gregoriadis, G.; Leathwood, P.D.; Ryman, B.E. Enzyme entrapment in liposomes. FEBS Lett. 1971, 14, 95–99. [Google Scholar] [CrossRef]
- Gregoriadis, G.; Ryman, B.E. Fate of Protein-Containing Liposomes Injected into Rats. Eur. J. Biochem. 1972, 24, 485–491. [Google Scholar] [CrossRef] [PubMed]
- Gregoriadis, G. The Carrier Potential of Liposomes in Biology and Medicine (Part One). N. Engl. J. Med. 1976, 295, 704–710. [Google Scholar] [CrossRef]
- Gregoriadis, G. The Carrier Potential of Liposomes in Biology and Medicine (Part Two). N. Engl. J. Med. 1976, 295, 765–770. [Google Scholar] [CrossRef] [PubMed]
- Felgner, P.L.; Gadek, T.R.; Holm, M.; Roman, R.; Chan, H.W.; Wenz, M.; Northrop, J.P.; Ringold, G.M.; Danielsen, M. Lipofection: A highly efficient, lipid-mediated DNA-transfection procedure. Proc. Natl. Acad. Sci. USA 1987, 84, 7413–7417. [Google Scholar] [CrossRef]
- Malone, R.W.; Felgner, P.L.; Verma, I.M. Cationic liposome-mediated RNA transfection. Proc. Natl. Acad. Sci. USA 1989, 86, 6077–6081. [Google Scholar] [CrossRef] [PubMed]
- Mislick, K.A.; Baldeschwieler, J.D. Evidence for the role of proteoglycans in cation-mediated gene transfer. Proc. Natl. Acad. Sci. USA 1996, 93, 12349–12354. [Google Scholar] [CrossRef]
- Sternberg, B.; Sorgi, F.L.; Huang, L. New structures in complex formation between DNA and cationic liposomes visualized by freeze—fracture electron microscopy. FEBS Lett. 1994, 356, 361–366. [Google Scholar] [CrossRef]
- Rädler, J.O.; Koltover, I.; Salditt, T.; Safinya, C.R. Structure of DNA-cationic liposome complexes: DNA intercalation in multilamellar membranes in distinct interhelical packing regimes. Science 1997, 275, 810–814. [Google Scholar] [CrossRef]
- Koltover, I.; Salditt, T.; Rädler, J.O.; Safinya, C.R. An inverted hexagonal phase of cationic liposome-DNA complexes related to DNA release and delivery. Science 1998, 281, 78–81. [Google Scholar] [CrossRef]
- Ewert, K.K.; Evans, H.M.; Zidovska, A.; Bouxsein, N.F.; Ahmad, A.; Safinya, C.R. A columnar phase of dendritic lipid-based cationic liposome-DNA complexes for gene delivery: Hexagonally ordered cylindrical micelles embedded in a DNA honeycomb lattice. J. Am. Chem. Soc. 2006, 128, 3998–4006. [Google Scholar] [CrossRef]
- Leal, C.; Bouxsein, N.F.; Ewert, K.K.; Safinya, C.R. Highly Efficient Gene Silencing Activity of siRNA Embedded in a Nanostructured Gyroid Cubic Lipid Matrix. J. Am. Chem. Soc. 2010, 132, 16841–16847. [Google Scholar] [CrossRef] [PubMed]
- Lin, A.J.; Slack, N.L.; Ahmad, A.; George, C.X.; Samuel, C.E.; Safinya, C.R. Three-dimensional imaging of lipid gene-carriers: Membrane charge density controls universal transfection behavior in lamellar cationic liposome-DNA complexes. Biophys. J. 2003, 84, 3307–3316. [Google Scholar] [CrossRef]
- Ahmad, A.; Evans, H.M.; Ewert, K.; George, C.X.; Samuel, C.E.; Safinya, C.R. New multivalent cationic lipids reveal bell curve for transfection efficiency versus membrane charge density: Lipid-DNA complexes for gene delivery. J. Gene Med. 2005, 7, 739–748. [Google Scholar] [CrossRef] [PubMed]
- Bouxsein, N.F.; McAllister, C.S.; Ewert, K.K.; Samuel, C.E.; Safinya, C.R. Structure and gene silencing activities of monovalent and pentavalent cationic lipid vectors complexed with siRNA. Biochemistry 2007, 46, 4785–4792. [Google Scholar] [CrossRef] [PubMed]
- Ponti, F.; Campolungo, M.; Melchiori, C.; Bono, N.; Candiani, G. Cationic lipids for gene delivery: Many players, one goal. Chem. Phys. Lipids 2021, 235, 105032. [Google Scholar] [CrossRef]
- Semple, S.C.; Akinc, A.; Chen, J.; Sandhu, A.P.; Mui, B.L.; Cho, C.K.; Sah, D.W.Y.; Stebbing, D.; Crosley, E.J.; Yaworski, E.; et al. Rational design of cationic lipids for siRNA delivery. Nat. Biotechnol. 2010, 28, 172–176. [Google Scholar] [CrossRef]
- Koynova, R.; Tenchov, B.; Wang, L.; MacDonald, R.C. Hydrophobic Moiety of Cationic Lipids Strongly Modulates Their Transfection Activity. Mol. Pharm. 2009, 6, 951–958. [Google Scholar] [CrossRef]
- Tranchant, I.; Thompson, B.; Nicolazzi, C.; Mignet, N.; Scherman, D. Physicochemical optimisation of plasmid delivery by cationic lipids. J. Gene Med. 2004, 6, S24–S35. [Google Scholar] [CrossRef] [PubMed]
- Labas, R.; Beilvert, F.; Barteau, B.; David, S.; Chèvre, R.; Pitard, B. Nature as a source of inspiration for cationic lipid synthesis. Genetica 2010, 138, 153–168. [Google Scholar] [CrossRef]
- Zhi, D.; Zhang, S.; Cui, S.; Zhao, Y.; Wang, Y.; Zhao, D. The Headgroup Evolution of Cationic Lipids for Gene Delivery. Bioconjug. Chem. 2013, 24, 487–519. [Google Scholar] [CrossRef]
- Wölk, C.; Janich, C.; Bakowsky, U.; Langner, A.; Brezesinski, G. Malonic acid based cationic lipids—The way to highly efficient DNA-carriers. Adv. Colloid Interface Sci. 2017, 248, 20–34. [Google Scholar] [CrossRef]
- Miller, A.D. Cationic Liposomes for Gene Therapy. Angew. Chem. Int. Ed. Engl. 1998, 37, 1768–1785. [Google Scholar] [CrossRef]
- Carrière, M.; Tranchant, I.; Niore, P.-A.; Byk, G.; Mignet, N.; Escriou, V.; Scherman, D.; Herscovici, J. Optimization of Cationic Lipid Mediated Gene Transfer: Structure-Function, Physico-Chemical, and Cellular Studies. J. Liposome Res. 2002, 12, 95–106. [Google Scholar] [CrossRef]
- Bhattacharya, S.; Bajaj, A. Advances in gene delivery through molecular design of cationic lipids. Chem. Commun. 2009, 31, 4632–4656. [Google Scholar] [CrossRef] [PubMed]
- Ewert, K.; Slack, N.L.; Ahmad, A.; Evans, H.M.; Lin, A.J.; Samuel, C.E.; Safinya, C.R. Cationic lipid-DNA complexes for gene therapy: Understanding the relationship between complex structure and gene delivery pathways at the molecular level. Curr. Med. Chem. 2004, 11, 133–149. [Google Scholar] [CrossRef] [PubMed]
- Ewert, K.K.; Evans, H.M.; Bouxsein, N.F.; Safinya, C.R. Dendritic cationic lipids with highly charged headgroups for efficient gene delivery. Bioconjug. Chem. 2006, 17, 877–888. [Google Scholar] [CrossRef] [PubMed]
- Maier, M.A.; Jayaraman, M.; Matsuda, S.; Liu, J.; Barros, S.; Querbes, W.; Tam, Y.K.; Ansell, S.M.; Kumar, V.; Qin, J.; et al. Biodegradable Lipids Enabling Rapidly Eliminated Lipid Nanoparticles for Systemic Delivery of RNAi Therapeutics. Mol. Ther. 2013, 21, 1570–1578. [Google Scholar] [CrossRef] [PubMed]
- Chesnoy, S.; Huang, L. Structure and function of lipid-DNA complexes for gene delivery. Annu. Rev. Biophys. Biomol. Struct. 2000, 29, 27–47. [Google Scholar] [CrossRef]
- Ewert, K.; Evans, H.M.; Ahmad, A.; Slack, N.L.; Lin, A.J.; Martin-Herranz, A.; Safinya, C.R. Lipoplex Structures and Their Distinct Cellular Pathways. In Advances in Genetics, Vol. 53: Non-Viral Vectors for Gene Therapy, 2nd ed.; Huang, L., Hung, M.C., Wagner, E., Eds.; Elsevier; Academic Press: San Diego, CA, USA, 2005; pp. 119–155. [Google Scholar] [CrossRef]
- Ewert, K.; Ahmad, A.; Evans, H.; Safinya, C. Cationic lipid-DNA complexes for non-viral gene therapy: Relating supramolecular structures to cellular pathways. Expert Opin. Biol. Ther. 2005, 5, 33–53. [Google Scholar] [CrossRef] [PubMed]
- Bielke, W.; Erbacher, C. (Eds.) Nucleic Acid Transfection; Springer: Berlin, Germany, 2010. [Google Scholar]
- Dan, N.; Danino, D. Structure and kinetics of lipid–nucleic acid complexes. Adv. Colloid Interface Sci. 2014, 205, 230–239. [Google Scholar] [CrossRef] [PubMed]
- Ewert, K.K.; Zidovska, A.; Ahmad, A.; Bouxsein, N.F.; Evans, H.M.; McAllister, C.S.; Samuel, C.E.; Safinya, C.R. Cationic Liposome–Nucleic Acid Complexes for Gene Delivery and Silencing: Pathways and Mechanisms for Plasmid DNA and siRNA. Top. Curr. Chem. 2010, 296, 191–226. [Google Scholar] [CrossRef] [PubMed]
- Safinya, C.R.; Ewert, K.K.; Majzoub, R.N.; Leal, C. Cationic liposome-nucleic acid complexes for gene delivery and gene silencing. New J. Chem. 2014, 38, 5164–5172. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Huang, L. Recent Advances in Nonviral Vectors for Gene Delivery. Acc. Chem. Res. 2012, 45, 971–979. [Google Scholar] [CrossRef]
- Huang, L.; Hung, M.C.; Wagner, E. (Eds.) Non-Viral Vectors for Gene Therapy, 2nd ed.; Elsevier; Academic Press: San Diego, CA, USA, 2005. [Google Scholar]
- Huang, L.; Hung, M.-C.; Wagner, E. (Eds.) Nonviral Vectors for Gene Therapy; Academic Press: San Diego, CA, USA, 1999. [Google Scholar]
- Huang, L.; Liu, D.; Wagner, E. (Eds.) Nonviral Vectors for Gene Therapy: Lipid- and Polymer-Based Gene Transfer; Academic Press: New York, NY, USA, 2014. [Google Scholar]
- Lin, P.J.C.; Tam, Y.Y.C.; Hafez, I.; Sandhu, A.; Chen, S.; Ciufolini, M.A.; Nabi, I.R.; Cullis, P.R. Influence of cationic lipid composition on uptake and intracellular processing of lipid nanoparticle formulations of siRNA. Nanomedicine 2013, 9, 233–246. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Kanasty, R.L.; Eltoukhy, A.A.; Vegas, A.J.; Dorkin, J.R.; Anderson, D.G. Non-viral vectors for gene-based therapy. Nat. Rev. Genet. 2014, 15, 541–555. [Google Scholar] [CrossRef]
- Foldvari, M.; Chen, D.W.; Nafissi, N.; Calderon, D.; Narsineni, L.; Rafiee, A. Non-viral gene therapy: Gains and challenges of non-invasive administration methods. J. Control. Release 2016, 240, 165–190. [Google Scholar] [CrossRef]
- Kulkarni, J.A.; Witzigmann, D.; Thomson, S.B.; Chen, S.; Leavitt, B.R.; Cullis, P.R.; van der Meel, R. The current landscape of nucleic acid therapeutics. Nat. Nanotechnol. 2021. [Google Scholar] [CrossRef]
- Ginn, S.L.; Amaya, A.K.; Alexander, I.E.; Edelstein, M.; Abedi, M.R. Gene therapy clinical trials worldwide to 2017: An update. J. Gene Med. 2018, 20, e3015. [Google Scholar] [CrossRef] [PubMed]
- The Journal of Gene Medicine: Gene Therapy Clinical Trials Worldwide. Available online: https://a873679.fmphost.com/fmi/webd/GTCT (accessed on 9 June 2021).
- Fire, A.; Xu, S.Q.; Montgomery, M.K.; Kostas, S.A.; Driver, S.E.; Mello, C.C. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 1998, 391, 806–811. [Google Scholar] [CrossRef] [PubMed]
- Cogoni, C.; Macino, G. Post-transcriptional gene silencing across kingdoms. Curr. Opin. Genet. Dev. 2000, 10, 638–643. [Google Scholar] [CrossRef]
- Elbashir, S.M.; Harborth, J.; Lendeckel, W.; Yalcin, A.; Weber, K.; Tuschl, T. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature 2001, 411, 494–498. [Google Scholar] [CrossRef]
- Caplen, N.J.; Parrish, S.; Imani, F.; Fire, A.; Morgan, R.A. Specific inhibition of gene expression by small double-stranded RNAs in invertebrate and vertebrate systems. Proc. Natl. Acad. Sci. USA 2001, 98, 9742–9747. [Google Scholar] [CrossRef] [PubMed]
- Sioud, M. Therapeutic siRNAs. Trends Pharmacol. Sci. 2004, 25, 22–28. [Google Scholar] [CrossRef]
- Karagiannis, T.C.; El-Osta, A. RNA interference and potential therapeutic applications of short interfering RNAs. Cancer Gene Ther. 2005, 12, 787–795. [Google Scholar] [CrossRef]
- Wan, C.; Allen, T.M.; Cullis, P.R. Lipid nanoparticle delivery systems for siRNA-based therapeutics. Drug Deliv. Transl. Res. 2014, 4, 74–83. [Google Scholar] [CrossRef]
- Spagnou, S.; Miller, A.D.; Keller, M. Lipidic Carriers of siRNA: Differences in the Formulation, Cellular Uptake, and Delivery with Plasmid DNA. Biochemistry 2004, 43, 13348–13356. [Google Scholar] [CrossRef]
- Ozcan, G.; Ozpolat, B.; Coleman, R.L.; Sood, A.K.; Lopez-Berestein, G. Preclinical and clinical development of siRNA-based therapeutics. Adv. Drug Deliv. Rev. 2015, 87, 108–119. [Google Scholar] [CrossRef]
- Lares, M.R.; Rossi, J.J.; Ouellet, D.L. RNAi and small interfering RNAs in human disease therapeutic applications. Trends Biotechnol. 2010, 28, 570–579. [Google Scholar] [CrossRef]
- Rupaimoole, R.; Slack, F.J. MicroRNA therapeutics: Towards a new era for the management of cancer and other diseases. Nat. Rev. Drug Discov. 2017, 16, 203–222. [Google Scholar] [CrossRef]
- Pineda, M.; Moghadam, F.; Ebrahimkhani, M.R.; Kiani, S. Engineered CRISPR Systems for Next Generation Gene Therapies. ACS Synth. Biol. 2017, 6, 1614–1626. [Google Scholar] [CrossRef]
- Yin, H.; Kauffman, K.J.; Anderson, D.G. Delivery technologies for genome editing. Nat. Rev. Drug Discov. 2017, 16, 387–399. [Google Scholar] [CrossRef]
- Finn, J.D.; Smith, A.R.; Patel, M.C.; Shaw, L.; Youniss, M.R.; van Heteren, J.; Dirstine, T.; Ciullo, C.; Lescarbeau, R.; Seitzer, J.; et al. A Single Administration of CRISPR/Cas9 Lipid Nanoparticles Achieves Robust and Persistent In Vivo Genome Editing. Cell Rep. 2018, 22, 2227–2235. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.; Ashwanikumar, N.; Robinson, E.; DuRoss, A.; Sun, C.; Murphy-Benenato, K.E.; Mihai, C.; Almarsson, Ö.; Sahay, G. Boosting Intracellular Delivery of Lipid Nanoparticle-Encapsulated mRNA. Nano Lett. 2017, 17, 5711–5718. [Google Scholar] [CrossRef] [PubMed]
- Weissman, D.; Karikó, K. mRNA: Fulfilling the Promise of Gene Therapy. Mol. Ther. 2015, 23, 1416–1417. [Google Scholar] [CrossRef] [PubMed]
- Sahin, U.; Karikó, K.; Türeci, Ö. mRNA-based therapeutics—Developing a new class of drugs. Nat. Rev. Drug Discov. 2014, 13, 759–780. [Google Scholar] [CrossRef]
- Karikó, K.; Muramatsu, H.; Welsh, F.A.; Ludwig, J.; Kato, H.; Akira, S.; Weissman, D. Incorporation of Pseudouridine into mRNA Yields Superior Nonimmunogenic Vector with Increased Translational Capacity and Biological Stability. Mol. Ther. 2008, 16, 1833–1840. [Google Scholar] [CrossRef]
- Karikó, K.; Muramatsu, H.; Ludwig, J.; Weissman, D. Generating the optimal mRNA for therapy: HPLC purification eliminates immune activation and improves translation of nucleoside-modified, protein-encoding mRNA. Nucleic Acids Res. 2011, 39, e142. [Google Scholar] [CrossRef]
- Karikó, K.; Buckstein, M.; Ni, H.; Weissman, D. Suppression of RNA Recognition by Toll-like Receptors: The Impact of Nucleoside Modification and the Evolutionary Origin of RNA. Immunity 2005, 23, 165–175. [Google Scholar] [CrossRef]
- Andries, O.; Mc Cafferty, S.; De Smedt, S.C.; Weiss, R.; Sanders, N.N.; Kitada, T. N1-methylpseudouridine-incorporated mRNA outperforms pseudouridine-incorporated mRNA by providing enhanced protein expression and reduced immunogenicity in mammalian cell lines and mice. J. Control. Release 2015, 217, 337–344. [Google Scholar] [CrossRef]
- Nance, K.D.; Meier, J.L. Modifications in an Emergency: The Role of N1-Methylpseudouridine in COVID-19 Vaccines. ACS Cent. Sci. 2021, 7, 748–756. [Google Scholar] [CrossRef] [PubMed]
- Hoy, S.M. Patisiran: First Global Approval. Drugs 2018, 78, 1625–1631. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, J.A.; Cullis, P.R.; van der Meel, R. Lipid Nanoparticles Enabling Gene Therapies: From Concepts to Clinical Utility. Nucleic Acid Ther. 2018, 28, 146–157. [Google Scholar] [CrossRef]
- Pardi, N.; Hogan, M.J.; Porter, F.W.; Weissman, D. mRNA vaccines—A new era in vaccinology. Nat. Rev. Drug Discov. 2018, 17, 261–279. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, J.A.; Witzigmann, D.; Chen, S.; Cullis, P.R.; van der Meel, R. Lipid Nanoparticle Technology for Clinical Translation of siRNA Therapeutics. Acc. Chem. Res. 2019, 52, 2435–2444. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, T.S.; Lee, A.C.H.; Akinc, A.; Bramlage, B.; Bumcrot, D.; Fedoruk, M.N.; Harborth, J.; Heyes, J.A.; Jeffs, L.B.; John, M.; et al. RNAi-mediated gene silencing in non-human primates. Nature 2006, 441, 111–114. [Google Scholar] [CrossRef] [PubMed]
- Mulligan, M.J.; Lyke, K.E.; Kitchin, N.; Absalon, J.; Gurtman, A.; Lockhart, S.; Neuzil, K.; Raabe, V.; Bailey, R.; Swanson, K.A.; et al. Phase I/II study of COVID-19 RNA vaccine BNT162b1 in adults. Nature 2020, 586, 589–593. [Google Scholar] [CrossRef]
- Polack, F.P.; Thomas, S.J.; Kitchin, N.; Absalon, J.; Gurtman, A.; Lockhart, S.; Perez, J.L.; Pérez Marc, G.; Moreira, E.D.; Zerbini, C.; et al. Safety and Efficacy of the BNT162b2 mRNA Covid-19 Vaccine. N. Engl. J. Med. 2020, 383, 2603–2615. [Google Scholar] [CrossRef] [PubMed]
- Gaspar, R.; Coelho, F.; Silva, B.F.B. Lipid-Nucleic Acid Complexes: Physicochemical Aspects and Prospects for Cancer Treatment. Molecules 2020, 25, 5006. [Google Scholar] [CrossRef] [PubMed]
- Baden, L.R.; El Sahly, H.M.; Essink, B.; Kotloff, K.; Frey, S.; Novak, R.; Diemert, D.; Spector, S.A.; Rouphael, N.; Creech, C.B.; et al. Efficacy and Safety of the mRNA-1273 SARS-CoV-2 Vaccine. N. Engl. J. Med. 2020, 384, 403–416. [Google Scholar] [CrossRef] [PubMed]
- Jackson, L.A.; Anderson, E.J.; Rouphael, N.G.; Roberts, P.C.; Makhene, M.; Coler, R.N.; McCullough, M.P.; Chappell, J.D.; Denison, M.R.; Stevens, L.J.; et al. An mRNA Vaccine against SARS-CoV-2—Preliminary Report. N. Engl. J. Med. 2020, 383, 1920–1931. [Google Scholar] [CrossRef]
- Corbett, K.S.; Edwards, D.; Leist, S.R.; Abiona, O.M.; Boyoglu-Barnum, S.; Gillespie, R.A.; Himansu, S.; Schäfer, A.; Ziwawo, C.T.; DiPiazza, A.T.; et al. SARS-CoV-2 mRNA Vaccine Development Enabled by Prototype Pathogen Preparedness. bioRxiv 2020. [Google Scholar] [CrossRef]
- United States Food and Drug Administration: Pfizer-BioNTech COVID-19 Vaccine Emergency Use Authorization Fact Sheet for Healthcare Providers Administering Vaccine. Available online: https://www.fda.gov/media/144413/download (accessed on 10 June 2021).
- Leventis, R.; Silvius, J.R. Interactions of mammalian cells with lipid dispersions containing novel metabolizable cationic amphiphiles. Biochim. Biophys. Acta Biomembr. 1990, 1023, 124–132. [Google Scholar] [CrossRef]
- Ewert, K.; Ahmad, A.; Evans, H.M.; Schmidt, H.-W.; Safinya, C.R. Efficient synthesis and cell-transfection properties of a new multivalent cationic lipid for nonviral gene delivery. J. Med. Chem. 2002, 45, 5023–5029. [Google Scholar] [CrossRef]
- Heyes, J.; Palmer, L.; Bremner, K.; MacLachlan, I. Cationic lipid saturation influences intracellular delivery of encapsulated nucleic acids. J. Control. Release 2005, 107, 276–287. [Google Scholar] [CrossRef]
- Hacein-Bey-Abina, S.; Garrigue, A.; Wang, G.P.; Soulier, J.; Lim, A.; Morillon, E.; Clappier, E.; Caccavelli, L.; Delabesse, E.; Beldjord, K.; et al. Insertional oncogenesis in 4 patients after retrovirus-mediated gene therapy of SCID-X1. J. Clin. Investig. 2008, 118, 3132–3142. [Google Scholar] [CrossRef]
- Thomas, C.E.; Ehrhardt, A.; Kay, M.A. Progress and problems with the use of viral vectors for gene therapy. Nat. Rev. Genet. 2003, 4, 346–358. [Google Scholar] [CrossRef]
- Raper, S.E.; Chirmule, N.; Lee, F.S.; Wivel, N.A.; Bagg, A.; Gao, G.-p.; Wilson, J.M.; Batshaw, M.L. Fatal systemic inflammatory response syndrome in a ornithine transcarbamylase deficient patient following adenoviral gene transfer. Mol. Genet. Metab. 2003, 80, 148–158. [Google Scholar] [CrossRef]
- Wilson, J.M. Lessons learned from the gene therapy trial for ornithine transcarbamylase deficiency. Mol. Genet. Metab. 2009, 96, 151–157. [Google Scholar] [CrossRef]
- Kouprina, N.; Tomilin, A.N.; Masumoto, H.; Earnshaw, W.C.; Larionov, V. Human artificial chromosome-based gene delivery vectors for biomedicine and biotechnology. Expert Opin. Drug Deliv. 2014, 11, 517–535. [Google Scholar] [CrossRef]
- Larin, Z.; Mejía, J.E. Advances in human artificial chromosome technology. Trends Genet. 2002, 18, 313–319. [Google Scholar] [CrossRef]
- Harrington, J.J.; Van Bokkelen, G.; Mays, R.W.; Gustashaw, K.; Willard, H.F. Formation of de novo centromeres and construction of first-generation human artificial microchromosomes. Nat. Genet. 1997, 15, 345–355. [Google Scholar] [CrossRef] [PubMed]
- Li, S.-D.; Huang, L. Non-viral is superior to viral gene delivery. J. Control. Release 2007, 123, 181–183. [Google Scholar] [CrossRef] [PubMed]
- Gindy, M.E.; Leone, A.M.; Cunningham, J.J. Challenges in the pharmaceutical development of lipid-based short interfering ribonucleic acid therapeutics. Expert Opin. Drug Deliv. 2012, 9, 171–182. [Google Scholar] [CrossRef] [PubMed]
- Ewert, K.; Zidovska, A.; Ahmad, A.; Bouxsein, N.; Evans, H.; McAllister, C.; Samuel, C.; Safinya, C. Cationic Liposome–Nucleic Acid Complexes for Gene Delivery and Silencing: Pathways and Mechanisms for Plasmid DNA and siRNA. In Nucleic Acid Transfection; Bielke, W., Erbacher, C., Eds.; Springer: Berlin, Germany, 2010; pp. 191–226. [Google Scholar] [CrossRef]
- Mahato, R.I.; Kim, S.W. Pharmaceutical Perspectives of Nucleic Acid-Based Therapy; CRC Press: Boca Raton, FL, USA, 2002. [Google Scholar]
- Cullis, P.R.; Hope, M.J. Lipid Nanoparticle Systems for Enabling Gene Therapies. Mol. Ther. 2017, 25, 1467–1475. [Google Scholar] [CrossRef]
- Israelachvili, J.N.; Mitchell, D.J.; Ninham, B.W. Theory of self-assembly of hydrocarbon amphiphiles into micelles and bilayers. J. Chem. Soc. Faraday Trans. 2 1976, 72, 1525–1568. [Google Scholar] [CrossRef]
- Israelachvili, J.N.; Mitchell, D.J.; Ninham, B.W. Theory of self-assembly of lipid bilayers and vesicles. Biochim. Biophys. Acta Biomembr. 1977, 470, 185–201. [Google Scholar] [CrossRef]
- Cullis, P.R.; Hope, M.J.; Tilcock, C.P.S. Lipid polymorphism and the roles of lipids in membranes. Chem. Phys. Lipids 1986, 40, 127–144. [Google Scholar] [CrossRef]
- Tilcock, C.P.S. Lipid polymorphism. Chem. Phys. Lipids 1986, 40, 109–125. [Google Scholar] [CrossRef]
- Helfrich, W.Z. Elastic Properties of Lipid Bilayers: Theory and Possible Experiments. Z. Naturforsch. C J. Biosci. 1973, 28, 693–703. [Google Scholar] [CrossRef]
- Lipowsky, R.; Sackmann, E. (Eds.) Structure and Dynamics of Membranes; Elsevier: Amsterdam, The Netherlands, 1995. [Google Scholar]
- Safran, S.A. Statistical Thermodynamics of Surfaces, Interfaces, and Membranes; Addison-Wesley: Reading, UK, 1994. [Google Scholar]
- Landau, L.D.; Lifshitz, E.M. Theory of Elasticity, Volume 7 of Course of Theoretical Physics; Pergamon Press: Oxford, UK, 1970. [Google Scholar]
- Szleifer, I.; Kramer, D.; Ben-Shaul, A.; Roux, D.; Gelbart, W.M. Curvature Elasticity of Pure and Mixed Surfactant Films. Phys. Rev. Lett. 1988, 60, 1966–1969. [Google Scholar] [CrossRef]
- Seddon, J.M. Structure of the inverted hexagonal (HII) phase, and non-lamellar phase transitions of lipids. Biochim. Biophys. Acta 1990, 1031, 1–69. [Google Scholar] [CrossRef]
- Gruner, S.M. Stability of lyotropic phases with curved interfaces. J. Phys. Chem. 1989, 93, 7562–7570. [Google Scholar] [CrossRef]
- Zhen, Y.; Ewert, K.K.; Fisher, W.S.; Steffes, V.M.; Li, Y.; Safinya, C.R. Paclitaxel loading in cationic liposome vectors is enhanced by replacement of oleoyl with linoleoyl tails with distinct lipid shapes. Sci. Rep. 2021, 11, 7311. [Google Scholar] [CrossRef] [PubMed]
- Behr, J.; Demeneix, B.; Loeffler, J.; Perez-Mutul, J. Efficient gene transfer into mammalian primary endocrine cells with lipopolyamine-coated DNA. Proc. Natl. Acad. Sci. USA 1989, 86, 6982–6986. [Google Scholar] [CrossRef] [PubMed]
- Zidovska, A.; Ewert, K.K.; Quispe, J.; Carragher, B.; Potter, C.S.; Safinya, C.R. Block liposome and nanotube formation is a general phenomenon of two-component membranes containing multivalent lipids. Soft Matter 2011, 7, 8363–8369. [Google Scholar] [CrossRef] [PubMed]
- Steffes, V.M.; Zhang, Z.; MacDonald, S.; Crowe, J.; Ewert, K.K.; Carragher, B.; Potter, C.S.; Safinya, C.R. PEGylation of Paclitaxel-Loaded Cationic Liposomes Drives Steric Stabilization of Bicelles and Vesicles thereby Enhancing Delivery and Cytotoxicity to Human Cancer Cells. ACS Appl. Mater. Interfaces 2020, 12, 151–162. [Google Scholar] [CrossRef] [PubMed]
- Johnsson, M.; Edwards, K. Phase Behavior and Aggregate Structure in Mixtures of Dioleoylphosphatidylethanolamine and Poly(Ethylene Glycol)-Lipids. Biophys. J. 2001, 80, 313–323. [Google Scholar] [CrossRef]
- Sandström, M.C.; Johansson, E.; Edwards, K. Structure of Mixed Micelles Formed in PEG-Lipid/Lipid Dispersions. Langmuir 2007, 23, 4192–4198. [Google Scholar] [CrossRef] [PubMed]
- Zidovska, A.; Evans, H.M.; Ewert, K.K.; Quispe, J.; Carragher, B.; Potter, C.S.; Safinya, C.R. Liquid crystalline phases of dendritic lipid-DNA self-assemblies: Lamellar, hexagonal, and DNA bundles. J. Phys. Chem. B 2009, 113, 3694–3703. [Google Scholar] [CrossRef]
- Siegel, D.P. The Modified Stalk Mechanism of Lamellar/Inverted Phase Transitions and Its Implications for Membrane Fusion. Biophys. J. 1999, 76, 291–313. [Google Scholar] [CrossRef]
- Porte, G. Lamellar phases and disordered phases of fluid bilayer membranes. J. Phys. Condens. Matter 1992, 4, 8649–8670. [Google Scholar] [CrossRef]
- Yang, L.; Huang, H.W. Observation of a Membrane Fusion Intermediate Structure. Science 2002, 297, 1877–1879. [Google Scholar] [CrossRef] [PubMed]
- Conn, C.E.; Ces, O.; Squires, A.M.; Mulet, X.; Winter, R.; Finet, S.M.; Templer, R.H.; Seddon, J.M. A pressure-jump time-resolved x-ray diffraction study of cubic-cubic transition kinetics in monoolein. Langmuir 2008, 24, 2331–2340. [Google Scholar] [CrossRef]
- Lindblom, G.; Larsson, K.; Johansson, L.; Fontell, K.; Forsen, S. The cubic phase of monoglyceride-water systems. Arguments for a structure based upon lamellar bilayer units. J. Am. Chem. Soc. 1979, 101, 5465–5470. [Google Scholar] [CrossRef]
- Leal, C.; Ewert, K.K.; Shirazi, R.S.; Bouxsein, N.F.; Safinya, C.R. Nanogyroids Incorporating Multivalent Lipids: Enhanced Membrane Charge Density and Pore Forming Ability for Gene Silencing. Langmuir 2011, 27, 7691–7697. [Google Scholar] [CrossRef] [PubMed]
- Manning, G.S. Limiting Laws and Counterion Condensation in Polyelectrolyte Solutions I. Colligative Properties. J. Chem. Phys. 1969, 51, 924–933. [Google Scholar] [CrossRef]
- Piotrowski-Daspit, A.S.; Kauffman, A.C.; Bracaglia, L.G.; Saltzman, W.M. Polymeric vehicles for nucleic acid delivery. Adv. Drug Deliv. Rev. 2020, 156, 119–132. [Google Scholar] [CrossRef]
- Lasic, D.D.; Strey, H.; Stuart, M.C.A.; Podgornik, R.; Frederik, P.M. The Structure of DNA−Liposome Complexes. J. Am. Chem. Soc. 1997, 119, 832–833. [Google Scholar] [CrossRef]
- Huebner, S.; Battersby, B.J.; Grimm, R.; Cevc, G. Lipid-DNA Complex Formation: Reorganization and Rupture of Lipid Vesicles in the Presence of DNA as Observed by Cryoelectron Microscopy. Biophys. J. 1999, 76, 3158–3166. [Google Scholar] [CrossRef]
- Majzoub, R.N.; Ewert, K.K.; Jacovetty, E.L.; Carragher, B.; Potter, C.S.; Li, Y.; Safinya, C.R. Patterned Threadlike Micelles and DNA-Tethered Nanoparticles: A Structural Study of PEGylated Cationic Liposome–DNA Assemblies. Langmuir 2015, 31, 7073–7083. [Google Scholar] [CrossRef] [PubMed]
- Safinya, C.R.; Roux, D.; Smith, G.S.; Sinha, S.K.; Dimon, P.; Clark, N.A.; Bellocq, A.M. Steric Interactions in a Model Multimembrane System: A Synchrotron X-ray Study. Phys. Rev. Lett. 1986, 57, 2718–2721. [Google Scholar] [CrossRef]
- Roux, D.; Safinya, C.R. A Synchrotron X-ray study of Competing Undulation and Electrostatic Interlayer Interactions in Fluid Multimembrane Lyotropic Phases. J. Phys. 1988, 49, 307–318. [Google Scholar] [CrossRef]
- Safinya, C.R.; Sirota, E.B.; Roux, D.; Smith, G.S. Universality in interacting membranes: The effect of cosurfactants on the interfacial rigidity. Phys. Rev. Lett. 1989, 62, 1134–1137. [Google Scholar] [CrossRef]
- Lei, N.; Safinya, C.R.; Bruinsma, R.F. Discrete Harmonic Model for Stacked Membranes: Theory and Experiment. J. Phys. II 1995, 5, 1155–1163. [Google Scholar] [CrossRef]
- Koltover, I.; Salditt, T.; Safinya, C.R. Phase diagram, stability, and overcharging of lamellar cationic lipid-DNA self-assembled complexes. Biophys. J. 1999, 77, 915–924. [Google Scholar] [CrossRef]
- Safinya, C.R. Structures of lipid-DNA complexes: Supramolecular assembly and gene delivery. Curr. Opin. Struct. Biol. 2001, 11, 440–448. [Google Scholar] [CrossRef]
- Salditt, T.; Koltover, I.; Rädler, J.O.; Safinya, C.R. Two-dimensional smectic ordering of linear DNA chains in self-assembled DNA-cationic liposome mixtures. Phys. Rev. Lett. 1997, 79, 2582–2585. [Google Scholar] [CrossRef]
- Salditt, T.; Koltover, I.; Rädler, J.O.; Safinya, C.R. Self-assembled DNA--cationic-lipid complexes: Two-dimensional smectic ordering, correlations, and interactions. Phys. Rev. E 1998, 58, 889–904. [Google Scholar] [CrossRef]
- Zidovska, A.; Evans, H.M.; Ahmad, A.; Ewert, K.K.; Safinya, C.R. The Role of Cholesterol and Structurally Related Molecules in Enhancing Transfection of Cationic Liposome–DNA Complexes. J. Phys. Chem. B 2009, 113, 5208–5216. [Google Scholar] [CrossRef][Green Version]
- Bouxsein, N.F.; Leal, C.l.; McAllister, C.S.; Ewert, K.K.; Li, Y.; Samuel, C.E.; Safinya, C.R. Two-Dimensional Packing of Short DNA with Nonpairing Overhangs in Cationic Liposome–DNA Complexes: From Onsager Nematics to Columnar Nematics with Finite-Length Columns. J. Am. Chem. Soc. 2011, 133, 7585–7595. [Google Scholar] [CrossRef]
- Koynova, R.; Tenchov, B. Cationic Lipids: Molecular Structure/Transfection Activity Relationships and Interactions with Biomembranes. Top. Curr. Chem. 2010, 296, 51–93. [Google Scholar] [CrossRef] [PubMed]
- Tenchov, B.G.; Wang, L.; Koynova, R.; MacDonald, R.C. Modulation of a membrane lipid lamellar-nonlamellar phase transition by cationic lipids: A measure for transfection efficiency. Biochim. Biophys. Acta Biomembr. 2008, 1778, 2405–2412. [Google Scholar] [CrossRef]
- Koynova, R.; Tenchov, B. Cationic phospholipids: Structure–transfection activity relationships. Soft Matter 2009, 5, 3187–3200. [Google Scholar] [CrossRef]
- Ziller, A.; Nogueira, S.S.; Hühn, E.; Funari, S.S.; Brezesinski, G.; Hartmann, H.; Sahin, U.; Haas, H.; Langguth, P. Incorporation of mRNA in Lamellar Lipid Matrices for Parenteral Administration. Mol. Pharm. 2018, 15, 642–651. [Google Scholar] [CrossRef] [PubMed]
- Michanek, A.; Kristen, N.; Höök, F.; Nylander, T.; Sparr, E. RNA and DNA interactions with zwitterionic and charged lipid membranes—A DSC and QCM-D study. Biochim. Biophys. Acta Biomembr. 2010, 1798, 829–838. [Google Scholar] [CrossRef]
- Artzner, F.; Zantl, R.; Rapp, G.; Rädler, J.O. Observation of a Rectangular Columnar Phase in Condensed Lamellar Cationic Lipid-DNA Complexes. Phys. Rev. Lett. 1998, 81, 5015–5018. [Google Scholar] [CrossRef]
- Koynova, R.; MacDonald, R.C. Columnar DNA Superlattices in Lamellar o-Ethylphosphatidylcholine Lipoplexes: Mechanism of the Gel-Liquid Crystalline Lipid Phase Transition. Nano Lett. 2004, 4, 1475–1479. [Google Scholar] [CrossRef]
- McManus, J.J.; Rädler, J.O.; Dawson, K.A. Observation of a Rectangular Columnar Phase in a DNA−Calcium−Zwitterionic Lipid Complex. J. Am. Chem. Soc. 2004, 126, 15966–15967. [Google Scholar] [CrossRef]
- Bouxsein, N.F.; Leal, C.; McAllister, C.S.; Li, Y.; Ewert, K.K.; Samuel, C.E.; Safinya, C.R. 3D Columnar Phase of Stacked Short DNA Organized by Coherent Membrane Undulations. Langmuir 2019, 35, 11891–11901. [Google Scholar] [CrossRef] [PubMed]
- Golubović, L.; Golubović, M. Fluctuations of Quasi-Two-Dimensional Smectics Intercalated between Membranes in Multilamellar Phases of DNA-Cationic Lipid Complexes. Phys. Rev. Lett. 1998, 80, 4341–4344. [Google Scholar] [CrossRef]
- Golubović, L.; Lubensky, T.C.; O’Hern, C.S. Structural properties of the sliding columnar phase in layered liquid crystalline systems. Phys. Rev. E 2000, 62, 1069–1094. [Google Scholar] [CrossRef]
- O’Hern, C.S.; Lubensky, T.C. Sliding Columnar Phase of DNA-Lipid Complexes. Phys. Rev. Lett. 1998, 80, 4345–4348. [Google Scholar] [CrossRef]
- Kulkarni, C.V.; Wachter, W.; Iglesias-Salto, G.; Engelskirchen, S.; Ahualli, S. Monoolein: A magic lipid? Phys. Chem. Chem. Phys. 2011, 13, 3004–3021. [Google Scholar] [CrossRef]
- Kang, M.; Kim, H.; Leal, C. Self-organization of nucleic acids in lipid constructs. Curr. Opin. Colloid Interface Sci. 2016, 26, 58–65. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Negro, M.; Kumar, K.; Barrán-Berdón, A.L.; Datta, S.; Kondaiah, P.; Junquera, E.; Bhattacharya, S.; Aicart, E. Efficient Cellular Knockdown Mediated by siRNA Nanovectors of Gemini Cationic Lipids Having Delocalizable Headgroups and Oligo-Oxyethylene Spacers. ACS Appl. Mater. Interfaces 2016, 8, 22113–22126. [Google Scholar] [CrossRef]
- Kim, H.; Leal, C. Cuboplexes: Topologically Active siRNA Delivery. ACS Nano 2015, 9, 10214–10226. [Google Scholar] [CrossRef]
- Kim, H.; Sung, J.; Chang, Y.; Alfeche, A.; Leal, C. Microfluidics Synthesis of Gene Silencing Cubosomes. ACS Nano 2018, 12, 9196–9205. [Google Scholar] [CrossRef] [PubMed]
- Leal, C.; Ewert, K.K.; Bouxsein, N.F.; Shirazi, R.S.; Li, Y.; Safinya, C.R. Stacking of short DNA induces the gyroid cubic-to-inverted hexagonal phase transition in lipid-DNA complexes. Soft Matter 2013, 9, 795–804. [Google Scholar] [CrossRef] [PubMed]
- Farhood, H.; Serbina, N.; Huang, L. The role of dioleoyl phosphatidylethanolamine in cationic liposome mediated gene transfer. Biochim. Biophys. Acta Biomembr. 1995, 1235, 289–295. [Google Scholar] [CrossRef]
- Degors, I.M.S.; Wang, C.; Rehman, Z.U.; Zuhorn, I.S. Carriers Break Barriers in Drug Delivery: Endocytosis and Endosomal Escape of Gene Delivery Vectors. Acc. Chem. Res. 2019. [Google Scholar] [CrossRef] [PubMed]
- Martens, T.F.; Remaut, K.; Demeester, J.; De Smedt, S.C.; Braeckmans, K. Intracellular delivery of nanomaterials: How to catch endosomal escape in the act. Nano Today 2014, 9, 344–364. [Google Scholar] [CrossRef]
- Smith, S.A.; Selby, L.I.; Johnston, A.P.R.; Such, G.K. The Endosomal Escape of Nanoparticles: Toward More Efficient Cellular Delivery. Bioconjug. Chem. 2019, 30, 263–272. [Google Scholar] [CrossRef]
- Poste, G.; Bucana, C.; Raz, A.; Bugelski, P.; Kirsh, R.; Fidler, I.J. Analysis of the Fate of Systemically Administered Liposomes and Implications for Their Use in Drug Delivery. Cancer Res. 1982, 42, 1412–1422. [Google Scholar]
- Lasic, D.D. Liposomes: From Physics to Applications; Elsevier: San Diego, CA, USA, 1993. [Google Scholar]
- Yan, X.; Scherphof, G.L.; Kamps, J.A.A.M. Liposome Opsonization. J. Liposome Res. 2005, 15, 109–139. [Google Scholar] [CrossRef]
- Lasic, D.D.; Needham, D. The “Stealth” Liposome: A Prototypical Biomaterial. Chem. Rev. 1995, 95, 2601–2628. [Google Scholar] [CrossRef]
- Lasic, D.D.; Martin, F. (Eds.) Stealth Liposomes; CRC Press: Boca Raton, FL, USA, 1995. [Google Scholar]
- Woodle, M.C.; Newman, M.; Collins, L.; Redemann, C.; Martin, F. Improved long circulating (Stealth®) liposomes using synthetic lipids. Proc. Intern. Symp. Control. Rel. Bioact. Mater. 1990, 17, 77–78. [Google Scholar]
- Klibanov, A.L.; Maruyama, K.; Torchilin, V.P.; Huang, L. Amphipathic polyethyleneglycols effectively prolong the circulation time of liposomes. FEBS Lett. 1990, 268, 235–237. [Google Scholar] [CrossRef]
- Blume, G.; Cevc, G. Liposomes for the sustained drug release in vivo. Biochim. Biophys. Acta Biomembr. 1990, 1029, 91–97. [Google Scholar] [CrossRef]
- Papahadjopoulos, D.; Allen, T.M.; Gabizon, A.; Mayhew, E.; Matthay, K.; Huang, S.K.; Lee, K.D.; Woodle, M.C.; Lasic, D.D.; Redemann, C. Sterically stabilized liposomes: Improvements in pharmacokinetics and antitumor therapeutic efficacy. Proc. Natl. Acad. Sci. USA 1991, 88, 11460–11464. [Google Scholar] [CrossRef] [PubMed]
- Allen, T.M.; Hansen, C.; Martin, F.; Redemann, C.; Yau-Young, A. Liposomes containing synthetic lipid derivatives of poly(ethylene glycol) show prolonged circulation half-lives in vivo. Biochim. Biophys. Acta Biomembr. 1991, 1066, 29–36. [Google Scholar] [CrossRef]
- Woodle, M.C.; Lasic, D.D. Sterically stabilized liposomes. Biochim. Biophys. Acta 1992, 1113, 171–199. [Google Scholar] [CrossRef]
- Silvander, M. Steric stabilization of liposomes—A review. Prog. Colloid Polym. Sci. 2002, 120, 35–40. [Google Scholar] [CrossRef]
- Gabizon, A.; Papahadjopoulos, D. Liposome formulations with prolonged circulation time in blood and enhanced uptake by tumors. Proc. Natl. Acad. Sci. USA 1988, 85, 6949–6953. [Google Scholar] [CrossRef] [PubMed]
- Allen, T.M.; Hansen, C.; Rutledge, J. Liposomes with prolonged circulation times: Factors affecting uptake by reticuloendothelial and other tissues. Biochim. Biophys. Acta Biomembr. 1989, 981, 27–35. [Google Scholar] [CrossRef]
- De Gennes, P.-G. Scaling Concepts in Polymer Physics; Cornell University Press: New York, NY, USA, 1979. [Google Scholar]
- Kuhl, T.L.; Leckband, D.E.; Lasic, D.D.; Israelachvili, J.N. Modulation of interaction forces between bilayers exposing short-chained ethylene oxide headgroups. Biophys. J. 1994, 66, 1479–1488. [Google Scholar] [CrossRef]
- Kenworthy, A.K.; Hristova, K.; Needham, D.; McIntosh, T.J. Range and magnitude of the steric pressure between bilayers containing phospholipids with covalently attached poly(ethylene glycol). Biophys. J. 1995, 68, 1921–1936. [Google Scholar] [CrossRef]
- Hong, K.; Zheng, W.; Baker, A.; Papahadjopoulos, D. Stabilization of cationic liposome-plasmid DNA complexes by polyamines and poly(ethylene glycol)-phospholipid conjugates for efficient in vivo gene delivery. FEBS Lett. 1997, 400, 233–237. [Google Scholar] [CrossRef]
- Barenholz, Y. Doxil®—The first FDA-approved nano-drug: Lessons learned. J. Control. Release 2012, 160, 117–134. [Google Scholar] [CrossRef]
- Silva, B.F.B.; Majzoub, R.N.; Chan, C.-L.; Li, Y.; Olsson, U.; Safinya, C.R. PEGylated cationic liposome–DNA complexation in brine is pathway-dependent. Biochim. Biophys. Acta Biomembr. 2014, 1838, 398–412. [Google Scholar] [CrossRef] [PubMed]
- Chan, C.-L.; Majzoub, R.N.; Shirazi, R.S.; Ewert, K.K.; Chen, Y.-J.; Liang, K.S.; Safinya, C.R. Endosomal escape and transfection efficiency of PEGylated cationic liposome–DNA complexes prepared with an acid-labile PEG-lipid. Biomaterials 2012, 33, 4928–4935. [Google Scholar] [CrossRef] [PubMed]
- Majzoub, R.N.; Chan, C.-L.; Ewert, K.K.; Silva, B.F.B.; Liang, K.S.; Jacovetty, E.L.; Carragher, B.; Potter, C.S.; Safinya, C.R. Uptake and transfection efficiency of PEGylated cationic liposome–DNA complexes with and without RGD-tagging. Biomaterials 2014, 35, 4996–5005. [Google Scholar] [CrossRef]
- Belliveau, N.M.; Huft, J.; Lin, P.J.C.; Chen, S.; Leung, A.K.K.; Leaver, T.J.; Wild, A.W.; Lee, J.B.; Taylor, R.J.; Tam, Y.K.; et al. Microfluidic Synthesis of Highly Potent Limit-size Lipid Nanoparticles for In Vivo Delivery of siRNA. Mol. Ther. Nucleic Acids 2012, 1, e37. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, J.A.; Myhre, J.L.; Chen, S.; Tam, Y.Y.C.; Danescu, A.; Richman, J.M.; Cullis, P.R. Design of lipid nanoparticles for in vitro and in vivo delivery of plasmid DNA. Nanomedicine 2017, 13, 1377–1387. [Google Scholar] [CrossRef]
- Kulkarni, J.A.; Darjuan, M.M.; Mercer, J.E.; Chen, S.; van der Meel, R.; Thewalt, J.L.; Tam, Y.Y.C.; Cullis, P.R. On the Formation and Morphology of Lipid Nanoparticles Containing Ionizable Cationic Lipids and siRNA. ACS Nano 2018, 12, 4787–4795. [Google Scholar] [CrossRef] [PubMed]
- Martin-Herranz, A.; Ahmad, A.; Evans, H.M.; Ewert, K.; Schulze, U.; Safinya, C.R. Surface Functionalized Cationic Lipid-DNA Complexes for Gene Delivery: PEGylated Lamellar Complexes Exhibit Distinct DNA-DNA Interaction Regimes. Biophys. J. 2004, 86, 1160–1168. [Google Scholar] [CrossRef][Green Version]
- Hafez, I.M.; Maurer, N.; Cullis, P.R. On the mechanism whereby cationic lipids promote intracellular delivery of polynucleic acids. Gene Ther. 2001, 8, 1188–1196. [Google Scholar] [CrossRef]
- Leung, A.K.K.; Hafez, I.M.; Baoukina, S.; Belliveau, N.M.; Zhigaltsev, I.V.; Afshinmanesh, E.; Tieleman, D.P.; Hansen, C.L.; Hope, M.J.; Cullis, P.R. Lipid Nanoparticles Containing siRNA Synthesized by Microfluidic Mixing Exhibit an Electron-Dense Nanostructured Core. J. Phys. Chem. C 2012, 116, 18440–18450. [Google Scholar] [CrossRef]
- Needham, D.; McIntosh, T.J.; Lasic, D.D. Repulsive interactions and mechanical stability of polymer-grafted lipid membranes. Biochim. Biophys. Acta Biomembr. 1992, 1108, 40–48. [Google Scholar] [CrossRef]
- Kong, L.; Campbell, F.; Kros, A. DePEGylation strategies to increase cancer nanomedicine efficacy. Nanoscale Horiz. 2019, 4, 378–387. [Google Scholar] [CrossRef]
- Romberg, B.; Hennink, W.E.; Storm, G. Sheddable Coatings for Long-Circulating Nanoparticles. Pharm. Res. 2008, 25, 55–71. [Google Scholar] [CrossRef] [PubMed]
- Mui, B.L.; Tam, Y.K.; Jayaraman, M.; Ansell, S.M.; Du, X.; Tam, Y.Y.C.; Lin, P.J.C.; Chen, S.; Narayanannair, J.K.; Rajeev, K.G.; et al. Influence of Polyethylene Glycol Lipid Desorption Rates on Pharmacokinetics and Pharmacodynamics of siRNA Lipid Nanoparticles. Mol. Ther. Nucleic Acids 2013, 2, e139. [Google Scholar] [CrossRef]
- Walker, G.F.; Fella, C.; Pelisek, J.; Fahrmeir, J.; Boeckle, S.; Ogris, M.; Wagner, E. Toward synthetic viruses: Endosomal pH-triggered deshielding of targeted polyplexes greatly enhances gene transfer in vitro and in vivo. Mol. Ther. 2005, 11, 418–425. [Google Scholar] [CrossRef] [PubMed]
- Pierschbacher, M.D.; Hayman, E.G.; Ruoslahti, E. Location of the cell-attachment site in fibronectin with monoclonal antibodies and proteolytic fragments of the molecule. Cell 1981, 26, 259–267. [Google Scholar] [CrossRef]
- Pierschbacher, M.D.; Ruoslahti, E. Cell attachment activity of fibronectin can be duplicated by small synthetic fragments of the molecule. Nature 1984, 309, 30–33. [Google Scholar] [CrossRef] [PubMed]
- Pierschbacher, M.D.; Ruoslahti, E. Variants of the cell recognition site of fibronectin that retain attachment-promoting activity. Proc. Natl. Acad. Sci. USA 1984, 81, 5985–5988. [Google Scholar] [CrossRef]
- Pytela, R.; Pierschbacher, M.D.; Ruoslahti, E. Identification and isolation of a 140 kd cell surface glycoprotein with properties expected of a fibronectin receptor. Cell 1985, 40, 191–198. [Google Scholar] [CrossRef]
- Ewert, K.K.; Kotamraju, V.R.; Majzoub, R.N.; Steffes, V.M.; Wonder, E.A.; Teesalu, T.; Ruoslahti, E.; Safinya, C.R. Synthesis of linear and cyclic peptide–PEG–lipids for stabilization and targeting of cationic liposome–DNA complexes. Bioorg. Med. Chem. Lett. 2016, 26, 1618–1623. [Google Scholar] [CrossRef]
- Ruoslahti, E.; Pierschbacher, M.D. Arg-Gly-Asp: A versatile cell recognition signal. Cell 1986, 44, 517–518. [Google Scholar] [CrossRef]
- Ruoslahti, E.; Bhatia, S.N.; Sailor, M.J. Targeting of drugs and nanoparticles to tumors. J. Cell Biol. 2010, 188, 759–768. [Google Scholar] [CrossRef]
- Clackson, T.; Lowmand, H.B. (Eds.) Phage Display: A Practical Approach; Oxford University Press: Oxford, UK, 2004. [Google Scholar]
- Teesalu, T.; Sugahara, K.N.; Ruoslahti, E. Mapping of Vascular ZIP Codes by Phage Display. Meth. Enzymol. 2012, 503, 35–56. [Google Scholar] [CrossRef]
- Ruoslahti, E. Specialization of Tumour Vasculature. Nat. Rev. Cancer 2002, 2, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Ruoslahti, E. Vascular zip codes in angiogenesis and metastasis. Biochem. Soc. Trans. 2004, 32, 397–402. [Google Scholar] [CrossRef] [PubMed]
- Laakkonen, P.; Åkerman, M.E.; Biliran, H.; Yang, M.; Ferrer, F.; Karpanen, T.; Hoffman, R.M.; Ruoslahti, E. Antitumor activity of a homing peptide that targets tumor lymphatics and tumor cells. Proc. Natl. Acad. Sci. USA 2004, 101, 9381–9386. [Google Scholar] [CrossRef]
- Ruoslahti, E.; Duza, T.; Zhang, L. Vascular Homing Peptides with Cell-Penetrating Properties. Curr. Pharm. Des. 2005, 11, 3655–3660. [Google Scholar] [CrossRef]
- Arap, W.; Pasqualini, R.; Ruoslahti, E. Cancer Treatment by Targeted Drug Delivery to Tumor Vasculature in a Mouse Model. Science 1998, 279, 377–380. [Google Scholar] [CrossRef]
- Laakkonen, P.; Porkka, K.; Hoffman, J.A.; Ruoslahti, E. A tumor-homing peptide with a targeting specificity related to lymphatic vessels. Nat. Med. 2002, 8, 751–755. [Google Scholar] [CrossRef]
- Park, J.-H.; von Maltzahn, G.; Xu, M.J.; Fogal, V.; Kotamraju, V.R.; Ruoslahti, E.; Bhatia, S.N.; Sailor, M.J. Cooperative nanomaterial system to sensitize, target, and treat tumors. Proc. Natl. Acad. Sci. USA 2010, 107, 981–986. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Giraudo, E.; Hoffman, J.A.; Hanahan, D.; Ruoslahti, E. Lymphatic Zip Codes in Premalignant Lesions and Tumors. Cancer Res. 2006, 66, 5696–5706. [Google Scholar] [CrossRef]
- Lee, S.-M.; Lee, E.-J.; Hong, H.-Y.; Kwon, M.-K.; Kwon, T.-H.; Choi, J.-Y.; Park, R.-W.; Kwon, T.-G.; Yoo, E.-S.; Yoon, G.-S.; et al. Targeting Bladder Tumor Cells In vivo and in the Urine with a Peptide Identified by Phage Display. Mol. Cancer Res. 2007, 5, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Simberg, D.; Duza, T.; Park, J.H.; Essler, M.; Pilch, J.; Zhang, L.; Derfus, A.M.; Yang, M.; Hoffman, R.M.; Bhatia, S.; et al. Biomimetic amplification of nanoparticle homing to tumors. Proc. Natl. Acad. Sci. USA 2007, 104, 932–936. [Google Scholar] [CrossRef]
- Pleiko, K.; Põšnograjeva, K.; Haugas, M.; Paiste, P.; Tobi, A.; Kurm, K.; Riekstina, U.; Teesalu, T. In vivo phage display: Identification of organ-specific peptides using deep sequencing and differential profiling across tissues. Nucleic Acids Res. 2021, 49, e38. [Google Scholar] [CrossRef] [PubMed]
- Põsnograjeva, K.; Pleiko, K.; Haugas, M.; Teesalu, T. New tools for streamlined in vivo homing peptide identification. In Cell-Penetrating Peptides: Methods and Protocols, 3rd ed.; Langel, Ü., Ed.; Springer: New York, NY, USA, 2021. [Google Scholar]
- Scodeller, P.; Asciutto, E.K. Targeting Tumors Using Peptides. Molecules 2020, 25, 808. [Google Scholar] [CrossRef] [PubMed]
- Kessler, H.; Diefenbach, B.; Finsinger, D.; Geyer, A.; Gurrath, M.; Goodman, S.L.; Hölzemann, G.; Haubner, R.; Jonczyk, A.; Müller, G.; et al. Design of superactive and selective integrin receptor antagonists containing the RGD sequence. Lett. Pept. Sci. 1995, 2, 155–160. [Google Scholar] [CrossRef]
- Haubner, R.; Gratias, R.; Diefenbach, B.; Goodman, S.L.; Jonczyk, A.; Kessler, H. Structural and Functional Aspects of RGD-Containing Cyclic Pentapeptides as Highly Potent and Selective Integrin αVβ3 Antagonists. J. Am. Chem. Soc. 1996, 118, 7461–7472. [Google Scholar] [CrossRef]
- Mas-Moruno, C.; Rechenmacher, F.; Kessler, H. Cilengitide: The First Anti-Angiogenic Small Molecule Drug Candidate. Design, Synthesis and Clinical Evaluation. Anti-Cancer Agents Med. Chem. 2010, 10, 753–768. [Google Scholar] [CrossRef]
- Dechantsreiter, M.A.; Planker, E.; Mathä, B.; Lohof, E.; Hölzemann, G.; Jonczyk, A.; Goodman, S.L.; Kessler, H. N-Methylated Cyclic RGD Peptides as Highly Active and Selective αVβ3 Integrin Antagonists. J. Med. Chem. 1999, 42, 3033–3040. [Google Scholar] [CrossRef]
- Temming, K.; Schiffelers, R.M.; Molema, G.; Kok, R.J. RGD-based strategies for selective delivery of therapeutics and imaging agents to the tumour vasculature. Drug Resist. Updates 2005, 8, 381–402. [Google Scholar] [CrossRef]
- Zanuy, D.; Sayago, F.J.; Revilla-López, G.; Ballano, G.; Agemy, L.; Kotamraju, V.R.; Jiménez, A.I.; Cativiela, C.; Nussinov, R.; Sawvel, A.M.; et al. Engineering strategy to improve peptide analogs: From structure-based computational design to tumor homing. J. Comput. Aided Mol. Des. 2013, 27, 31–43. [Google Scholar] [CrossRef]
- Ruoslahti, E. Tumor penetrating peptides for improved drug delivery. Adv. Drug Deliv. Rev. 2017, 110, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Teesalu, T.; Sugahara, K.N.; Kotamraju, V.R.; Ruoslahti, E. C-end rule peptides mediate neuropilin-1-dependent cell, vascular, and tissue penetration. Proc. Natl. Acad. Sci. USA 2009, 106, 16157–16162. [Google Scholar] [CrossRef] [PubMed]
- Sugahara, K.N.; Teesalu, T.; Karmali, P.P.; Kotamraju, V.R.; Agemy, L.; Girard, O.M.; Hanahan, D.; Mattrey, R.F.; Ruoslahti, E. Tissue-Penetrating Delivery of Compounds and Nanoparticles into Tumors. Cancer Cell 2009, 16, 510–520. [Google Scholar] [CrossRef] [PubMed]
- Sugahara, K.N.; Teesalu, T.; Karmali, P.P.; Kotamraju, V.R.; Agemy, L.; Greenwald, D.R.; Ruoslahti, E. Coadministration of a Tumor-Penetrating Peptide Enhances the Efficacy of Cancer Drugs. Science 2010, 328, 1031–1035. [Google Scholar] [CrossRef]
- Teesalu, T.; Sugahara, K.N.; Ruoslahti, E. Tumor penetrating peptides. Front. Oncol. 2013, 3. [Google Scholar] [CrossRef] [PubMed]
- Simón-Gracia, L.; Hunt, H.; Scodeller, P.; Gaitzsch, J.; Kotamraju, V.R.; Sugahara, K.N.; Tammik, O.; Ruoslahti, E.; Battaglia, G.; Teesalu, T. iRGD peptide conjugation potentiates intraperitoneal tumor delivery of paclitaxel with polymersomes. Biomaterials 2016, 104, 247–257. [Google Scholar] [CrossRef]
- Sharma, S.; Kotamraju, V.R.; Mölder, T.; Tobi, A.; Teesalu, T.; Ruoslahti, E. Tumor-Penetrating Nanosystem Strongly Suppresses Breast Tumor Growth. Nano Lett. 2017, 17, 1356–1364. [Google Scholar] [CrossRef]
- Hunt, H.; Simón-Gracia, L.; Tobi, A.; Kotamraju, V.R.; Sharma, S.; Nigul, M.; Sugahara, K.N.; Ruoslahti, E.; Teesalu, T. Targeting of p32 in peritoneal carcinomatosis with intraperitoneal linTT1 peptide-guided pro-apoptotic nanoparticles. J. Control. Release 2017, 260, 142–153. [Google Scholar] [CrossRef]
- Paasonen, L.; Sharma, S.; Braun, G.B.; Kotamraju, V.R.; Chung, T.D.Y.; She, Z.-G.; Sugahara, K.N.; Yliperttula, M.; Wu, B.; Pellecchia, M.; et al. New p32/gC1qR Ligands for Targeted Tumor Drug Delivery. ChemBioChem 2016, 17, 570–575. [Google Scholar] [CrossRef]
- Wannasarit, S.; Wang, S.; Figueiredo, P.; Trujillo, C.; Eburnea, F.; Simón-Gracia, L.; Correia, A.; Ding, Y.; Teesalu, T.; Liu, D.; et al. A Virus-Mimicking pH-Responsive Acetalated Dextran-Based Membrane-Active Polymeric Nanoparticle for Intracellular Delivery of Antitumor Therapeutics. Adv. Funct. Mater. 2019, 29, 1905352. [Google Scholar] [CrossRef]
- Säälik, P.; Lingasamy, P.; Toome, K.; Mastandrea, I.; Rousso-Noori, L.; Tobi, A.; Simón-Gracia, L.; Hunt, H.; Paiste, P.; Kotamraju, V.R.; et al. Peptide-guided nanoparticles for glioblastoma targeting. J. Control. Release 2019, 308, 109–118. [Google Scholar] [CrossRef]
- Simon-Gracia, L.; Savier, E.; Parizot, C.; Brossas, J.Y.; Loisel, S.; Teesalu, T.; Conti, F.; Charlotte, F.; Scatton, O.; Aoudjehane, L.; et al. Bifunctional Therapeutic Peptides for Targeting Malignant B Cells and Hepatocytes: Proof of Concept in Chronic Lymphocytic Leukemia. Adv. Ther. 2020, 3, 2000131. [Google Scholar] [CrossRef]
- Simón-Gracia, L.; Sidorenko, V.; Uustare, A.; Ogibalov, I.; Tasa, A.; Tshubrik, O.; Teesalu, T. Novel Anthracycline Utorubicin for Cancer Therapy. Angew. Chem. Int. Ed. Engl. 2021. [Google Scholar] [CrossRef]
- Simón-Gracia, L.; Scodeller, P.; Fuentes, S.S.; Gómez, V.; Vallejo, X.R.; San Sebastián, E.; Sidorenko, V.; Di Silvio, D.; Suck, M.; De Lorenzi, F.; et al. Application of polymersomes engineered to target p32 protein for detection of small breast tumors in mice. Oncotarget 2018, 9, 18682–18697. [Google Scholar] [CrossRef] [PubMed]
- Scodeller, P.; Simón-Gracia, L.; Kopanchuk, S.; Tobi, A.; Kilk, K.; Säälik, P.; Kurm, K.; Squadrito, M.L.; Kotamraju, V.R.; Rinken, A.; et al. Precision Targeting of Tumor Macrophages with a CD206 Binding Peptide. Sci. Rep. 2017, 7, 14655. [Google Scholar] [CrossRef] [PubMed]
- Lepland, A.; Asciutto, E.K.; Malfanti, A.; Simón-Gracia, L.; Sidorenko, V.; Vicent, M.J.; Teesalu, T.; Scodeller, P. Targeting Pro-Tumoral Macrophages in Early Primary and Metastatic Breast Tumors with the CD206-Binding mUNO Peptide. Mol. Pharm. 2020, 17, 2518–2531. [Google Scholar] [CrossRef]
- Figueiredo, P.; Lepland, A.; Scodeller, P.; Fontana, F.; Torrieri, G.; Tiboni, M.; Shahbazi, M.A.; Casettari, L.; Kostiainen, M.A.; Hirvonen, J.; et al. Peptide-guided resiquimod-loaded lignin nanoparticles convert tumor-associated macrophages from M2 to M1 phenotype for enhanced chemotherapy. Acta Biomater. 2020. [Google Scholar] [CrossRef] [PubMed]
- Keler, T.; Ramakrishna, V.; Fanger, M.W. Mannose receptor-targeted vaccines. Expert Opin. Biol. Ther. 2004, 4, 1953–1962. [Google Scholar] [CrossRef]
- Conniot, J.; Scomparin, A.; Peres, C.; Yeini, E.; Pozzi, S.; Matos, A.I.; Kleiner, R.; Moura, L.I.F.; Zupančič, E.; Viana, A.S.; et al. Immunization with mannosylated nanovaccines and inhibition of the immune-suppressing microenvironment sensitizes melanoma to immune checkpoint modulators. Nat. Nanotechnol. 2019, 14, 891–901. [Google Scholar] [CrossRef]
- Silva, J.M.; Zupancic, E.; Vandermeulen, G.; Oliveira, V.G.; Salgado, A.; Videira, M.; Gaspar, M.; Graca, L.; Préat, V.; Florindo, H.F. In vivo delivery of peptides and Toll-like receptor ligands by mannose-functionalized polymeric nanoparticles induces prophylactic and therapeutic anti-tumor immune responses in a melanoma model. J. Control. Release 2015, 198, 91–103. [Google Scholar] [CrossRef]
- Lingasamy, P.; Tobi, A.; Haugas, M.; Hunt, H.; Paiste, P.; Asser, T.; Rätsep, T.; Kotamraju, V.R.; Bjerkvig, R.; Teesalu, T. Bi-specific tenascin-C and fibronectin targeted peptide for solid tumor delivery. Biomaterials 2019, 219, 119373. [Google Scholar] [CrossRef]
- Lingasamy, P.; Tobi, A.; Kurm, K.; Kopanchuk, S.; Sudakov, A.; Salumäe, M.; Rätsep, T.; Asser, T.; Bjerkvig, R.; Teesalu, T. Tumor-penetrating peptide for systemic targeting of Tenascin-C. Sci. Rep. 2020, 10, 5809. [Google Scholar] [CrossRef]
- Wonder, E.; Simón-Gracia, L.; Scodeller, P.; Majzoub, R.N.; Kotamraju, V.R.; Ewert, K.K.; Teesalu, T.; Safinya, C.R. Competition of charge-mediated and specific binding by peptide-tagged cationic liposome–DNA nanoparticles in vitro and in vivo. Biomaterials 2018, 166, 52–63. [Google Scholar] [CrossRef] [PubMed]
- Campbell, R.B.; Ying, B.; Kuesters, G.M.; Hemphill, R. Fighting cancer: From the bench to bedside using second generation cationic liposomal therapeutics. J. Pharm. Sci. 2009, 98, 411–429. [Google Scholar] [CrossRef]
- Strieth, S.; Eichhorn, M.E.; Sauer, B.; Schulze, B.; Teifel, M.; Michaelis, U.; Dellian, M. Neovascular targeting chemotherapy: Encapsulation of paclitaxel in cationic liposomes impairs functional tumor microvasculature. Int. J. Cancer 2004, 110, 117–124. [Google Scholar] [CrossRef]
- Strieth, S.; Nussbaum, C.F.; Eichhorn, M.E.; Fuhrmann, M.; Teifel, M.; Michaelis, U.; Berghaus, A.; Dellian, M. Tumor-selective vessel occlusions by platelets after vascular targeting chemotherapy using paclitaxel encapsulated in cationic liposomes. Int. J. Cancer 2008, 122, 452–460. [Google Scholar] [CrossRef] [PubMed]
- Schmitt-Sody, M.; Strieth, S.; Krasnici, S.; Sauer, B.; Schulze, B.; Teifel, M.; Michaelis, U.; Naujoks, K.; Dellian, M. Neovascular Targeting Therapy: Paclitaxel Encapsulated in Cationic Liposomes Improves Antitumoral Efficacy. Clin. Cancer Res. 2003, 9, 2335–2341. [Google Scholar] [PubMed]
- Kunstfeld, R.; Wickenhauser, G.; Michaelis, U.; Teifel, M.; Umek, W.; Naujoks, K.; Wolff, K.; Petzelbauer, P. Paclitaxel Encapsulated in Cationic Liposomes Diminishes Tumor Angiogenesis and Melanoma Growth in a “Humanized” SCID Mouse Model. J. Investig. Dermatol. 2003, 120, 476–482. [Google Scholar] [CrossRef] [PubMed]
- Fasol, U.; Frost, A.; Büchert, M.; Arends, J.; Fiedler, U.; Scharr, D.; Scheuenpflug, J.; Mross, K. Vascular and pharmacokinetic effects of EndoTAG-1 in patients with advanced cancer and liver metastasis. Ann. Oncol. 2012, 23, 1030–1036. [Google Scholar] [CrossRef] [PubMed]
- Koudelka, Š.; Turánek, J. Liposomal paclitaxel formulations. J. Control. Release 2012, 163, 322–334. [Google Scholar] [CrossRef]
- Wani, M.C.; Taylor, H.L.; Wall, M.E.; Coggon, P.; McPhail, A.T. Plant antitumor agents. VI. Isolation and structure of taxol, a novel antileukemic and antitumor agent from Taxus brevifolia. J. Am. Chem. Soc. 1971, 93, 2325–2327. [Google Scholar] [CrossRef]
- Jordan, M.A.; Wilson, L. Microtubules as a target for anticancer drugs. Nat. Rev. Cancer 2004, 4, 253–265. [Google Scholar] [CrossRef]
- Weaver, B.A. How Taxol/paclitaxel kills cancer cells. Mol. Biol. Cell 2014, 25, 2677–2681. [Google Scholar] [CrossRef]
- Rowinsky, E.K.; Donehower, R.C. Paclitaxel (Taxol). N. Engl. J. Med. 1995, 332, 1004–1014. [Google Scholar] [CrossRef] [PubMed]
- Markman, M.; Mekhail, T.M. Paclitaxel in cancer therapy. Expert Opin. Pharmacother. 2002, 3, 755–766. [Google Scholar] [CrossRef] [PubMed]
- Ramalingam, S.; Belani, C.P. Paclitaxel for non-small cell lung cancer. Expert Opin. Pharmacother. 2004, 5, 1771–1780. [Google Scholar] [CrossRef]
- Hironaka, S.; Zenda, S.; Boku, N.; Fukutomi, A.; Yoshino, T.; Onozawa, Y. Weekly paclitaxel as second-line chemotherapy for advanced or recurrent gastric cancer. Gastric Cancer 2006, 9, 14–18. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, J.; Matsui, T.; Kodera, Y. Paclitaxel chemotherapy for the treatment of gastric cancer. Gastric Cancer 2009, 12, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Moxley, K.M.; McMeekin, D.S. Endometrial Carcinoma: A Review of Chemotherapy, Drug Resistance, and the Search for New Agents. Oncologist 2010, 15, 1026–1033. [Google Scholar] [CrossRef]
- Teo, P.Y.; Cheng, W.; Hedrick, J.L.; Yang, Y.Y. Co-delivery of drugs and plasmid DNA for cancer therapy. Adv. Drug Deliv. Rev. 2016, 98, 41–63. [Google Scholar] [CrossRef] [PubMed]
- Dorr, R.T. Pharmacology and Toxicology of Cremophor EL Diluent. Ann. Pharmacother. 1994, 28, S11–S14. [Google Scholar] [CrossRef]
- Weiss, R.B.; Donehower, R.C.; Wiernik, P.H.; Ohnuma, T.; Gralla, R.J.; Trump, D.L.; Jr, J.R.B.; Echo, D.A.V.; Hoff, D.D.V.; Leyland-Jones, B. Hypersensitivity reactions from taxol. J. Clin. Oncol. 1990, 8, 1263–1268. [Google Scholar] [CrossRef]
- Gelderblom, H.; Verweij, J.; Nooter, K.; Sparreboom, A. Cremophor EL: The drawbacks and advantages of vehicle selection for drug formulation. Eur. J. Cancer 2001, 37, 1590–1598. [Google Scholar] [CrossRef]
- Sofias, A.M.; Dunne, M.; Storm, G.; Allen, C. The battle of “nano” paclitaxel. Adv. Drug Deliv. Rev. 2017, 122, 20–30. [Google Scholar] [CrossRef]
- Hong, S.-S.; Choi, J.Y.; Kim, J.O.; Lee, M.-K.; Kim, S.H.; Lim, S.-J. Development of paclitaxel-loaded liposomal nanocarrier stabilized by triglyceride incorporation. Int. J. Nanomed. 2016, 11, 4465–4477. [Google Scholar] [CrossRef]
- Zhou, R.; Mazurchuk, R.V.; Tamburlin, J.H.; Harrold, J.M.; Mager, D.E.; Straubinger, R.M. Differential Pharmacodynamic Effects of Paclitaxel Formulations in an Intracranial Rat Brain Tumor Model. J. Pharmacol. Exp. Ther. 2010, 332, 479–488. [Google Scholar] [CrossRef]
- Ait-Oudhia, S.; Mager, D.E.; Straubinger, R.M. Application of Pharmacokinetic and Pharmacodynamic Analysis to the Development of Liposomal Formulations for Oncology. Pharmaceutics 2014, 6, 137–174. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.A.; Anyarambhatla, G.; Ma, L.; Ugwu, S.; Xuan, T.; Sardone, T.; Ahmad, I. Development and characterization of a novel Cremophor® EL free liposome-based paclitaxel (LEP-ETU) formulation. Eur. J. Pharm. Biopharm. 2005, 59, 177–187. [Google Scholar] [CrossRef] [PubMed]
- Dellian, M.; Yuan, F.; Trubetskoy, V.S.; Torchilin, V.P.; Jain, R.K. Vascular permeability in a human tumour xenograft: Molecular charge dependence. Br. J. Cancer 2000, 82, 1513–1518. [Google Scholar] [CrossRef] [PubMed]
- Eichhorn, M.E.; Ischenko, I.; Luedemann, S.; Strieth, S.; Papyan, A.; Werner, A.; Bohnenkamp, H.; Guenzi, E.; Preissler, G.; Michaelis, U.; et al. Vascular targeting by EndoTAG™-1 enhances therapeutic efficacy of conventional chemotherapy in lung and pancreatic cancer. Int. J. Cancer 2010, 126, 1235–1245. [Google Scholar] [CrossRef] [PubMed]
- Koudelka, Š.; Turánek-Knötigová, P.; MaŠek, J.; Korvasová, Z.; Škrabalová, M.; Plocková, J.; Bartheldyová, E.; Turánek, J. Liposomes with high encapsulation capacity for paclitaxel: Preparation, characterisation and in vivo anticancer effect. J. Pharm. Sci. 2010, 99, 2309–2319. [Google Scholar] [CrossRef] [PubMed]


















Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ewert, K.K.; Scodeller, P.; Simón-Gracia, L.; Steffes, V.M.; Wonder, E.A.; Teesalu, T.; Safinya, C.R. Cationic Liposomes as Vectors for Nucleic Acid and Hydrophobic Drug Therapeutics. Pharmaceutics 2021, 13, 1365. https://doi.org/10.3390/pharmaceutics13091365
Ewert KK, Scodeller P, Simón-Gracia L, Steffes VM, Wonder EA, Teesalu T, Safinya CR. Cationic Liposomes as Vectors for Nucleic Acid and Hydrophobic Drug Therapeutics. Pharmaceutics. 2021; 13(9):1365. https://doi.org/10.3390/pharmaceutics13091365
Chicago/Turabian StyleEwert, Kai K., Pablo Scodeller, Lorena Simón-Gracia, Victoria M. Steffes, Emily A. Wonder, Tambet Teesalu, and Cyrus R. Safinya. 2021. "Cationic Liposomes as Vectors for Nucleic Acid and Hydrophobic Drug Therapeutics" Pharmaceutics 13, no. 9: 1365. https://doi.org/10.3390/pharmaceutics13091365
APA StyleEwert, K. K., Scodeller, P., Simón-Gracia, L., Steffes, V. M., Wonder, E. A., Teesalu, T., & Safinya, C. R. (2021). Cationic Liposomes as Vectors for Nucleic Acid and Hydrophobic Drug Therapeutics. Pharmaceutics, 13(9), 1365. https://doi.org/10.3390/pharmaceutics13091365