
Formulation Considerations for Autologous T Cell Drug Products


Abstract
:1. Introduction
2. The T Cell Dose
3. Towards Automated Formulation
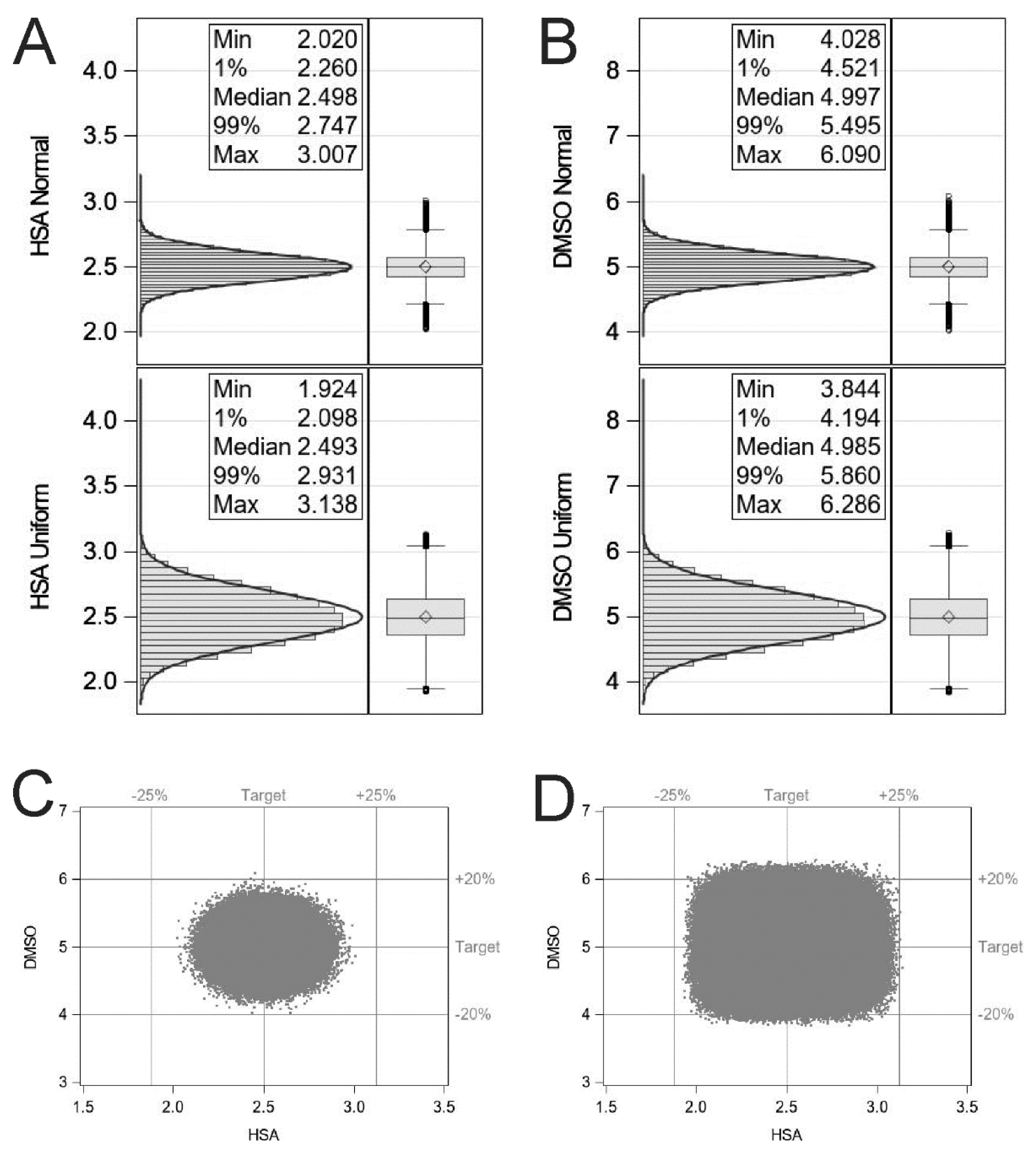
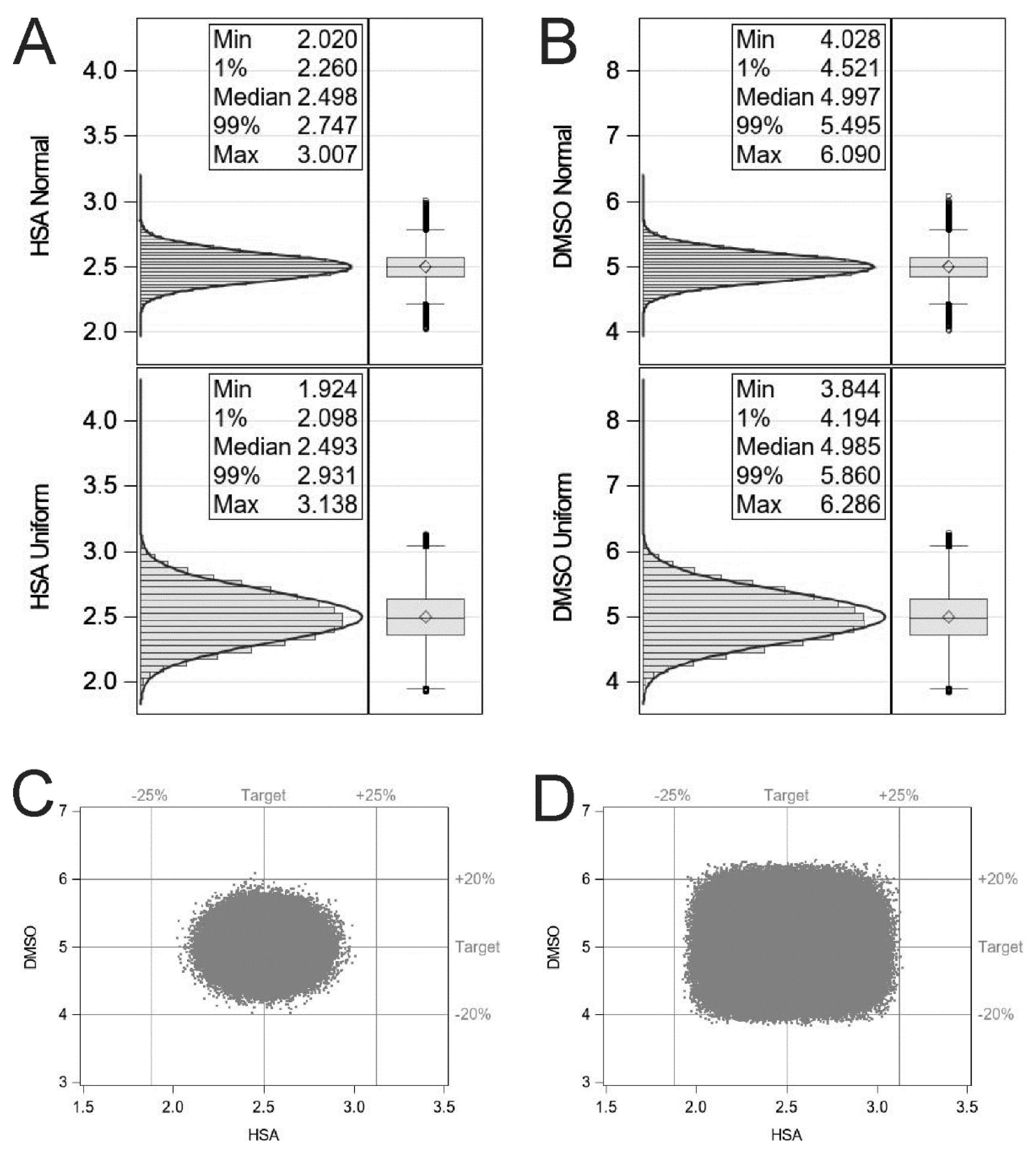
4. Fill–Finish and Monte Carlo Simulation
5. Primary Container Selection
Failure of the Primary Container
6. Particulates, Extractables and Leachables
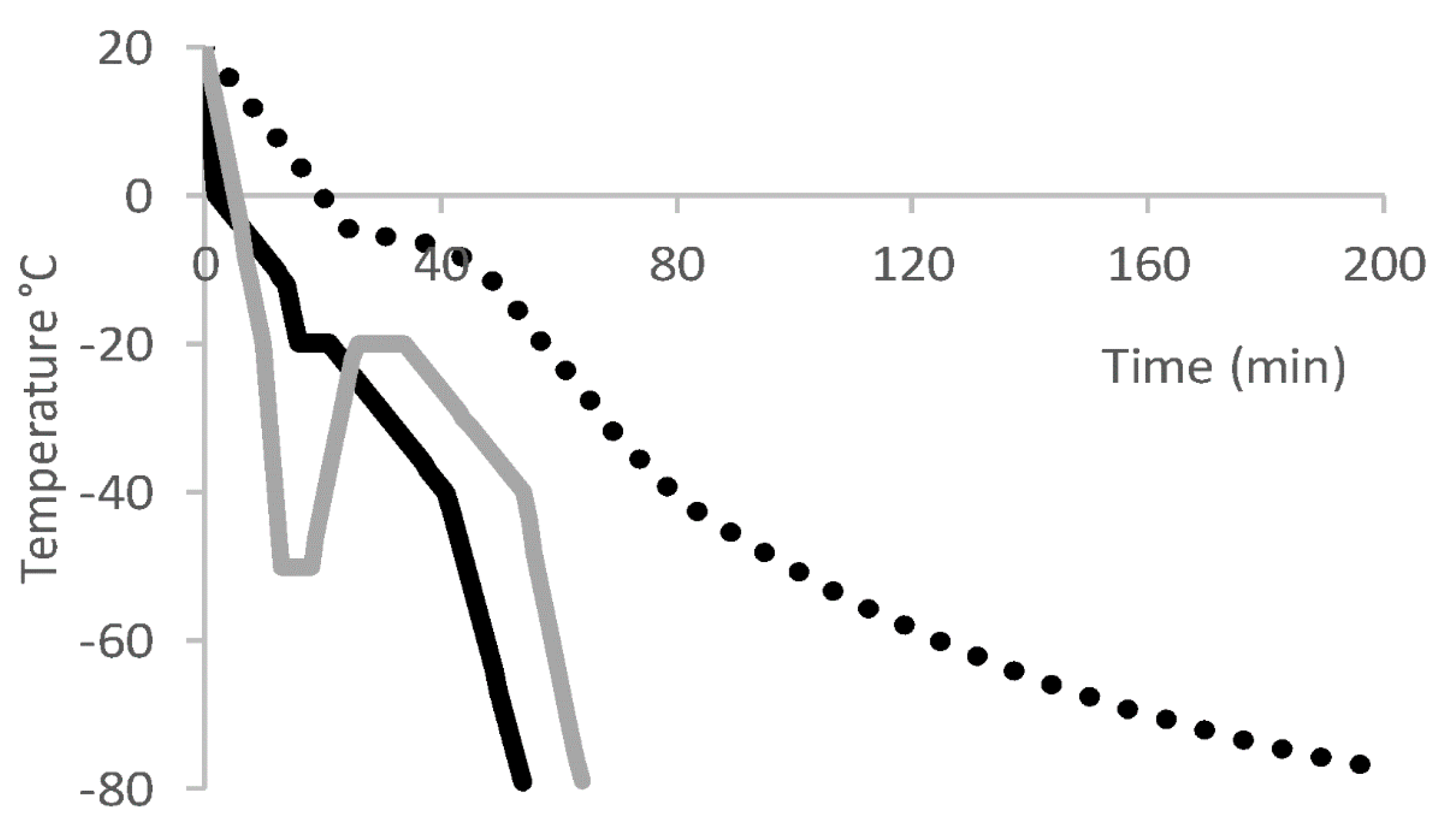
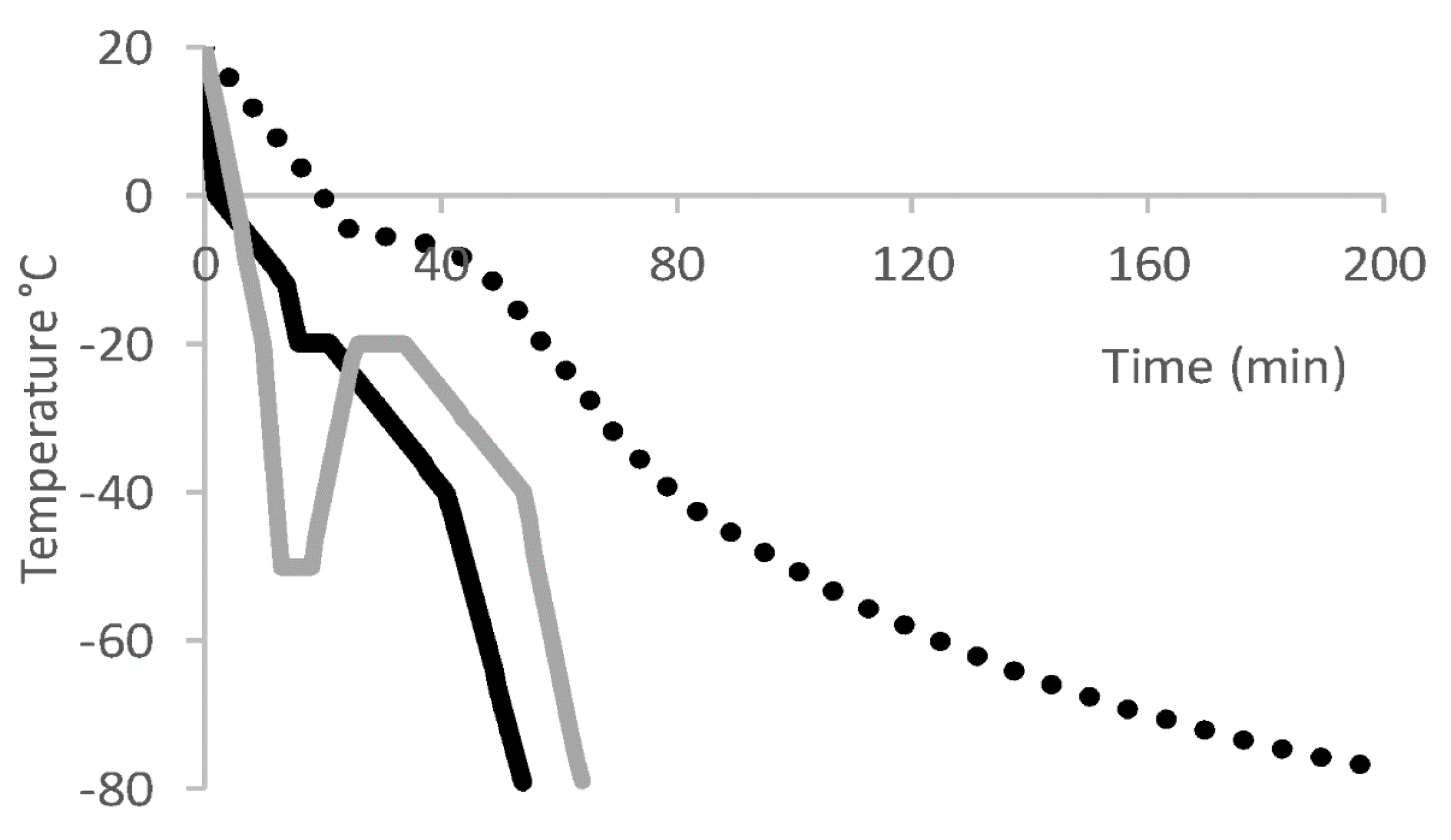
7. Freeze and Thaw of T Cell Drug Product
8. The Physical Effects of DMSO on Cells
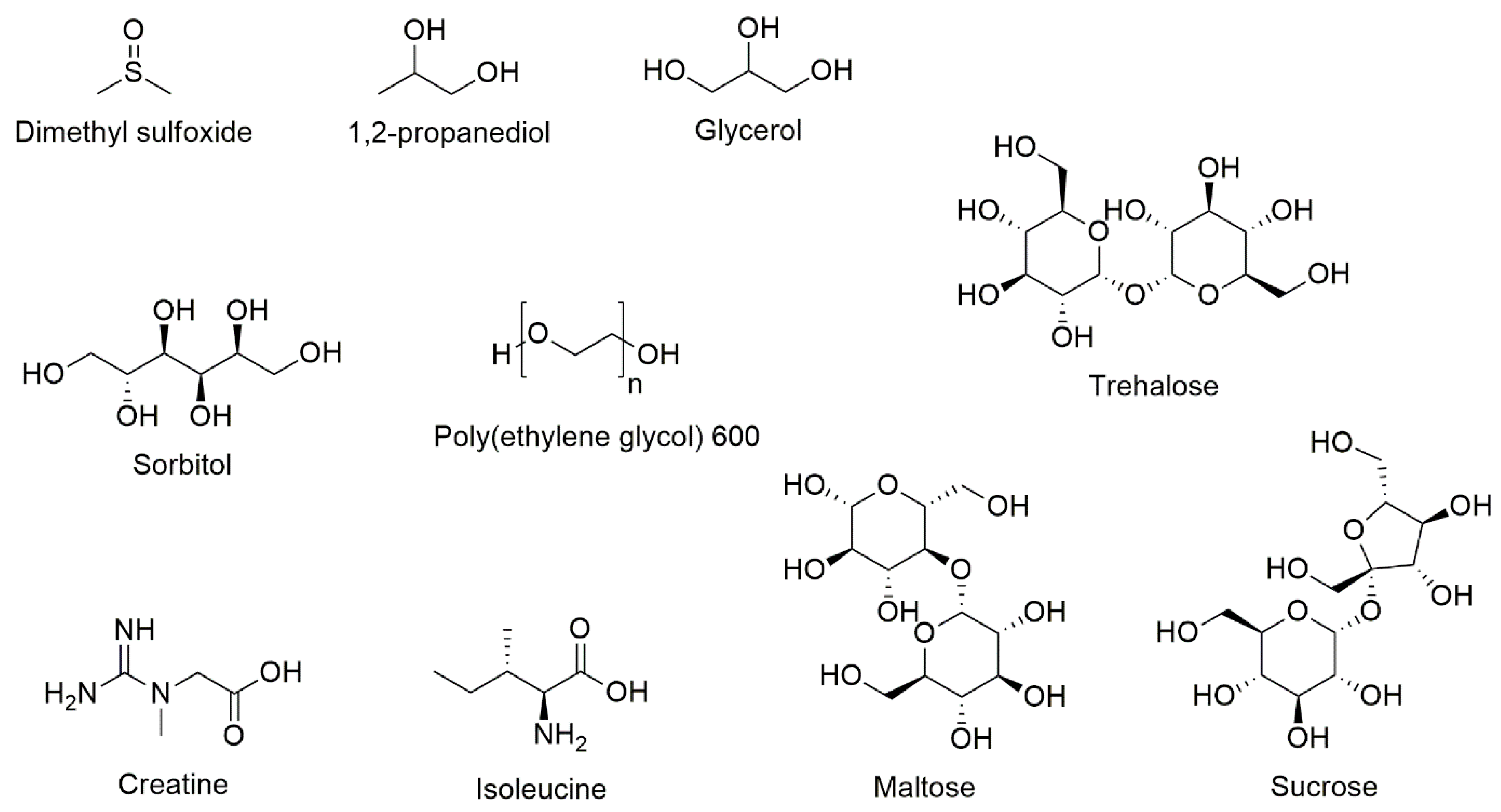
9. Potential Formulation Strategies for Reducing DMSO in Drug Product
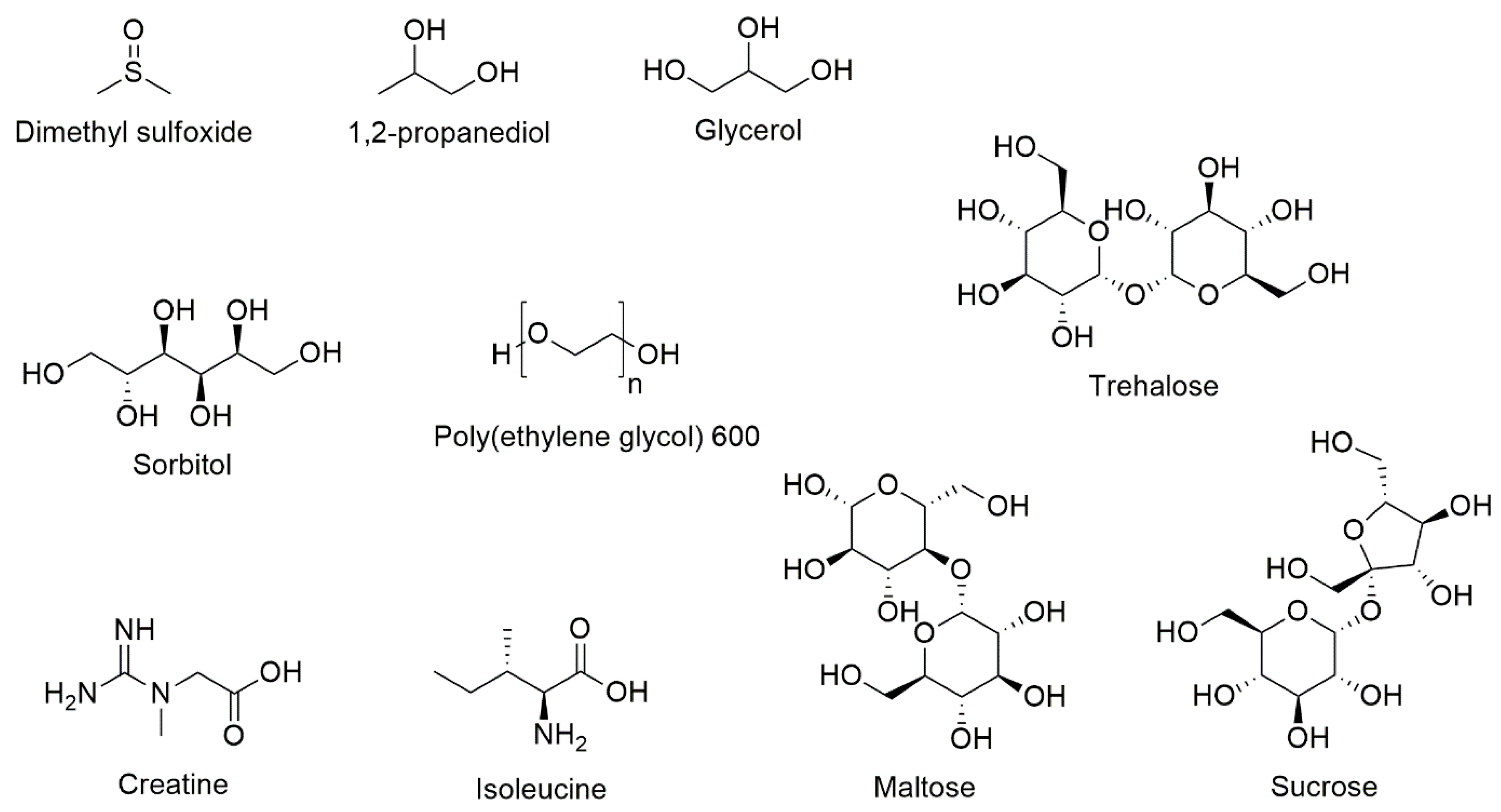
10. Novel Excipients and Formulations of T Cells
11. Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Harris, D.T.; Kranz, D.M. Adoptive T Cell Therapies: A Comparison of T Cell Receptors and Chimeric Antigen Receptors. Trends Pharmacol. Sci. 2016, 37, 220–230. [Google Scholar] [CrossRef] [Green Version]
- Grupp, S.A.; Kalos, M.; Barrett, D.; Aplenc, R.; Porter, D.L.; Rheingold, S.R.; Teachey, D.T.; Chew, A.; Hauck, B.; Wright, J.F.; et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N. Engl. J. Med. 2013, 368, 1509–1518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porter, D.L.; Levine, B.L.; Kalos, M.; Bagg, A.; June, C.H. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N. Engl. J. Med. 2011, 365, 725–733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Wang, L. The Emerging World of TCR-T Cell Trials Against Cancer: A Systematic Review. Technol. Cancer Res. Treat. 2019, 18, 1533033819831068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreno, E.; Forero, J.V.; Lengerke-Diaz, P.A.; Castro, J.E. Chimeric Antigen Receptor T Cell Therapy in Oncology—Pipeline at a glance: Analysis of the ClinicalTrials.gov database. Crit. Rev. Oncol. Hematol. 2021, 159, 103239. [Google Scholar] [CrossRef]
- Dai, X.; Mei, Y.; Nie, J.; Bai, Z. Scaling up the Manufacturing Process of Adoptive T Cell Immunotherapy. Biotechnol. J. 2019, 14, e1800239. [Google Scholar] [CrossRef]
- Perez, C.; Gruber, I.; Arber, C. Off-the-Shelf Allogeneic T Cell Therapies for Cancer: Opportunities and Challenges Using Naturally Occurring "Universal" Donor T Cells. Front. Immunol. 2020, 11, 583716. [Google Scholar] [CrossRef]
- International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH). Q8(R2) Pharmaceutical Development. Available online: https://database.ich.org/sites/default/files/Q8_R2_Guideline.pdf (accessed on 19 February 2021).
- Porter, D.L.; Hwang, W.T.; Frey, N.V.; Lacey, S.F.; Shaw, P.A.; Loren, A.W.; Bagg, A.; Marcucci, K.T.; Shen, A.; Gonzalez, V.; et al. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci. Transl. Med. 2015, 7, 303ra139. [Google Scholar] [CrossRef] [Green Version]
- Celgene Corporation—Bristol-Myers Squibb. Package Insert—ABECMA. Available online: https://www.fda.gov/media/147055/download (accessed on 4 February 2021).
- Novartis Pharmaceuticals Corporation. Package Insert-KYMRIAH. Available online: https://www.fda.gov/media/107296/download (accessed on 4 February 2021).
- Kite Pharma Inc. Package Insert—TECARTUS. Available online: https://www.fda.gov/media/140409/download (accessed on 4 February 2021).
- Juno Therapeutics Inc.—Bristol-Myers Squibb. Package Insert—BREYANZI. Available online: https://www.fda.gov/media/145711/download (accessed on 4 February 2021).
- Kite Pharma Inc. Package Insert—YESCARTA. Available online: https://www.fda.gov/media/108377/download (accessed on 4 February 2021).
- Dasyam, N.; George, P.; Weinkove, R. Chimeric antigen receptor T-cell therapies: Optimising the dose. Br. J. Clin. Pharmacol. 2020, 86, 1678–1689. [Google Scholar] [CrossRef]
- Yan, Z.X.; Li, L.; Wang, W.; OuYang, B.S.; Cheng, S.; Wang, L.; Wu, W.; Xu, P.P.; Muftuoglu, M.; Hao, M.; et al. Clinical Efficacy and Tumor Microenvironment Influence in a Dose-Escalation Study of Anti-CD19 Chimeric Antigen Receptor T Cells in Refractory B-Cell Non-Hodgkin’s Lymphoma. Clin. Cancer Res. 2019, 25, 6995–7003. [Google Scholar] [CrossRef] [Green Version]
- Xia, Y.; Tian, X.; Wang, J.; Qiao, D.; Liu, X.; Xiao, L.; Liang, W.; Ban, D.; Chu, J.; Yu, J.; et al. Treatment of metastatic non-small cell lung cancer with NY-ESO-1 specific TCR engineered-T cells in a phase I clinical trial: A case report. Oncol. Lett. 2018, 16, 6998–7007. [Google Scholar] [CrossRef] [Green Version]
- Spink, K.; Steinsapir, A. The long road to affordability: A cost of goods analysis for an autologous CAR-T process. Cell Gene Ther. Insights 2018, 4, 1105–1116. [Google Scholar] [CrossRef]
- Jin, J.; Gkitsas, N.; Fellowes, V.S.; Ren, J.; Feldman, S.A.; Hinrichs, C.S.; Stroncek, D.F.; Highfill, S.L. Enhanced clinical-scale manufacturing of TCR transduced T-cells using closed culture system modules. J. Transl. Med. 2018, 16, 13. [Google Scholar] [CrossRef] [Green Version]
- Vedvyas, Y.; McCloskey, J.E.; Yang, Y.; Min, I.M.; Fahey, T.J.; Zarnegar, R.; Hsu, Y.S.; Hsu, J.M.; Van Besien, K.; Gaudet, I.; et al. Manufacturing and preclinical validation of CAR T cells targeting ICAM-1 for advanced thyroid cancer therapy. Sci. Rep. 2019, 9, 10634. [Google Scholar] [CrossRef] [Green Version]
- Meneghel, J.; Kilbride, P.; Morris, G.J. Cryopreservation as a Key Element in the Successful Delivery of Cell-Based Therapies-A Review. Front. Med. 2020, 7, 592242. [Google Scholar] [CrossRef]
- Chong, E.A.; Levine, B.L.; Schuster, S. Clinical outcomes for anti-CD19 CAR T cell (CTL019) products not meeting commercial release specifications. Cytotherapy 2020, 22, S29. [Google Scholar] [CrossRef]
- Kite Pharma Inc. Summary Basis for Regulatory Action, BLA #:125643; Kite Pharma Inc.: Los Angeles, CA, USA, 2017. [Google Scholar]
- U.S. Food and Drug Administration. Sterile Drug Products Produced by Aseptic Processing—Current Good Manufacturing Practice. Available online: https://www.fda.gov/media/71026/download (accessed on 19 February 2021).
- Jorgensen, F.; Lambert, P. Accurate Biopharmaceutical Dispensing: Peristaltic or Piston Pumps? Innov. Pharm. Technol. 2008, 26, 78–80. [Google Scholar]
- Peterson, A. Checkweighing Fill Weight of Parenteral Product Is the Heart of Process Quality. In Practical Aseptic Processing: Fill and Finish; Lysfjord, J., Ed.; Parenteral Drug Association: Bethesda, MD, USA, 2015; Volume 1. [Google Scholar]
- Svay, K.; Urrea, C.; Shamlou, P.A.; Zhang, H. Computational fluid dynamics analysis of mixing and gas-liquid mass transfer in wave bag bioreactor. Biotechnol. Prog. 2020, 36, e3049. [Google Scholar] [CrossRef]
- Amer, M.; Feng, Y.; Ramsey, J.D. Using CFD simulations and statistical analysis to correlate oxygen mass transfer coefficient to both geometrical parameters and operating conditions in a stirred-tank bioreactor. Biotechnol. Prog. 2019, 35, e2785. [Google Scholar] [CrossRef]
- Nie, L.; Hu, M.; Yan, X.; Guo, T.; Wang, H.; Zhang, S.; Qu, H. Optimization of a Coupling Process for Insulin Degludec According to a Quality by Design (QbD) Paradigm. AAPS PharmSciTech 2018, 19, 2185–2194. [Google Scholar] [CrossRef]
- Committee for Medicinal Products for Human Use. Assessment Report, Yescarta, International Non-Proprietary Name: Axicabtagene Ciloleucel. Available online: https://www.ema.europa.eu/en/documents/assessment-report/yescarta-epar-public-assessment-report_en.pdf (accessed on 12 February 2021).
- Toms, D.; Deardon, R.; Ungrin, M. Climbing the mountain: Experimental design for the efficient optimization of stem cell bioprocessing. J. Biol. Eng. 2017, 11, 35. [Google Scholar] [CrossRef]
- Shieu, W.; Lamar, D.; Stauch, O.B.; Maa, Y.F. Filling of High-Concentration Monoclonal Antibody Formulations: Investigating Underlying Mechanisms That Affect Precision of Low-Volume Fill by Peristaltic Pump. PDA J. Pharm. Sci. Technol. 2016, 70, 143–156. [Google Scholar] [CrossRef]
- Simmchen, J.; Ventura, R.; Segura, J. Progress in the removal of di-[2-ethylhexyl]-phthalate as plasticizer in blood bags. Transfus. Med. Rev. 2012, 26, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Rethwisch, D.G.; Callister, W.D. Characteristics, Applications and Processing of Polymers. In Materials Science and Engineering: An Introduction, 7th ed.; University of Minnesota: Minneapolis, MN, USA; Wiley: Hoboken, NJ, USA, 2007; p. 546. [Google Scholar]
- Beirnes, K.J.; Burns, C.M. Thermal analysis of the glass transition of plasticized poly(vinyl chloride). J. Appl. Polym. Sci. 1986, 31, 2561–2567. [Google Scholar] [CrossRef]
- Ratner, B.D.; Hoffman, A.S.; Schoen, F.J.; Lemons, J.E. Classes of Materials Used in Medicine. In Biomaterials Science: An Introduction to Materials in Medicine, 2nd ed.; Elsevier: Amsterdam, The Netherlands, 2004; p. 78. [Google Scholar]
- Fakirov, S.; Krasteva, B. On the Glass Transition Temperature of Polyethylene as Revealed by Microhardness Measurements. J. Macromol. Sci. B 2000, 39, 297–301. [Google Scholar] [CrossRef]
- Wang, K.; Deng, Q. The Thermal and Mechanical Properties of Poly(ethylene-co-vinyl acetate) Random Copolymers (PEVA) and its Covalently Crosslinked Analogues (cPEVA). Polymers 2019, 11, 1055. [Google Scholar] [CrossRef] [Green Version]
- Baboo, J.; Kilbride, P.; Delahaye, M.; Milne, S.; Fonseca, F.; Blanco, M.; Meneghel, J.; Nancekievill, A.; Gaddum, N.; Morris, G.J. The Impact of Varying Cooling and Thawing Rates on the Quality of Cryopreserved Human Peripheral Blood T Cells. Sci. Rep. 2019, 9, 3417. [Google Scholar] [CrossRef]
- Church, S.E.; Gunther, J.C.; Pollock, K. Methods for Cryogenic Storage. U.S. Patent Application Publication US 2020/0077644 A1, 12 March 2020. [Google Scholar]
- Woods, E.J.; Thirumala, S. Packaging Considerations for Biopreservation. Transfus. Med. Hemother. 2011, 38, 149–156. [Google Scholar] [CrossRef] [Green Version]
- Marais, S.; Bureau, E.; Gouanve, F.; Ben Salem, E.; Hirata, Y.; Andrio, A.; Cabot, C.; Atmani, H. Transport of water and gases through EVA/PVC blend films—permeation and DSC investigations. Polym. Test. 2004, 23, 475–486. [Google Scholar] [CrossRef] [Green Version]
- Marais, S.; Hirata, Y.; Langevin, D.; Chappey, C.; Nguyen, T.Q.; Metayer, M. Permeation and Sorption of Water and Gases Through EVA Copolymers Films. Mat. Res. Innovat. 2002, 6, 79–88. [Google Scholar] [CrossRef]
- Maes, C.; Luyten, W.; Herremans, G.; Peeters, R.; Carleer, R.; Buntinx, M. Recent Updates on the Barrier Properties of Ethylene Vinyl Alcohol Copolymer (EVOH): A Review. Polym. Rev. 2018, 58, 209–246. [Google Scholar] [CrossRef] [Green Version]
- Mele, L.; Dallavalle, F.M.; Verri, M.G.; Balza, G.; Allione, B.; Salvi, F.; Levis, A.; Caraccio, V.; Borzini, P. Safety control of peripheral blood progenitor cell processing-eight year-survey of microbiological contamination and bag ruptures in a single institution. Transfus. Apher. Sci. 2005, 33, 269–274. [Google Scholar] [CrossRef]
- Thyagarajan, B.; Berger, M.; Sumstad, D.; McKenna, D.H. Loss of integrity of umbilical cord blood unit freezing bags: Description and consequences. Transfusion 2008, 48, 1138–1142. [Google Scholar] [CrossRef]
- Khuu, H.M.; Cowley, H.; David-Ocampo, V.; Carter, C.S.; Kasten-Sportes, C.; Wayne, A.S.; Solomon, S.R.; Bishop, M.R.; Childs, R.M.; Read, E.J. Catastrophic failures of freezing bags for cellular therapy products: Description, cause, and consequences. Cytotherapy 2002, 4, 539–549. [Google Scholar] [CrossRef]
- Levine, B.L.; Miskin, J.; Wonnacott, K.; Keir, C. Global Manufacturing of CAR T Cell Therapy. Mol. Ther. Methods Clin. Dev. 2017, 4, 92–101. [Google Scholar] [CrossRef] [Green Version]
- Committee for Medicinal Products for Human Use. Guideline on Plastic Immediate Packaging Materials. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-plastic-immediate-packaging-materials_en.pdf (accessed on 26 February 2021).
- BioPhorum Operations Group Ltd. BioPhorum Best Practices Guide for Extractables Testing of Polymeric Single-Use Components Used in Biopharmaceutical Manufacturing. Available online: https://www.biophorum.com/download/extractables-testing-of-polymeric-single-use-components-used-in-biopharmaceutical-manufacturing/ (accessed on 20 February 2021).
- The United States Pharmacopeial Convention. <790> Visible Particulates in Injections, Issue 1 ed.; USP–NF 2021; United States Pharmacopeia (USP): Rockville, MD, USA, 2021. [Google Scholar]
- Wormuth, K.; Gauthier, M.; Labedan, M.; Cantin, V.; Gaston, F.; Thaust, M.; Montenay, N.; Barbaroux, M. Visible Particulate Matter in Single-Use Bags: From Measurement to Prevention. BioProcess Int. 2019, 17, 50–53. [Google Scholar]
- The United States Pharmacopeial Convention. <788> Particulate Matter in Injections, Issue 1 ed.; USP–NF 2021; United States Pharmacopeia (USP): Rockville, MD, USA, 2021. [Google Scholar]
- Johnson, M.W. Understanding Particulates in Single-Use Bags: Their Relationship to USP Chapter <788>. BioProcess Int. 2014, 12, 22–28. [Google Scholar]
- ASTM E3230-20. Standard Practice for Extraction of Particulate Matter from the Surfaces of Single-Use Components and Assemblies Designed for Use in Biopharmaceutical Manufacturing; ASTM International: West Conshohocken, PA, USA, 2020. [Google Scholar] [CrossRef]
- Grabarek, A.D.; Senel, E.; Menzen, T.; Hoogendoorn, K.H.; Pike-Overzet, K.; Hawe, A.; Jiskoot, W. Particulate impurities in cell-based medicinal products traced by flow imaging microscopy combined with deep learning for image analysis. Cytotherapy 2020, 23, 339–347. [Google Scholar] [CrossRef]
- Morris, G.J.; Acton, E. Controlled ice nucleation in cryopreservation—A review. Cryobiology 2013, 66, 85–92. [Google Scholar] [CrossRef]
- Levin, R.L. A generalized method for the minimization of cellular osmotic stresses and strains during the introduction and removal of permeable cryoprotectants. J. Biomech. Eng. 1982, 104, 81–86. [Google Scholar] [CrossRef]
- Ragoonanan, V.; Hubel, A.; Aksan, A. Response of the cell membrane-cytoskeleton complex to osmotic and freeze/thaw stresses. Cryobiology 2010, 61, 335–344. [Google Scholar] [CrossRef]
- Năstase, G.; Lyu, C.; Ukpai, G.; Şerban, A.; Rubinsky, B. Isochoric and isobaric freezing of fish muscle. Biochem. Biophys. Res. Commun. 2017, 485, 279–283. [Google Scholar] [CrossRef]
- Hunt, C.J. Cryopreservation: Vitrification and Controlled Rate Cooling. Methods Mol. Biol. 2017, 1590, 41–77. [Google Scholar] [CrossRef]
- Guest, R.D.; Rothwell, D.; Kirillov, N.; Mowbray, S.; Sheard, V.; Gibbons, S.; Acton, E.; Gilham, D.; Morris, J.; Hawkins, R. 059 Role of cryopreservation in the clinical delivery of T cell based adoptive cell therapies (ACT). Cryobiology 2013, 67, 414. [Google Scholar] [CrossRef]
- Foussat, A.; Rietze, R.; Pottier, E.; Schryver, B.; Thompson, M.; Ehrhardt, R. Effective Cryopreservation and Recovery of Human Regulatory T Cells. BioProcess Int. 2014, 12, 34–38. [Google Scholar]
- Chamberlain, C.; Ahsan, G.; James, J.; Gilmour, K. 88 Validation of controlled rate freezing of T-Cells for KYMRIAH® production. Arch. Dis. Child. 2019, 104, A34. [Google Scholar]
- Buhl, T.; Legler, T.J.; Rosenberger, A.; Schardt, A.; Schön, M.P.; Haenssle, H.A. Controlled-rate freezer cryopreservation of highly concentrated peripheral blood mononuclear cells results in higher cell yields and superior autologous T-cell stimulation for dendritic cell-based immunotherapy. Cancer Immunol. Immunother. 2012, 61, 2021–2031. [Google Scholar] [CrossRef] [Green Version]
- Singh, H.; Figliola, M.J.; Dawson, M.J.; Olivares, S.; Zhang, L.; Yang, G.; Maiti, S.; Manuri, P.; Senyukov, V.; Jena, B.; et al. Manufacture of clinical-grade CD19-specific T cells stably expressing chimeric antigen receptor using Sleeping Beauty system and artificial antigen presenting cells. PLoS ONE 2013, 8, e64138. [Google Scholar] [CrossRef]
- Schulz, J.C.; Germann, A.; Kemp-Kamke, B.; Mazzotta, A.; von Briesen, H.; Zimmermann, H. Towards a xeno-free and fully chemically defined cryopreservation medium for maintaining viability, recovery, and antigen-specific functionality of PBMC during long-term storage. J. Immunol. Methods 2012, 382, 24–31. [Google Scholar] [CrossRef]
- Germann, A.; Oh, Y.J.; Schmidt, T.; Schön, U.; Zimmermann, H.; von Briesen, H. Temperature fluctuations during deep temperature cryopreservation reduce PBMC recovery, viability and T-cell function. Cryobiology 2013, 67, 193–200. [Google Scholar] [CrossRef]
- Boutron, P.; Mehl, P. Theoretical prediction of devitrification tendency: Determination of critical warming rates without using finite expansions. Cryobiology 1990, 27, 359–377. [Google Scholar] [CrossRef]
- Steif, P.S.; Palastro, M.C.; Rabin, Y. The Effect of Temperature Gradients on Stress Development During Cryopreservation via Vitrification. Cell Preserv. Technol. 2007, 5, 104–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Notman, R.; Noro, M.; O’Malley, B.; Anwar, J. Molecular basis for dimethylsulfoxide (DMSO) action on lipid membranes. J. Am. Chem. Soc. 2006, 128, 13982–13983. [Google Scholar] [CrossRef]
- de Ménorval, M.A.; Mir, L.M.; Fernández, M.L.; Reigada, R. Effects of dimethyl sulfoxide in cholesterol-containing lipid membranes: A comparative study of experiments in silico and with cells. PLoS ONE 2012, 7, e41733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kovrigin, E.L.; Potekhin, S.A. Preferential solvation changes upon lysozyme heat denaturation in mixed solvents. Biochemistry 1997, 36, 9195–9199. [Google Scholar] [CrossRef]
- Roy, S.; Bagchi, B. Comparative study of protein unfolding in aqueous urea and dimethyl sulfoxide solutions: Surface polarity, solvent specificity, and sequence of secondary structure melting. J. Phys. Chem. B 2014, 118, 5691–5697. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, K.R.; Goyal, B.; Kumar, A.; Durani, S. Scrutiny of electrostatic-driven conformational ordering of polypeptide chains in DMSO: A study with a model oligopeptide. RSC Adv. 2017, 7, 27981–27991. [Google Scholar] [CrossRef]
- Fukui, Y.; Katsumaru, H. Dynamics of nuclear actin bundle induction by dimethyl sulfoxide and factors affecting its development. J. Cell Biol. 1980, 84, 131–140. [Google Scholar] [CrossRef] [Green Version]
- Lampugnani, M.G.; Pedenovi, M.; Niewiarowski, A.; Casali, B.; Donati, M.B.; Corbascio, G.C.; Marchisio, P.C. Effects of dimethyl sulfoxide (DMSO) on microfilament organization, cellular adhesion, and growth of cultured mouse B16 melanoma cells. Exp. Cell Res. 1987, 172, 385–396. [Google Scholar] [CrossRef]
- Tunçer, S.; Gurbanov, R.; Sheraj, I.; Solel, E.; Esenturk, O.; Banerjee, S. Low dose dimethyl sulfoxide driven gross molecular changes have the potential to interfere with various cellular processes. Sci. Rep. 2018, 8, 14828. [Google Scholar] [CrossRef]
- The United States Pharmacopeial Convention. <785> Osmolality and Osmolarity, Issue 1 ed.; USP–NF 2021; United States Pharmacopeia (USP): Rockville, MD, USA, 2021. [Google Scholar]
- Obeidy, P.; Ju, L.A.; Oehlers, S.H.; Zulkhernain, N.S.; Lee, Q.; Galeano Niño, J.L.; Kwan, R.Y.; Tikoo, S.; Cavanagh, L.L.; Mrass, P.; et al. Partial loss of actin nucleator actin-related protein 2/3 activity triggers blebbing in primary T lymphocytes. Immunol. Cell Biol. 2020, 98, 93–113. [Google Scholar] [CrossRef]
- Ruan, R.; Zou, L.; Sun, S.; Liu, J.; Wen, L.; Gao, D.; Ding, W. Cell blebbing upon addition of cryoprotectants: A self-protection mechanism. PLoS ONE 2015, 10, e0125746. [Google Scholar] [CrossRef]
- Ragoonanan, V.; Less, R.; Aksan, A. Response of the cell membrane-cytoskeleton complex to osmotic and freeze/thaw stresses. Part 2: The link between the state of the membrane-cytoskeleton complex and the cellular damage. Cryobiology 2013, 66, 96–104. [Google Scholar] [CrossRef]
- Kollerup Madsen, B.; Hilscher, M.; Zetner, D.; Rosenberg, J. Adverse reactions of dimethyl sulfoxide in humans: A systematic review. F1000Res. 2018, 7, 1746. [Google Scholar] [CrossRef]
- Committee for Advanced Therapies. Minutes for the Meeting on 19–20 February 2015. Available online: https://www.ema.europa.eu/en/documents/minutes/minutes-cat-meeting-19-20-february-2015_en.pdf (accessed on 16 February 2021).
- Cox, M.A.; Kastrup, J.; Hrubiško, M. Historical perspectives and the future of adverse reactions associated with haemopoietic stem cells cryopreserved with dimethyl sulfoxide. Cell Tissue Bank. 2012, 13, 203–215. [Google Scholar] [CrossRef]
- Raes, A.; Van Aken, S.; Craen, M.; Donckerwolcke, R.; Vande Walle, J. A reference frame for blood volume in children and adolescents. BMC Pediatr. 2006, 6, 3. [Google Scholar] [CrossRef] [Green Version]
- Mfarrej, B.; Bouchet, G.; Couquiaud, J.; Regimbaud, L.; Binninger, S.; Mercier, M.; Lemarié, C.; Houzé, P.; Chabannon, C.; Calmels, B. Pre-clinical assessment of the Lovo device for dimethyl sulfoxide removal and cell concentration in thawed hematopoietic progenitor cell grafts. Cytotherapy 2017, 19, 1501–1508. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Chan, L.L.; Grimaud, M.; Fayed, A.; Zhu, Q.; Marasco, W.A. High-Throughput Image Cytometry Detection Method for CAR-T Transduction, Cell Proliferation, and Cytotoxicity Assays. Cytometry A 2020, 99, 689–697. [Google Scholar] [CrossRef]
- Wiezorek, J. Methods of Administering Chimeric Antigen Receptor Immunotherapy. U.S. Patent Application Publication US 2019/0151361 A1, 23 May 2019. [Google Scholar]
- Mirabel, C.; Puente-Massaguer, E.; Del Mazo-Barbara, A.; Reyes, B.; Morton, P.; Gòdia, F.; Vives, J. Stability enhancement of clinical grade multipotent mesenchymal stromal cell-based products. J. Transl. Med. 2018, 16, 291. [Google Scholar] [CrossRef] [PubMed]
- Rubinstein, P.; Dobrila, L.; Rosenfield, R.E.; Adamson, J.W.; Migliaccio, G.; Migliaccio, A.R.; Taylor, P.E.; Stevens, C.E. Processing and cryopreservation of placental/umbilical cord blood for unrelated bone marrow reconstitution. Proc. Natl. Acad. Sci. USA 1995, 92, 10119–10122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hospira Healthcare Corporation. Dextran 40 in Dextrose Injection & Dextran 40 in Sodium Chloride Injection—Potential for Crystallization—Notice to Hospitals. Available online: https://healthycanadians.gc.ca/recall-alert-rappel-avis/hc-sc/2013/36461a-eng.php (accessed on 15 March 2021).
- Weng, L.; Beauchesne, P.R. Dimethyl sulfoxide-free cryopreservation for cell therapy: A review. Cryobiology 2020, 94, 9–17. [Google Scholar] [CrossRef]
- Stubbs, C.; Bailey, T.L.; Murray, K.; Gibson, M.I. Polyampholytes as Emerging Macromolecular Cryoprotectants. Biomacromolecules 2020, 21, 7–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharp, D.M.; Picken, A.; Morris, T.J.; Hewitt, C.J.; Coopman, K.; Slater, N.K. Amphipathic polymer-mediated uptake of trehalose for dimethyl sulfoxide-free human cell cryopreservation. Cryobiology 2013, 67, 305–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.; Wu, L.; Ren, J.; Bemmer, V.; Zajicek, R.; Chen, R. Comb-like Pseudopeptides Enable Very Rapid and Efficient Intracellular Trehalose Delivery for Enhanced Cryopreservation of Erythrocytes. ACS Appl. Mater. Interfaces 2020, 12, 28941–28951. [Google Scholar] [CrossRef] [PubMed]
- International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH). The Common Technical Document for the for the Registration of Pharmaceuticals for Human Use: Quality—MQ4(R1). Available online: https://database.ich.org/sites/default/files/M4Q_R1_Guideline.pdf (accessed on 8 March 2021).
- Center for Drug Evaluation and Research—Office of Pharmaceutical Quality. Inactive Ingredient Search for Approved Drug Products. Available online: https://www.accessdata.fda.gov/scripts/cder/iig/index.cfm# (accessed on 23 January 2021).
- Sheskey, P.J.; Hancock, B.C.; Moss, G.P.; Goldfarb, D.J. Handbook of Pharmaceutical Excipients, 9th ed.; Pharmaceutical Press: London, UK, 2020. [Google Scholar]
- DeMerlis, C.; Goldring, J.; Velagaleti, R.; Brock, W.; Osterberg, R. Regulatory Update: The IPEC Novel Excipient Safety Evaluation Procedure. Pharm. Technol. 2009, 33, 72–82. [Google Scholar]
- Center for Drug Evaluation and Research (CDER) and Center for Biologics Evaluation and Research (CBER). Guidance for Industry Nonclinical Studies for the Safety Evaluation of Pharmaceutical Excipients. Available online: https://www.fda.gov/media/72260/download (accessed on 16 March 2021).
- Committee for Medicinal Products for Human Use. Guideline on Excipients in the Dossier for Application for Marketing Authorisation of a Medicinal Product. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-excipients-dossier-application-marketing-authorisation-medicinal-product-revision-2_en.pdf (accessed on 17 March 2021).
- U.S. Food and Drug Administration. Novel Excipient Review Program Proposal; Request for Information and Comments. Federal Register 2019, 84, 66669–66671. [Google Scholar]
- Pi, C.H.; Yu, G.; Dosa, P.I.; Hubel, A. Characterizing modes of action and interaction for multicomponent osmolyte solutions on Jurkat cells. Biotechnol. Bioeng. 2019, 116, 631–643. [Google Scholar] [CrossRef]
- Li, F.J.; Zhang, S.D.; Liang, J.Z.; Wang, J.Z. Effect of polyethylene glycol on the crystallization and impact properties of polylactide-based blends. Polym. Adv. Technol. 2015, 26, 465–475. [Google Scholar] [CrossRef]
- Pi, C.H.; Yu, G.; Petersen, A.; Hubel, A. Characterizing the “sweet spot” for the preservation of a T-cell line using osmolytes. Sci. Rep. 2018, 8, 16223. [Google Scholar] [CrossRef]
- Pi, C.H.; Hornberger, K.; Dosa, P.; Hubel, A. Understanding the freezing responses of T cells and other subsets of human peripheral blood mononuclear cells using DSMO-free cryoprotectants. Cytotherapy 2020, 22, 291–300. [Google Scholar] [CrossRef]
- Wei, B.; Berning, K.; Quan, C.; Zhang, Y.T. Glycation of antibodies: Modification, methods and potential effects on biological functions. MAbs 2017, 9, 586–594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Awan, M.; Buriak, I.; Fleck, R.; Fuller, B.; Goltsev, A.; Kerby, J.; Lowdell, M.; Mericka, P.; Petrenko, A.; Petrenko, Y.; et al. Dimethyl sulfoxide: A central player since the dawn of cryobiology, is efficacy balanced by toxicity? Regen. Med. 2020, 15, 1463–1491. [Google Scholar] [CrossRef] [PubMed]
- American Association of Blood Banks. Standards for Blood Banks and Transfusion Services, 32nd ed.; American Association of Blood Banks (AABB): Bethesda, MD, USA, 2020. [Google Scholar]
- Committee for Human Medicinal Products. Propylene Glycol Used as an Excipient. Available online: https://www.ema.europa.eu/en/documents/report/propylene-glycol-used-excipient-report-published-support-questions-answers-propylene-glycol-used_en.pdf (accessed on 6 March 2021).
- Gottlieb, S. Statement from FDA Commissioner Scott Gottlieb, M.D. and Peter Marks, M.D., Ph.D., Director of the Center for Biologics Evaluation and Research on New Policies to Advance Development of Safe and Effective Cell and Gene Therapies. Available online: https://www.fda.gov/news-events/press-announcements/statement-fda-commissioner-scott-gottlieb-md-and-peter-marks-md-phd-director-center-biologics (accessed on 22 March 2021).

{kind=link}
{kind=link}
{kind=link}
| T Cell Drug Product (Proprietary Name) | Dose, Adults (CAR-Positive Viable T Cells) | Primary Container, Number Supplied per Dose | Fill Volume Per Bag/Vial, Number Bags/Vials Supplied per Dose |
|---|---|---|---|
| brexucabtagene autoleucel (Tecartus™) | 2 × 106 per kg body weight, max. 2 × 108 | cryogenic infusion bag | ~68 mL, one |
| axicabtagene ciloleucel (Yescarta®) | 2 × 106 per kg body weight, max. 2 × 108 | cryogenic infusion bag | ~68 mL, one |
| Tisagenlecleucel (Kymriah®) | 0.6–6.0 × 108 | cryogenic infusion bag | 10–50 mL, one to three |
| lisocabtagene maraleucel (Breyanzi®) | 0.5–1.1 × 108 (CD8 and CD4 components, 1:1) | cryogenic vials | 4.6 mL, one to four (for each CD8 and CD4 component) |
| idecabtagene vicleucel (Abecma®) | 3.0–4.6 × 108 | cryogenic infusion bag, 50/250/500 mL nominal volume | within validated range, one or more |
| ClinicalTrials.gov Identifier | Phase | Study Complete | Sponsor | Transgene (CAR/TCR) | Indication | Dose (CAR/TCR-T Cells) |
|---|---|---|---|---|---|---|
| NCT03289455 | I/II | May 2020 | Autolus Limited | CD19/22 CAR | B Cell ALL | 1 to 5.0 × 106/kg |
| NCT02030847 | II | Apr 2018 | University of Pennsylvania | CD19 CAR | B Cell ALL | 1–5 × 108 CART-19 cells administered via split dosing: 10% day 1 (1–5 × 107), 30% day 2 (3 × 107–1.5 × 108), 60% day 3 (6 × 107–3 × 108) |
| NCT01626495 | I/IIa | Jul 2019 | University of Pennsylvania | CD19 CAR | R/R 2 Leukaemia/Lymphoma | 10% day 0, 30% day 1, possibly 60% day 14+; total dose goal ~1.5 × 107–5 × 109 (~0.3 × 106–1.0 × 108/kg) |
| NCT02030834 | IIa | Sep 2020 | University of Pennsylvania | humanised CD19 CAR | Diffuse Large B Cell Lymphoma | total dose of 1–5 × 108 |
| NCT03232619 | I/II | Sep 2020 | Bioray Laboratories, Second Xiangya Hospital of Central South University | humanised CD19 CAR | B Cell ALL | 0.5–5 × 106/kg |
| NCT02550535/ NCT01621724 | I/II | May 2018 | Cell Medica Ltd., University College London, Cell Therapy Catapult | Wilms tumour antigen 1 TCR | Myelodys-plastic syndrome, acute myeloid leukaemia | ≤2 × 107/kg (Cohort 1) ≤1 × 108/kg (Cohort 2) |
| NCT01567891 | I/IIa | Jun 2017 | Adaptimmune | NY-ESO-1 TCR | R/R Ovarian Cancer | 1–6 × 109 |
| NCT00509288 | II | Jul 2012 | National Cancer Institute | HLA-A 0201 TCR | Metastatic Melanoma | 5 × 108 to 3 × 1011 |
| Parameter | Specifications 1 | Method 2 |
|---|---|---|
| Identity | CAR/TCR presence | qPCR 3 |
| Appearance | Colour | Ph. Eur. 2.2.2 (USP <631>) |
| Dose | Number CAR/TCR positive cells | Calculated (from cell viability and transduction efficiency) |
| Potency | T cell function Transduction Efficiency | Cell-based assay Flow Cytometry (% T cells expressing transgene) |
| Purity | Total cell count CD3+ cell count Cell viability Residual beads 4 | Cell count Flow Cytometry (% CD3+ cells) qPCR Microscopy |
| Safety | Copies of transgene RCL 5 Sterility Mycoplasma Endotoxin | qPCR qPCR/cell-based assay Ph. Eur. 2.6.27 (USP <71>) Ph. Eur. 2.6.7 (USP <63>) Ph. Eur. 2.6.14 (USP <85>) |
| Polymer/Property | EVA19 | EVA50 | EVA70 | PVC | LDPE 1 |
|---|---|---|---|---|---|
| % vinyl acetate | 19 | 50 | 70 | - | - |
| % crystallinity | 29 | 6 | 0 | 0 | 0 |
| P H2O(g) (barrer) 2 | 1134 | 9357 | 12,287 | 239 | 73 |
| P O2 (barrer) | 5.3 | 8.1 | 3.6 | 0.08 | 2.3 |
| P CO2 (barrer) | 57 | 70 | 30 | 0.28 | 16 |
| 3 αH2O/O2 | 214 | 1155 | 3413 | 3034 | 32 |
| αH2O/CO2 | 20 | 134 | 410 | 856 | 4.6 |
| αCO2/O2 | 10.8 | 8.6 | 8.3 | 3.5 | 7.0 |
| Component | Solvent | Time, at 40 °C | |||||
|---|---|---|---|---|---|---|---|
| 50% Ethanol | 0.5 N NaOH | 0.1 M Phosphoric Acid | WFI 2 | 24 h | 21 days | 70 days | |
| Bags for long-term storage | X | X | X | X | X | X | X |
| Storage bag tubing | X | X | X | X | X | X | X |
| Storage bag ports | X | X | X | X | X | X | X |
| Bag ports | X | X | X | X | X | X | |
| Tubing | X | X | X | X | X | X | |
| Tubing connectors, overmolded junctions | X | X | X | X | X | ||
| Aseptic connectors | X | X | X | X | X | ||
| Sterilising/process filters | X | X | X | X | X | ||
| O-rings, gaskets | X | X | |||||
| T Cell Drug Product (Proprietary Name) | HSA Conc. (%) | DMSO Conc. (%) | Other Excipients |
|---|---|---|---|
| brexucabtagene autoleucel (Tecartus™) | - | 5 | sodium chloride, injection |
| axicabtagene ciloleucel (Yescarta®) | 2.5 | 5 | sodium chloride, injection |
| Tisagenlecleucel (Kymriah®) | 5.0 | 7.5 | 31.25% Plasma-Lyte A, 31.25% of 5% Dextrose/0.45% sodium chloride, 10% Dextran 40 (LMD)/5% Dextrose |
| lisocabtagene maraleucel (Breyanzi®) | 0.25 | 7.5 | 24% (v/v) Multiple Electrolytes for Injection, Type 1 |
| idecabtagene vicleucel (Abecma®) | 0.0 | 5 | 50% Plasma-Lyte A |
| Material | MW (g/mol) | Density (g/mL) (20 °C) | Viscosity (20 °C) (mPa·s) | LogP | Polar Surface Area (Å2) | Solubility in H2O (mg/mL) (20 °C) |
|---|---|---|---|---|---|---|
| DMSO | 78.13 | 1.10 | 1.996 | −1.35 | 17.07 | miscible |
| 1,2-propanediol | 76.09 | 1.036 | 42 | −0.92 | 40.5 | miscible |
| Glycerol | 92.09 | 1.261 | 1412 | −1.76 | 60.7 | miscible |
| Sorbitol | 182.17 | 1.489 | - | −2.2 | 121.38 | 2350 |
| Poly(ethylene glycol) 600 1 | 600 | 1.12 | 150–190 | - | - | miscible |
| Creatine | 131.13 | 1.33 | - | −0.2 | 90.4 | 13.3 (18 °C) |
| Isoleucine | 131.17 | - | - | −1.7 | 63.32 | 34.4 |
| Trehalose | 342.3 | 1.58 (24 °C) | - | −3 2 | 190 | 592 |
| Maltose | 342.3 | 1.54 | - | −3 2 | 189.53 | 780 |
| Sucrose | 342.3 | 1.59 | - | −3.7 | 189.53 | 2120 (25 °C) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
van der Walle, C.F.; Godbert, S.; Saito, G.; Azhari, Z. Formulation Considerations for Autologous T Cell Drug Products. Pharmaceutics 2021, 13, 1317. https://doi.org/10.3390/pharmaceutics13081317
van der Walle CF, Godbert S, Saito G, Azhari Z. Formulation Considerations for Autologous T Cell Drug Products. Pharmaceutics. 2021; 13(8):1317. https://doi.org/10.3390/pharmaceutics13081317
Chicago/Turabian Stylevan der Walle, Christopher F., Sonya Godbert, Gabriele Saito, and Zein Azhari. 2021. "Formulation Considerations for Autologous T Cell Drug Products" Pharmaceutics 13, no. 8: 1317. https://doi.org/10.3390/pharmaceutics13081317
APA Stylevan der Walle, C. F., Godbert, S., Saito, G., & Azhari, Z. (2021). Formulation Considerations for Autologous T Cell Drug Products. Pharmaceutics, 13(8), 1317. https://doi.org/10.3390/pharmaceutics13081317