
Microfluidic Formulation of DNA-Loaded Multicomponent Lipid Nanoparticles for Gene Delivery

 , , ,
, , , 
 , , , ,
, , , , 
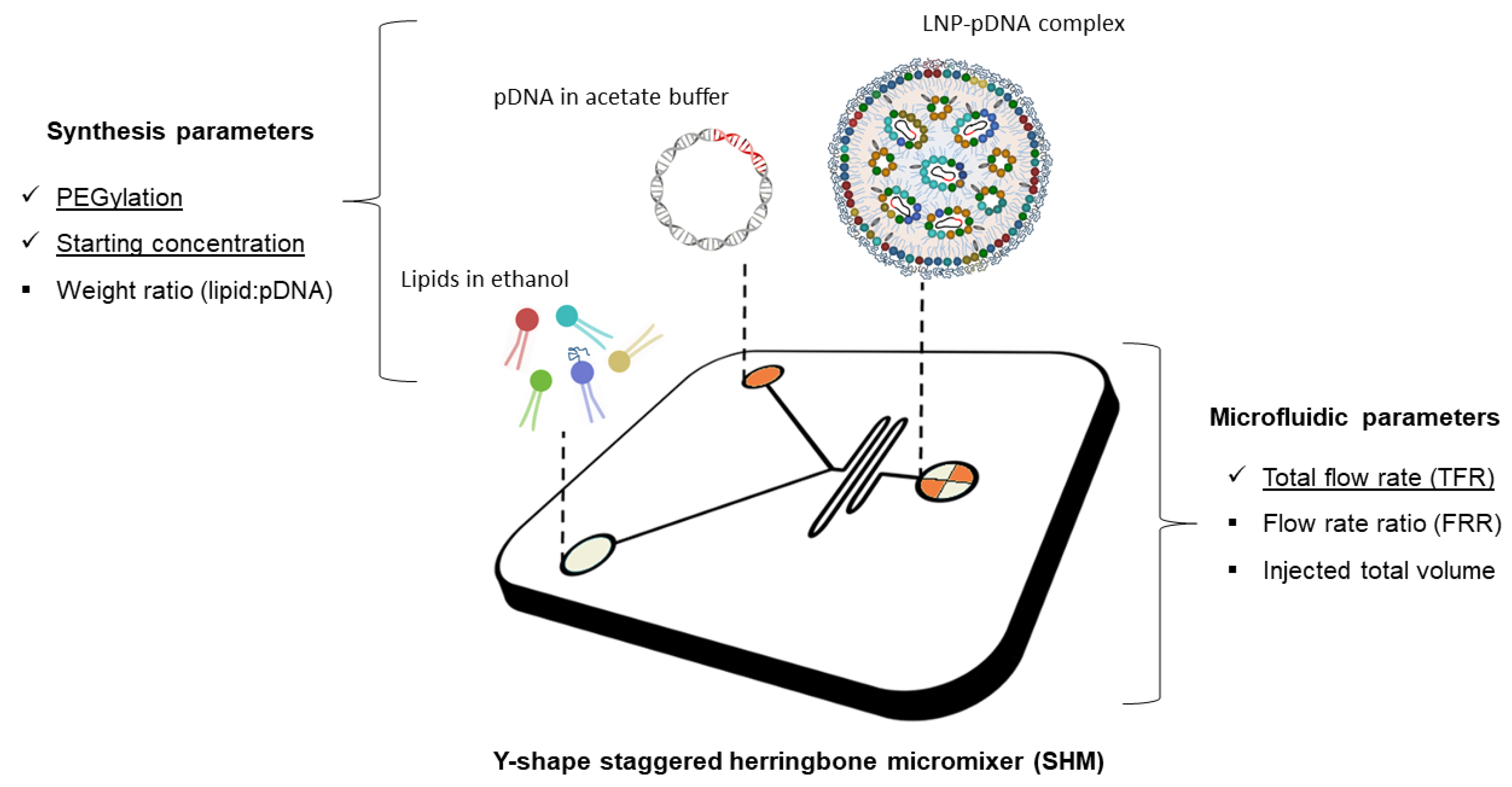{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. Lipid-Based Gene Delivery Systems
1.2. Microfluidic Manufacturing of Lipid Nanoparticles
1.3. Role of Microfluidic Parameters and Effects on Cellular Response
2. Results and Discussion
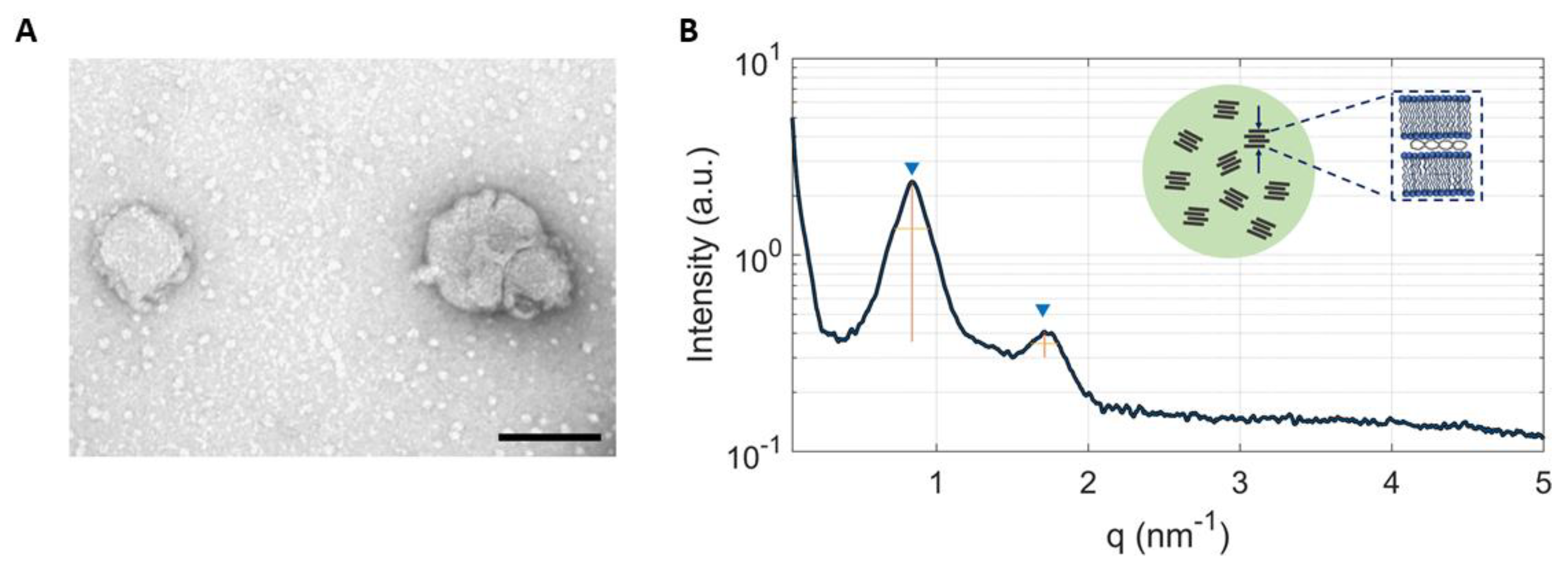
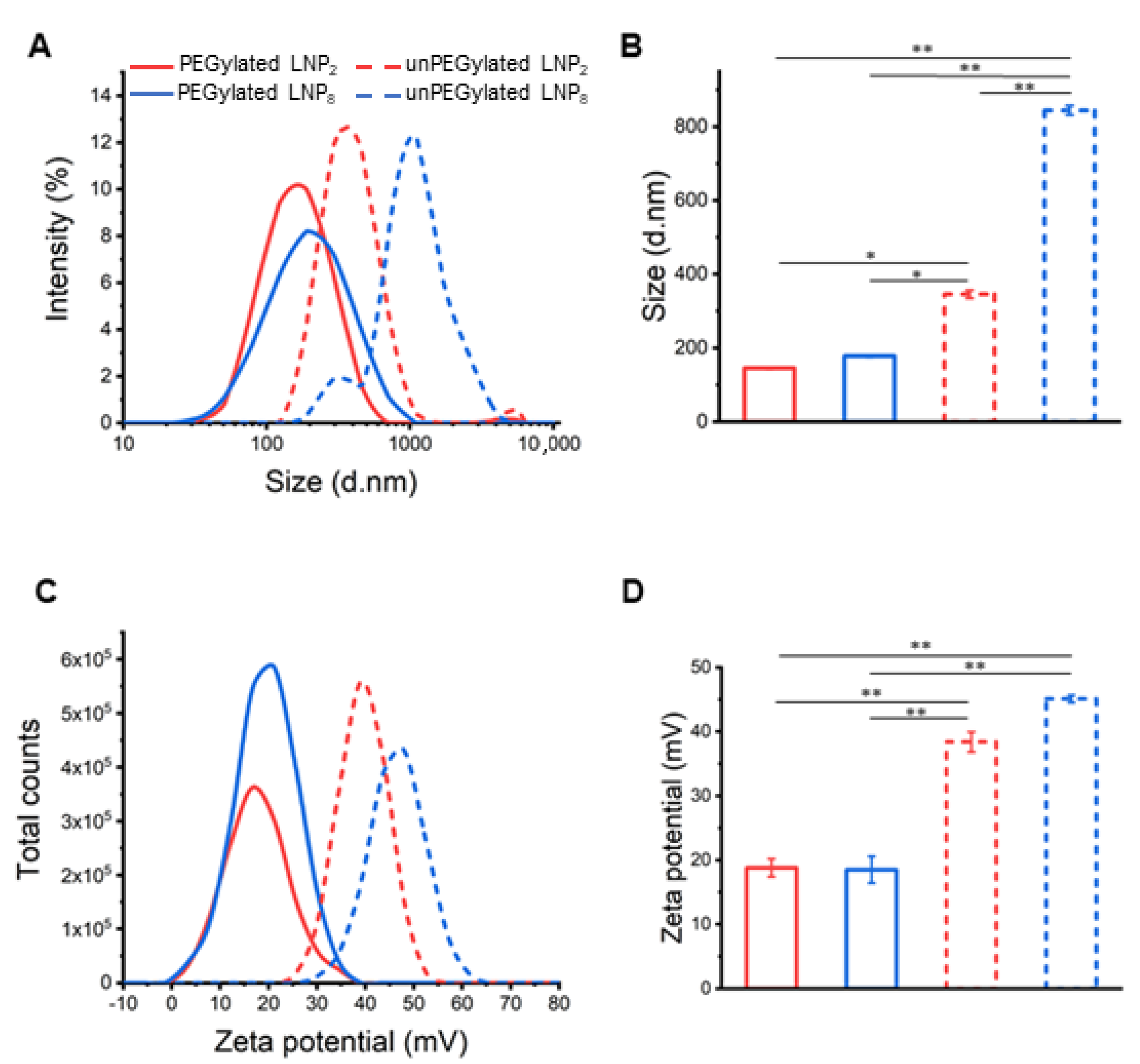
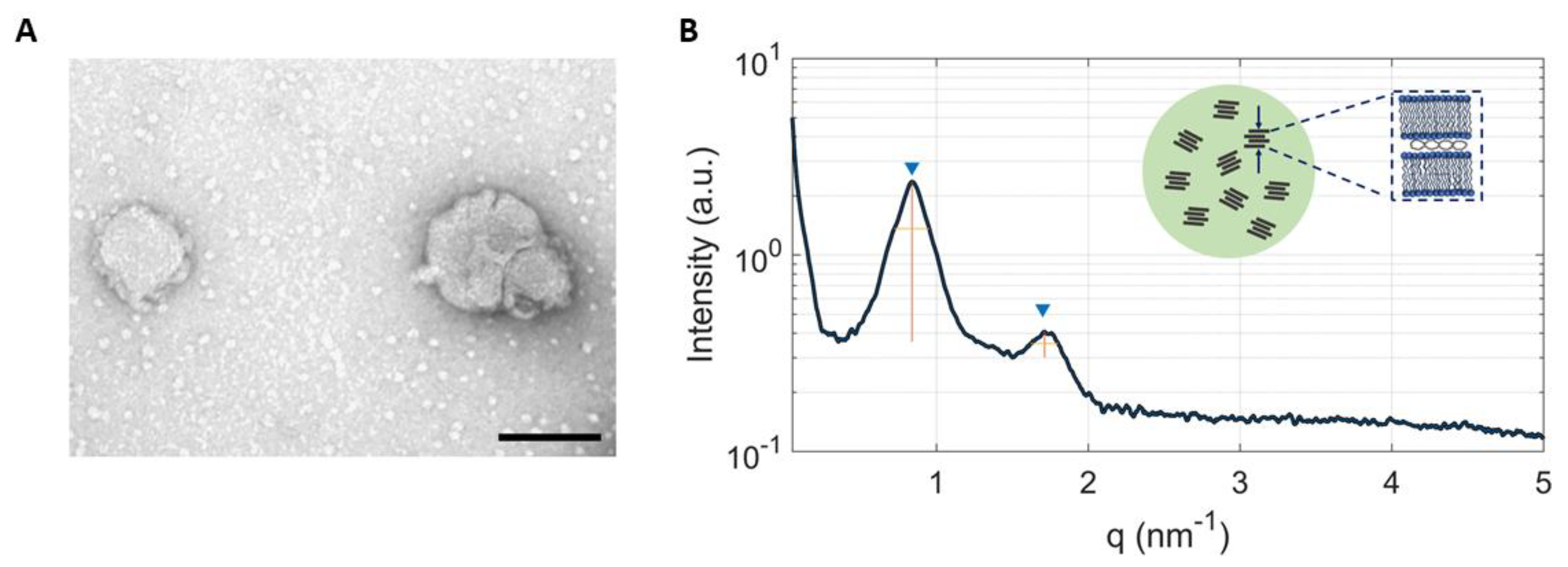
2.1. Physicochemical Characterization of LNPs
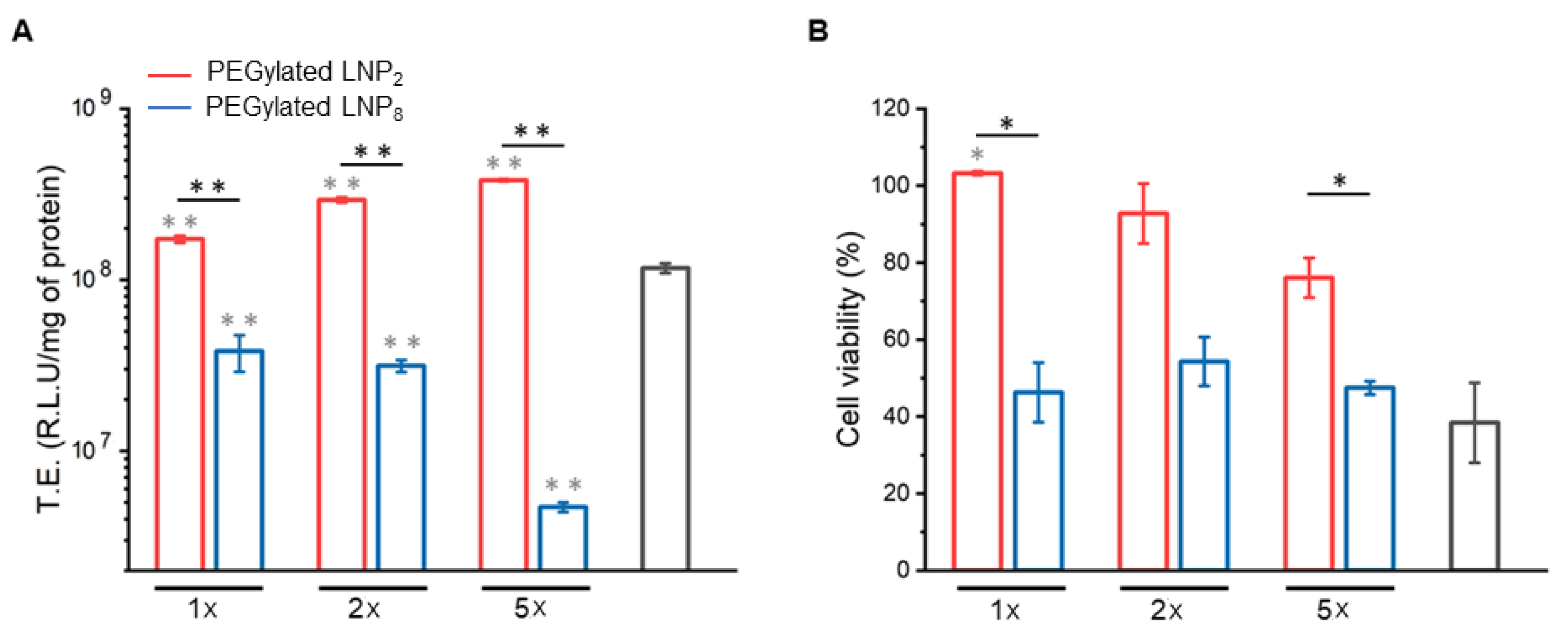
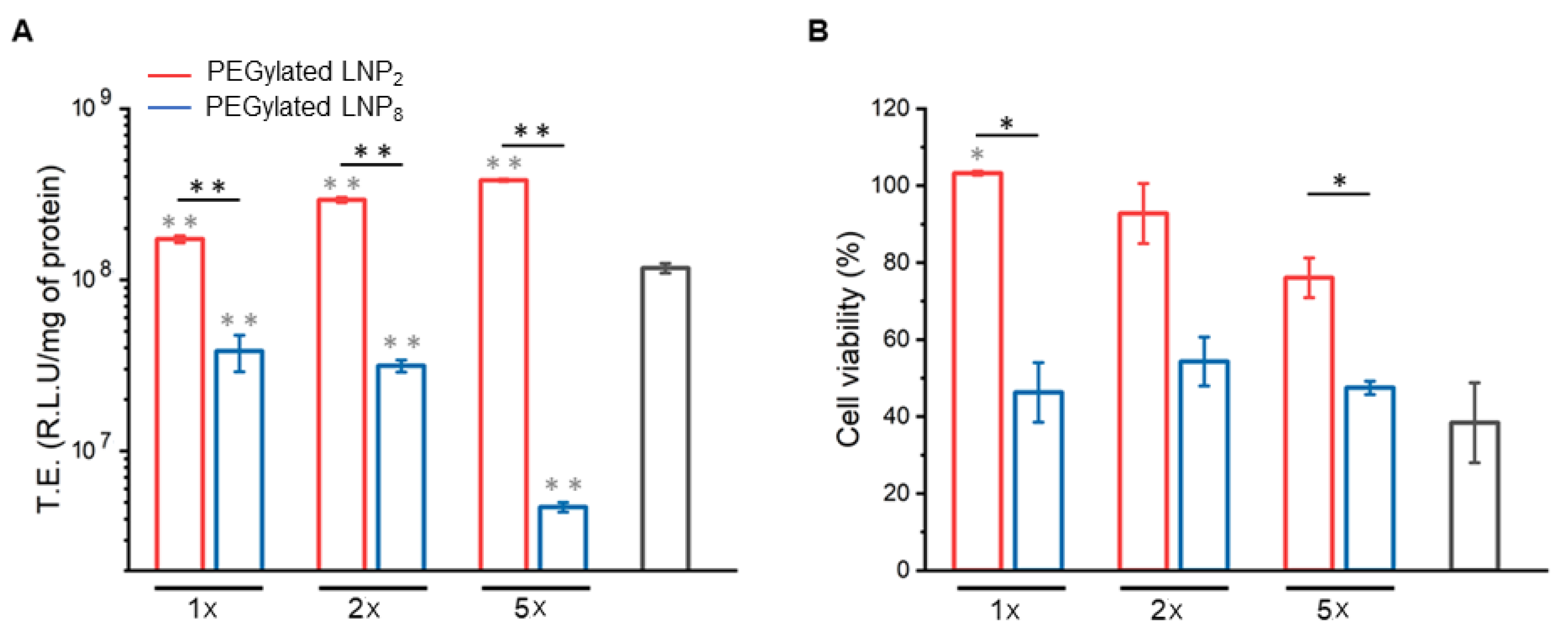
2.2. Cell Transfection and Viability
3. Materials and Methods
3.1. Microfluidic Preparation of Plasmid Containing LNPs
3.2. DLS Characterization of LNPs
3.3. Quantification of Plasmid DNA Loading
3.4. Synchrotron Small Angle X-ray Scattering
3.5. Transfection Efficiency Assay
3.6. Cell Viability Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- MacLachlan, I.; Cullis, P. Diffusible-PEG-Lipid Stabilized Plasmid Lipid Particles. Adv. Genet. 2005, 53, 157–188. [Google Scholar]
- Capecchi, M.R. High efficiency transformation by direct microinjection of DNA into cultured mammalian cells. Cell 1980, 22, 479–488. [Google Scholar] [CrossRef]
- Yin, H.; Kanasty, R.L.; Eltoukhy, A.A.; Vegas, A.J.; Dorkin, J.R.; Anderson, D.G. Non-viral vectors for gene-based therapy. Nat. Rev. Genet. 2014, 15, 541–555. [Google Scholar] [CrossRef]
- Jahn, A.; Vreeland, W.N.; Gaitan, M.; Locascio, L.E. Controlled vesicle self-assembly in microfluidic channels with hydrodynamic focusing. J. Am. Chem. Soc. 2004, 126, 2674–2675. [Google Scholar] [CrossRef] [PubMed]
- Wagner, A.; Vorauer-Uhl, K.; Kreismayr, G.; Katinger, H. The crossflow injection technique: An improvement of the ethanol injection method. J. Liposome Res. 2002, 12, 259–270. [Google Scholar] [CrossRef] [PubMed]
- Sanghani, A.; Kafetzis, K.; Sato, Y.; Elboraie, S.; Fajardo-Sanchez, J.; Harashima, H.; Tagalakis, A.; Yu-Wai-Man, C. Novel PEGylated lipid nanoparticles have a high encapsulation efficiency and effectively deliver MRTF-B siRNA in conjunctival fibroblasts. Pharmaceutics 2021, 13, 382. [Google Scholar] [CrossRef] [PubMed]
- Elouahabi, A.; Ruysschaert, J.-M. Formation and intracellular trafficking of lipoplexes and polyplexes. Mol. Ther. 2005, 11, 336–347. [Google Scholar] [CrossRef] [PubMed]
- Digiacomo, L.; Palchetti, S.; Pozzi, D.; Amici, A.; Caracciolo, G.; Marchini, C. Cationic lipid/DNA complexes manufactured by microfluidics and bulk self-assembly exhibit different transfection behavior. Biochem. Biophys. Res. Commun. 2018, 503, 508–512. [Google Scholar] [CrossRef] [PubMed]
- de la Torre, L.G.; Pessoa, A.C.S.N.; de Carvalho, B.G.; Taketa, T.B.; Eş, I.; Perli, G. Bulk and Microfluidic Synthesis of Stealth and Cationic Liposomes for Gene Delivery Applications. In DNA Vaccines; Humana: New York, NY, USA, 2021; pp. 253–269. [Google Scholar]
- Grimaldi, N.; Andrade, F.; Segovia, N.; Ferrer-Tasies, L.; Sala, S.; Veciana, J.; Ventosa, N. Lipid-based nanovesicles for nanomedicine. Chem. Soc. Rev. 2016, 45, 6520–6545. [Google Scholar] [CrossRef] [Green Version]
- MacLachlan, I. Liposomal formulations for nucleic acid delivery. Antisense drug technology: Principles, strategies, and applications. Antisense Drug Technol. 2007, 2, 237–270. [Google Scholar]
- Evers, M.J.W.; Kulkarni, J.; Van Der Meel, R.; Cullis, P.R.; Vader, P.; Schiffelers, R.M. State-of-the-Art Design and Rapid-Mixing Production Techniques of Lipid Nanoparticles for Nucleic Acid Delivery. Small Methods 2018, 2, 1700375. [Google Scholar] [CrossRef]
- Kimura, N.; Maeki, M.; Sato, Y.; Ishida, A.; Tani, H.; Harashima, H.; Tokeshi, M. Development of a Microfluidic-Based Post-Treatment Process for Size-Controlled Lipid Nanoparticles and Application to siRNA Delivery. ACS Appl. Mater. Interfaces 2020, 12, 34011–34020. [Google Scholar] [CrossRef] [PubMed]
- Kauffman, K.J.; Dorkin, J.R.; Yang, J.H.; Heartlein, M.W.; DeRosa, F.; Mir, F.F.; Fenton, O.S.; Anderson, D.G. Optimization of lipid nanoparticle formulations for mRNA delivery in vivo with fractional factorial and definitive screening designs. Nano Lett. 2015, 15, 7300–7306. [Google Scholar] [CrossRef]
- Pozzi, D.; Marchini, C.; Cardarelli, F.; Rossetta, A.; Colapicchioni, V.; Amici, A.; Montani, M.; Motta, S.; Brocca, P.; Cantù, L.; et al. Mechanistic understanding of gene delivery mediated by highly efficient multicomponent envelope-type nanoparticle systems. Mol. Pharm. 2013, 10, 4654–4665. [Google Scholar] [CrossRef]
- Samaridou, E.; Heyes, J.; Lutwyche, P. Lipid nanoparticles for nucleic acid delivery: Current perspectives. Adv. Drug Deliv. Rev. 2020, 154, 37–63. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, J.; Myhre, J.L.; Chen, S.; Tam, Y.Y.C.; Danescu, A.; Richman, J.; Cullis, P.R. Design of lipid nanoparticles for in vitro and in vivo delivery of plasmid DNA. Nanomed. Nanotechnol. Biol. Med. 2017, 13, 1377–1387. [Google Scholar] [CrossRef]
- Eygeris, Y.; Patel, S.; Jozic, A.; Sahay, G. Deconvoluting lipid nanoparticle structure for messenger RNA delivery. Nano Lett. 2020, 20, 4543–4549. [Google Scholar] [CrossRef]
- Di Santo, R.; Digiacomo, L.; Palchetti, S.; Palmieri, V.; Perini, G.; Pozzi, D.; Papi, M.; Caracciolo, G. Microfluidic manufacturing of surface-functionalized graphene oxide nanoflakes for gene delivery. Nanoscale 2019, 11, 2733–2741. [Google Scholar] [CrossRef]
- Di Santo, R.; Quagliarini, E.; Palchetti, S.; Pozzi, D.; Palmieri, V.; Perini, G.; Papi, M.; Capriotti, A.L.; Laganà, A.; Caracciolo, G. Microfluidic-generated lipid-graphene oxide nanoparticles for gene delivery. Appl. Phys. Lett. 2019, 114, 233701. [Google Scholar] [CrossRef]
- Belliveau, N.; Huft, J.; Lin, P.J.; Chen, S.; Leung, A.K.; Leaver, T.J.; Wild, A.W.; Lee, J.B.; Taylor, R.J.; Tam, Y.K.; et al. Microfluidic synthesis of highly potent limit-size lipid nanoparticles for in vivo delivery of siRNA. Mol. Ther. Nucleic Acids 2012, 1, e37. [Google Scholar] [CrossRef] [PubMed]
- Zhigaltsev, I.V.; Belliveau, N.; Hafez, I.; Leung, A.K.K.; Huft, J.; Hansen, C.; Cullis, P.R. Bottom-up design and synthesis of limit size lipid nanoparticle systems with aqueous and triglyceride cores using millisecond microfluidic mixing. Langmuir 2012, 28, 3633–3640. [Google Scholar] [CrossRef]
- Leung, A.K.K.; Tam, Y.Y.C.; Chen, S.; Hafez, I.M.; Cullis, P.R. Microfluidic mixing: A general method for encapsulating macromolecules in lipid nanoparticle systems. J. Phys. Chem. B 2015, 119, 8698–8706. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Love, K.T.; Chen, Y.; Eltoukhy, A.A.; Kastrup, C.; Sahay, G.; Jeon, A.; Dong, Y.; Whitehead, K.A.; Anderson, D.G. Rapid discovery of potent siRNA-containing lipid nanoparticles enabled by controlled microfluidic formulation. J. Am. Chem. Soc. 2012, 134, 6948–6951. [Google Scholar] [CrossRef] [PubMed]
- Maeki, M.; Fujishima, Y.; Sato, Y.; Yasui, T.; Kaji, N.; Ishida, A.; Tani, H.; Baba, Y.; Harashima, H.; Tokeshi, M. Understanding the formation mechanism of lipid nanoparticles in microfluidic devices with chaotic micromixers. PLoS ONE 2017, 12, e0187962. [Google Scholar] [CrossRef] [PubMed]
- Roces, C.B.; Lou, G.; Jain, N.; Abraham, S.; Thomas, A.; Halbert, G.W.; Perrie, Y. Manufacturing considerations for the development of lipid nanoparticles using microfluidics. Pharmaceutics 2020, 12, 1095. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, J.A.; Darjuan, M.M.; Mercer, J.E.; Chen, S.; van der Meel, R.; Thewalt, J.L.; Tam, Y.Y.C.; Cullis, P.R. On the formation and morphology of lipid nanoparticles containing ionizable cationic lipids and siRNA. ACS Nano 2018, 12, 4787–4795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leung, A.K.K.; Hafez, I.M.; Baoukina, S.; Belliveau, N.; Zhigaltsev, I.V.; Afshinmanesh, E.; Tieleman, D.P.; Hansen, C.L.; Hope, M.J.; Cullis, P.R. Lipid nanoparticles containing siRNA synthesized by microfluidic mixing exhibit an electron-dense nanostructured core. J. Phys. Chem. C 2012, 116, 18440–18450. [Google Scholar] [CrossRef]
- Walsh, C.; Ou, K.; Belliveau, N.M.; Leaver, T.J.; Wild, A.W.; Huft, J.; Lin, P.J.; Chen, S.; Leung, A.K.; Lee, J.B.; et al. Microfluidic-Based Manufacture of siRNA-Lipid Nanoparticles for Therapeutic Applications, in Drug Delivery System; Humana Press: New York, NY, USA, 2014; pp. 109–120. [Google Scholar]
- Zukancic, D.; Suys, E.J.A.; Pilkington, E.H.; Algarni, A.; Al-Wassiti, H.; Truong, N.P. The importance of poly(Ethylene glycol) and lipid structure in targeted gene delivery to lymph nodes by lipid nanoparticles. Pharmaceutics 2020, 12, 1068. [Google Scholar] [CrossRef] [PubMed]
- Kimura, S.; Khalil, I.A.; Elewa, Y.H.; Harashima, H. Novel lipid combination for delivery of plasmid DNA to immune cells in the spleen. J. Control. Release 2021, 330, 753–764. [Google Scholar] [CrossRef]
- Marchini, C.; Pozzi, D.; Montani, M.; Alfonsi, C.; Amici, A.; Amenitsch, H.; De Sanctis, S.C.; Caracciolo, G. Tailoring lipoplex composition to the lipid composition of plasma membrane: A Trojan horse for cell entry? Langmuir 2010, 26, 13867–13873. [Google Scholar] [CrossRef]
- Caracciolo, G.; Pozzi, D.; Caminiti, R.; Marchini, C.; Montani, M.; Amici, A.; Amenitsch, H. Enhanced transfection efficiency of multicomponent lipoplexes in the regime of optimal membrane charge density. J. Phys. Chem. B 2008, 112, 11298–11304. [Google Scholar] [CrossRef]
- Shepherd, S.J.; Issadore, D.; Mitchell, M.J. Microfluidic formulation of nanoparticles for biomedical applications. Biomaterials 2021, 274, 120826. [Google Scholar] [CrossRef] [PubMed]
- Cardarelli, F.; Digiacomo, L.; Marchini, C.; Amici, A.; Salomone, F.; Fiume, G.; Rossetta, A.; Gratton, E.; Pozzi, D.; Caracciolo, G. The intracellular trafficking mechanism of Lipofectamine-based transfection reagents and its implication for gene delivery. Sci. Rep. 2016, 6, 25879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caracciolo, G.; Pozzi, D.; Caminiti, R.; Marchini, C.; Montani, M.; Amici, A.; Amenitsch, H. DNA release from cationic liposome/DNA complexes by anionic lipids. Appl. Phys. Lett. 2006, 89, 233903. [Google Scholar] [CrossRef]
- Caracciolo, G.; Caminiti, R.; Digman, M.A.; Gratton, E.; Sanchez, S. Efficient escape from endosomes determines the superior efficiency of multicomponent lipoplexes. J. Phys. Chem. B 2009, 113, 495–497. [Google Scholar] [CrossRef] [Green Version]
- Caracciolo, G.; Amenitsch, H. Cationic liposome/DNA complexes: From structure to interactions with cellular membranes. Eur. Biophys. J. 2012, 41, 815–829. [Google Scholar] [CrossRef]
- Pan, J.; Heberle, F.; Tristram-Nagle, S.; Szymanski, M.; Koepfinger, M.; Katsaras, J.; Kucerka, N. Molecular structures of fluid phase phosphatidylglycerol bilayers as determined by small angle neutron and X-ray scattering. Biochim. Biophys. Acta (BBA) Biomembr. 2012, 1818, 2135–2148. [Google Scholar] [CrossRef]
- Song, Y.; Hormes, J.; Kumar, C.S.S.R. Microfluidic Synthesis of Nanomaterials. Small 2008, 4, 698–711. [Google Scholar] [CrossRef]
- Prabha, S.; Zhou, W.-Z.; Panyam, J.; Labhasetwar, V. Size-dependency of nanoparticle-mediated gene transfection: Studies with fractionated nanoparticles. Int. J. Pharm. 2002, 244, 105–115. [Google Scholar] [CrossRef]
- Harashima, H.; Hiraiwa, T.; Ochi, Y.; Kiwada, H. Size Dependent Liposome Degradation in Blood: In vivo/In vitro Correlation by Kinetic Modeling. J. Drug Target. 1995, 3, 253–261. [Google Scholar] [CrossRef]
- Milton, H.J.; Martin, N.E.; Modi, M. PEGylation: A novel process for modifying pharmacokinetics. Clin. Pharm. 2001, 40, 539–551. [Google Scholar]
- Rapaport, H.; Kuzmenko, I.; Lafont, S.; Kjaer, K.; Howes, P.B.; Als-Nielsen, J.; Lahav, M.; Leiserowitz, L. Cholesterol Monohydrate Nucleation in Ultrathin Films on Water. Biophys. J. 2001, 81, 2729–2736. [Google Scholar] [CrossRef] [Green Version]
- Pozzi, D.; Caracciolo, G.; Caminiti, R.; De Sanctis, S.C.; Amenitsch, H.; Marchini, C.; Montani, M.; Amici, A. Toward the Rational Design of Lipid Gene Vectors: Shape Coupling between Lipoplex and Anionic Cellular Lipids Controls the Phase Evolution of Lipoplexes and the Efficiency of DNA Release. ACS Appl. Mater. Interfaces 2009, 1, 2237–2249. [Google Scholar] [CrossRef] [PubMed]
- Stepanenko, A.; Dmitrenko, V. HEK293 in cell biology and cancer research: Phenotype, karyotype, tumorigenicity, and stress-induced genome-phenotype evolution. Gene 2015, 569, 182–190. [Google Scholar] [CrossRef]
- Hudecova, S.; Lencesova, L.; Csaderova, L.; Sirova, M.; Cholujova, D.; Cagala, M. Chemically mimicked hypoxia modulates gene expression and protein levels of the sodium calcium exchanger in HEK 293 cell line via HIF-1α. Gen. Physiol. Biophys. 2011, 30, 196–206. [Google Scholar] [CrossRef]
- Le Ru, A.; Jacob, D.; Transfiguracion, J.; Ansorge, S.; Henry, O.; Kamen, A.A. Scalable production of influenza virus in HEK-293 cells for efficient vaccine manufacturing. Vaccine 2010, 28, 3661–3671. [Google Scholar] [CrossRef]
- Liu, X.; Shan, K.; Shao, X.; Shi, X.; He, Y.; Liu, Z.; Jacob, J.A.; Deng, L. Nanotoxic Effects of Silver Nanoparticles on Normal HEK-293 Cells in Comparison to Cancerous HeLa Cell Line. Int. J. Nanomed. 2021, 16, 753–761. [Google Scholar] [CrossRef]
- Thomas, P.; Smart, T.G. HEK293 cell line: A vehicle for the expression of recombinant proteins. J. Pharmacol. Toxicol. Methods 2005, 51, 187–200. [Google Scholar] [CrossRef]
- Palchetti, S.; Pozzi, D.; Marchini, C.; Amici, A.; Andreani, C.; Bartolacci, C.; Digiacomo, L.; Gambini, V.; Cardarelli, F.; Di Rienzo, C.; et al. Manipulation of lipoplex concentration at the cell surface boosts transfection efficiency in hard-to-transfect cells. Nanomed. Nanotechnol. Biol. Med. 2017, 13, 681–691. [Google Scholar] [CrossRef]
- Boukamp, P.; Petrussevska, R.T.; Breitkreutz, D.; Hornung, J.; Markham, A.; Fusenig, N.E. Normal keratinization in a spontaneously immortalized aneuploid human keratinocyte cell line. J. Cell Biol. 1988, 106, 761–771. [Google Scholar] [CrossRef] [Green Version]
- Carter, T.H.; Liu, K.; Ralph, W.; Chen, D.; Qi, M.; Fan, S.; Yuan, F.; Rosen, E.M.; Auborn, K.J. Diindolylmethane Alters Gene Expression in Human Keratinocytes In Vitro. J. Nutr. 2002, 132, 3314–3324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haider, R.; Sartori, B.; Radeticchio, A.; Wolf, M.; Dal Zilio, S.; Marmiroli, B.; Amenitsch, H. µDrop: A system for high-throughput small-angle X-ray scattering measurements of microlitre samples. J. Appl. Crystallogr. 2021, 54, 132–141. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Quagliarini, E.; Renzi, S.; Digiacomo, L.; Giulimondi, F.; Sartori, B.; Amenitsch, H.; Tassinari, V.; Masuelli, L.; Bei, R.; Cui, L.; et al. Microfluidic Formulation of DNA-Loaded Multicomponent Lipid Nanoparticles for Gene Delivery. Pharmaceutics 2021, 13, 1292. https://doi.org/10.3390/pharmaceutics13081292
Quagliarini E, Renzi S, Digiacomo L, Giulimondi F, Sartori B, Amenitsch H, Tassinari V, Masuelli L, Bei R, Cui L, et al. Microfluidic Formulation of DNA-Loaded Multicomponent Lipid Nanoparticles for Gene Delivery. Pharmaceutics. 2021; 13(8):1292. https://doi.org/10.3390/pharmaceutics13081292
Chicago/Turabian StyleQuagliarini, Erica, Serena Renzi, Luca Digiacomo, Francesca Giulimondi, Barbara Sartori, Heinz Amenitsch, Valentina Tassinari, Laura Masuelli, Roberto Bei, Lishan Cui, and et al. 2021. "Microfluidic Formulation of DNA-Loaded Multicomponent Lipid Nanoparticles for Gene Delivery" Pharmaceutics 13, no. 8: 1292. https://doi.org/10.3390/pharmaceutics13081292
APA StyleQuagliarini, E., Renzi, S., Digiacomo, L., Giulimondi, F., Sartori, B., Amenitsch, H., Tassinari, V., Masuelli, L., Bei, R., Cui, L., Wang, J., Amici, A., Marchini, C., Pozzi, D., & Caracciolo, G. (2021). Microfluidic Formulation of DNA-Loaded Multicomponent Lipid Nanoparticles for Gene Delivery. Pharmaceutics, 13(8), 1292. https://doi.org/10.3390/pharmaceutics13081292