
Nanoparticulate Drug Delivery Strategies to Address Intestinal Cytochrome P450 CYP3A4 Metabolism towards Personalized Medicine




Abstract
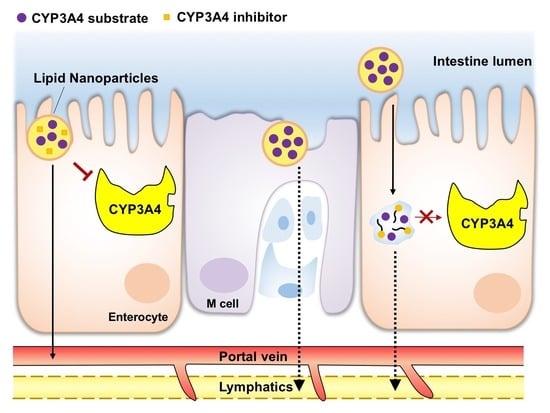
1. Introduction
2. Intestinal CYP3A4 as a Critical GIT Barrier
2.1. Contribution of Gut Wall to Drug Metabolism
2.2. Drug Metabolism Variation by Enterocytic CYP3A4
3. Select Compounds Involving CYP3A4 Interaction and Their Classification
4. LNS Strategies of Overcoming Pre-Systemic CYP3A4 Metabolism
4.1. Local Saturation or Inhibition of Enterocytic CYP3A4 Activity at Proximal GIT
4.2. Minimizing CYP3A4 Drug Metabolism via Intestinal Lymphatic Drug Transport
4.3. Targeting Distal GIT to Exploit the Least CYP3A4 Activity
5. Translating LNS for Personalized Medicine
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| API | Active pharmaceutical ingredient |
| AUC | Area under the plasma (blood) curves |
| BCS | Biopharmaceutics Classification System |
| BDDCS | Biopharmaceutics Drug Disposition Classification System |
| CAM | Complementary alternative medicine |
| CRASD | Controlled release amorphous solid dispersion systems |
| CYPs | Cytochrome P450 |
| Cmax | Maximum (peak) drug concentration in the plasma, serum or whole blood |
| ER | Endoplasmic reticulum |
| F | the extent of drug bioavailability |
| GIT | Gastrointestinal tract |
| HMG-CoA | Hydroxyl-methyl-glutaryl coenzyme A |
| LNS | Lipid-based nanosystems |
| Microfold cells | M cells |
| NP(s) | Nanoparticle(s) |
| NLC | Nanostructured lipid carriers |
| NPs | nanoparticles |
| PK | pharmacokinetics |
| PLN | Polymer-lipid hybrid nanoparticles |
| PXR | Pregnane X receptor |
| SLN | Solid lipid nanoparticles |
| SJW | St. John wort |
| SNEDDS | Self-nanoemulsifying drug delivery systems |
References
- Coleman, J.J. Prescribing in 2019: What are the safety concerns? Expert Opin. Drug Saf. 2019, 18, 69–74. [Google Scholar] [CrossRef]
- Pang, K.S.; Rodrigues, A.D.; Peter, R.M. Enzyme- and Transporter-Based Drug–Drug Interactions: Progress and Future Challenges, 1st ed.; Springer Science Business Media LLC: New York, NY, USA, 2010. [Google Scholar]
- Polasek, T.M.; Shakib, S.; Rostami-Hodjegan, A. Precision dosing in clinical medicine: Present and future. Expert Rev. Clin. Pharmacol. 2018, 11, 743–746. [Google Scholar] [CrossRef] [PubMed]
- WHO. Global Patient Safety Challenge: Medication Without Harm; World Health Organisation: Geneva, Switzerland, 2017; Available online: https://www.who.int/initiatives/medication-without-harm (accessed on 11 May 2021).
- Homayun, B.; Lin, X.; Choi, H.-J. Challenges and Recent Progress in Oral Drug Delivery Systems for Biopharmaceuticals. Pharmaceutics 2019, 11, 129. [Google Scholar] [CrossRef]
- Traverso, G.; Langer, R. Perspective: Special delivery for the gut. Nature 2015, 519, S19. [Google Scholar] [CrossRef] [PubMed]
- Vinarov, Z.; Abdallah, M.; Agundez, J.A.G.; Allegaert, K.; Basit, A.W.; Braeckmans, M.; Ceulemans, J.; Corsetti, M.; Griffin, B.T.; Grimm, M.; et al. Impact of gastroin-testinal tract variability on oral drug absorption and pharmacokinetics: An UNGAP review. Eur. J. Pharm. Sci. 2021, 162, 105812–105845. [Google Scholar] [CrossRef]
- Uetrecht, J.P.; Trager, W. Drug Metabolism: Chemical and Enzymatic Aspects; CRC Press: Boca Raton, FL, USA, 2007. [Google Scholar]
- Grimsrud, K.N.; Sherwin, C.M.T.; Constance, J.E.; Tak, C.; Zuppa, A.F.; Spigarelli, M.G.; Mihalopoulos, N.L. Special population considerations and regulatory affairs for clinical research. Clin. Res. Regul. Aff. 2015, 32, 45–54. [Google Scholar] [CrossRef]
- Gamboa, J.M.; Leong, K.W. In vitro and in vivo models for the study of oral delivery of nanoparticles. Adv. Drug Deliv. Rev. 2013, 65, 800–810. [Google Scholar] [CrossRef]
- Galipeau, H.; Verdu, E.F. The complex task of measuring intestinal permeability in basic and clinical science. Neurogastroenterol. Motil. 2016, 28, 957–965. [Google Scholar] [CrossRef]
- Nebert, D.W.; Russell, D. Clinical importance of the cytochromes P450. Lancet 2002, 360, 1155–1162. [Google Scholar] [CrossRef]
- Zanger, U.M.; Schwab, M. Cytochrome P450 enzymes in drug metabolism: Regulation of gene expression, enzyme activities, and impact of genetic variation. Pharmacol. Ther. 2013, 138, 103–141. [Google Scholar] [CrossRef] [PubMed]
- Morgan, E.T.; Goralski, K.B.; Piquette-Miller, M.; Renton, K.W.; Robertson, G.R.; Chaluvadi, M.R.; Charles, K.A.; Clarke, S.J.; Kacevska, M.; Liddle, C.; et al. Regulation of drug-metabolizing enzymes and trans-porters in infection, inflammation, and cancer. Drug Metab. Dispos. 2008, 36, 205–216. [Google Scholar] [CrossRef]
- Harris, R.Z.; Jang, G.R.; Tsunoda, S. Dietary Effects on Drug Metabolism and Transport. Clin. Pharmacokinet. 2003, 42, 1071–1088. [Google Scholar] [CrossRef] [PubMed]
- Lammers, L.A.; Achterbergh, R.; Romijn, J.A.; Mathôt, R.A.A. Nutritional Status Differentially Alters Cytochrome P450 3A4 (CYP3A4) and Uridine 5′-Diphospho-Glucuronosyltransferase (UGT) Mediated Drug Metabolism: Effect of Short-Term Fasting and High Fat Diet on Midazolam Metabolism. Eur. J. Drug Metab. Pharmacokinet. 2018, 43, 751–767. [Google Scholar] [CrossRef]
- Dresser, G.K.; Spence, J.D.; Bailey, D.G. Pharmacokinetic-pharmacodynamic consequences and clinical relevance of cyto-chrome p450 3a4 inhibition. Clin. Pharmacokinet. 2000, 38, 41–57. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.A.; Di, L.; Kerns, E.H. The effect of plasma protein binding on in vivo efficacy: Misconceptions in drug discovery. Nat. Rev. Drug Discov. 2010, 9, 929–939. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Wong, H. Food effects on oral drug absorption: Application of physiologically-based pharmacokinetic modeling as a predictive tool. Pharmaceutics 2020, 12, 672. [Google Scholar] [CrossRef] [PubMed]
- Custodio, J.M.; Wu, C.-Y.; Benet, L.Z. Predicting drug disposition, absorption/elimination/transporter interplay and the role of food on drug absorption. Adv. Drug Deliv. Rev. 2008, 60, 717–733. [Google Scholar] [CrossRef]
- Singh, B.N. Effects of Food on Clinical Pharmacokinetics. Clin. Pharmacokinet. 1999, 37, 213–255. [Google Scholar] [CrossRef]
- O’Shea, J.P.; Holm, R.; O’Driscoll, C.M.; Griffin, B.T. Food for thought: Formulating away the food effect—A PEARRL review. J. Pharm. Pharmacol. 2018, 71, 510–535. [Google Scholar] [CrossRef]
- Huizinga, J.D.; Chen, J.-H.; Zhu, Y.F.; Pawelka, A.J.; McGinn, R.; Bardakjian, B.L.; Parsons, S.; Kunze, W.A.; Wu, R.Y.; Bercik, P.; et al. The origin of segmentation motor activity in the intestine. Nat. Commun. 2014, 5, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Food-Effect Bioavailability and Fed Bioequivalence Studies. Available online: http://www.fda.gov/cder/guidance/index.htm (accessed on 3 May 2021).
- Dowden, H.; Munro, J. Trends in clinical success rates and therapeutic focus. Nat. Rev. Drug Discov. 2019, 18, 495–496. [Google Scholar] [CrossRef]
- Zhang, D.; Hop, C.E.; Patilea-Vrana, G.; Gampa, G.; Seneviratne, H.; Unadkat, J.D.; Kenny, J.R.; Nagapudi, K.; Di, L.; Zhou, L.; et al. Drug Concentration Asymmetry in Tissues and Plasma for Small Molecule–Related Therapeutic Modalities. Drug Metab. Dispos. 2019, 47, 1122–1135. [Google Scholar] [CrossRef]
- Drug Bioavailability. Available online: https://www.merckmanuals.com/professional/clinical-pharmacology/pharmacokinetics/drug-bioavailability (accessed on 7 June 2021).
- Mueller, E.A.; Jm, K.; Kutz, K. Minor influence of a fat-rich meal on the pharmacokinetics of a new oral formulation of cy-closporine. Transpl. Proc. 1994, 26, 2957–2958. [Google Scholar]
- Mueller, E.A.; Kovarik, J.M.; van Bree, J.B.; Grevel, J.; Lucker, P.W.; Kutz, K. Influence of a fat-rich meal on the pharmacokinetics of a new oral for-mulation of cyclosporine in a crossover comparison with the market formulation. Pharm. Res. 1994, 11, 151–155. [Google Scholar] [CrossRef]
- Nanjwade, B.K.; Patel, D.J.; Udhani, R.A.; Manvi, F.V. Functions of Lipids for Enhancement of Oral Bioavailability of Poorly Water-Soluble Drugs. Sci. Pharm. 2011, 79, 705–727. [Google Scholar] [CrossRef]
- Humberstone, A.J.; Charman, W.N. Lipid-based vehicles for the oral delivery of poorly water-soluble drugs. Adv. Drug Deliv. Rev. 1997, 25, 103–128. [Google Scholar] [CrossRef]
- Poovi, G.; Damodharan, N. Lipid nanoparticles: A challenging approach for oral delivery of BCS Class-II drugs. Futur. J. Pharm. Sci. 2018, 4, 191–205. [Google Scholar] [CrossRef]
- Ganesan, P.; Narayanasamy, D. Lipid nanoparticles: Different preparation techniques, characterization, hurdles, and strategies for the production of solid lipid nanoparticles and nanostructured lipid carriers for oral drug delivery. Sustain. Chem. Pharm. 2017, 6, 37–56. [Google Scholar] [CrossRef]
- He, H.; Lu, Y.; Qi, J.; Zhu, Q.; Chen, Z.; Wu, W. Adapting liposomes for oral drug delivery. Acta Pharm. Sin. B 2018, 9, 36–48. [Google Scholar] [CrossRef] [PubMed]
- Cherniakov, I.; Domb, A.J.; Hoffman, A. Self-nano-emulsifying drug delivery systems: An update of the biopharmaceutical aspects. Expert Opin. Drug Deliv. 2015, 12, 1121–1133. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.X.; Ahmed, T.; Li, L.Y.; Li, J.; Abbasi, A.Z.; Wu, X.Y. Design of nanocarriers for nanoscale drug delivery to enhance cancer treatment using hybrid polymer and lipid building blocks. Nanoscale 2016, 9, 1334–1355. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.X.; Wong, H.L.; Xue, H.Y.; Eoh, J.Y.; Wu, X.Y. Nanomedicine of synergistic drug combinations for cancer therapy – Strategies and perspectives. J. Control. Release 2016, 240, 489–503. [Google Scholar] [CrossRef] [PubMed]
- Rezhdo, O.; Speciner, L.; Carrier, R. Lipid-associated oral delivery: Mechanisms and analysis of oral absorption enhancement. J. Control. Release 2016, 240, 544–560. [Google Scholar] [CrossRef] [PubMed]
- Porter, C.J.H.; Trevaskis, N.L.; Charman, W.N. Lipids and lipid-based formulations: Optimizing the oral delivery of lipophilic drugs. Nat. Rev. Drug Discov. 2007, 6, 231–248. [Google Scholar] [CrossRef]
- Patel, R.; Barker, J.; ElShaer, A. Pharmaceutical Excipients and Drug Metabolism: A Mini-Review. Int. J. Mol. Sci. 2020, 21, 8224. [Google Scholar] [CrossRef]
- Ren, X.; Mao, X.; Cao, L.; Xue, K.; Si, L.; Qiu, J.; Schimmer, A.D.; Li, G. Nonionic surfactants are strong inhibitors of cytochrome P450 3A biotransformation activity in vitro and in vivo. Eur. J. Pharm. Sci. 2009, 36, 401–411. [Google Scholar] [CrossRef]
- Ren, X.; Mao, X.; Si, L.; Cao, L.; Xiong, H.; Qiu, J.; Schimmer, A.; Li, G. Pharmaceutical excipients inhibit cytochrome P450 activity in cell free systems and after systemic administration. Eur. J. Pharm. Biopharm. 2008, 70, 279–288. [Google Scholar] [CrossRef]
- Xu, Y.; Shrestha, N.; Préat, V.; Beloqui, A. Overcoming the intestinal barrier: A look into targeting approaches for improved oral drug delivery systems. J. Control. Release 2020, 322, 486–508. [Google Scholar] [CrossRef]
- The Human Protein ATLAS: CYP3A4. Available online: https://www.proteinatlas.org/ENSG00000160868-CYP3A4/tissue (accessed on 1 July 2021).
- Thummel, K.E. Gut instincts: CYP3A4 and intestinal drug metabolism. J. Clin. Investig. 2007, 117, 3173–3176. [Google Scholar] [CrossRef]
- Xie, F.; Ding, X.; Zhang, Q.-Y. An update on the role of intestinal cytochrome P450 enzymes in drug disposition. Acta Pharm. Sin. B 2016, 6, 374–383. [Google Scholar] [CrossRef]
- Von Richter, O.; Burk, O.; Fromm, M.F.; Thon, K.P.; Eichelbaum, M.; Kivistö, K.T. Cytochrome P450 3A4 and P-glycoprotein expression in human small intestinal enterocytes and hepatocytes: A comparative analysis in paired tissue specimens. Clin. Pharmacol. Ther. 2004, 75, 172–183. [Google Scholar] [CrossRef] [PubMed]
- Dresser, G.K.; Bailey, D.G. A basic conceptual and practical overview of interactions with highly prescribed drugs. Can. J. Clin. Pharmacol. 2002, 9, 191–198. [Google Scholar]
- Van Herwaarden, A.E.; Wagenaar, E.; van der Kruijssen, C.M.M.; van Waterschoot, R.A.B.; Smit, J.W.; Song, J.-Y.; van der Valk, M.A.; van Tellingen, O.; van der Hoorn, J.W.A.; Rosing, H.; et al. Knockout of cytochrome P450 3A yields new mouse models for understanding xenobiotic metabolism. J. Clin. Investig. 2007, 117, 3583–3592. [Google Scholar] [CrossRef] [PubMed]
- Lown, K.S.; Kolars, J.C.; Thummel, K.E.; Barnett, J.L.; Kunze, K.L.; Wrighton, S.A.; Watkins, P.B. Interpatient heterogeneity in expression of CYP3A4 and CYP3A5 in small bowel. Lack of prediction by the erythromycin breath test. Drug Metab. Dispos. 1994, 22. [Google Scholar]
- Ekins, S.; Stresser, D.M.; Williams, J.A. In vitro and pharmacophore insights into CYP3A enzymes. Trends Pharmacol. Sci. 2003, 24, 161–166. [Google Scholar] [CrossRef]
- Racha, J.K.; Zhao, Z.S.; Olejnik, N.; Warner, N.; Chan, R.; Moore, D.; Satoh, H. Substrate dependent inhibition profiles of fourteen drugs on CYP3A4 activity measured by a high throughput LC-MS/MS method with four probe drugs, midazolam, testosterone, nifedipine and terfenadine. Drug Metab. Pharmacokinet. 2003, 18, 128–138. [Google Scholar] [CrossRef] [PubMed]
- Paine, M.F.; Khalighi, M.; Fisher, J.M.; Shen, D.D.; Kunze, K.L.; Marsh, C.; Perkins, J.D.; Thummel, K.E. Characterization of interintestinal and intraintestinal variations in human CYP3A-dependent metabolism. J. Pharmacol. Expert Ther. 1997, 283, 1552–1562. [Google Scholar]
- Urquhart, B.L.; Tirona, R.G.; Kim, R.B. Nuclear Receptors and the Regulation of Drug-Metabolizing Enzymes and Drug Transporters: Implications for Interindividual Variability in Response to Drugs. J. Clin. Pharmacol. 2007, 47, 566–578. [Google Scholar] [CrossRef]
- Brewer, C.T.; Chen, T. PXR variants: The impact on drug metabolism and therapeutic responses. Acta Pharm. Sin. B 2016, 6, 441–449. [Google Scholar] [CrossRef]
- Fritz, A.; Busch, D.; Lapczuk, J.; Ostrowski, M.; Drozdzik, M.; Oswald, S. Expression of clinically relevant drug-metabolizing enzymes along the human intestine and their correlation to drug transporters and nuclear receptors: An intra-subject analysis. Basic Clin. Pharmacol. Toxicol. 2018, 124, 245–255. [Google Scholar] [CrossRef]
- Xing, Y.; Yan, J.; Niu, Y. PXR: A center of transcriptional regulation in cancer. Acta Pharm. Sin. B 2019, 10, 197–206. [Google Scholar] [CrossRef]
- Renton, K.W. Cytochrome P450 regulation and drug biotransformation during inflammation and infection. Curr. Drug Metab. 2004, 5, 235–243. [Google Scholar] [CrossRef] [PubMed]
- He, P.; Court, M.H.; Greenblatt, D.J.; Von Moltke, L.L. Factors influencing midazolam hydroxylation activity in human liver microsomes. Drug Metab. Dispos. 2006, 34, 1198–1207. [Google Scholar] [CrossRef]
- Philip, H.; John, H. Top 100 Drug Interactions 2015: A Guide to Patient Management; American College of Clinical Pharmacy: Lenexa, KS, USA, 2015; p. 188. [Google Scholar]
- Benet, L.Z.; Broccatelli, F.; Oprea, T. BDDCS Applied to Over 900 Drugs. AAPS J. 2011, 13, 519–547. [Google Scholar] [CrossRef] [PubMed]
- Australian Government. Australian Public Assessment Report for Vardenafil; Australian Government: Canberra, Australia, 2011. [Google Scholar]
- Basalious, E.B.; El-Sebaie, W.; El-Gazayerly, O. Rapidly absorbed orodispersible tablet containing molecularly dispersed fe-lodipine for management of hypertensive crisis: Development, optimization and in vitro/in vivo studies. Pharm. Dev. Technol. 2013, 18, 407–416. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.-S.; Park, J.-S. Design of PVP/VA S-630 based tadalafil solid dispersion to enhance the dissolution rate. Eur. J. Pharm. Sci. 2017, 97, 269–276. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Han, N.; Zhao, B.; Xie, Y.; Wang, S. Enhanced dissolution rate and oral bioavailability of simvastatin nanocrystal prepared by sonoprecipitation. Drug Dev. Ind. Pharm. 2012, 38, 1230–1239. [Google Scholar] [CrossRef]
- Kristin, F.; Rene, H.; Boontida, M.; Buraphacheep, J.V.; Maximilian, A.; Johanna, M.; Peter, L. Dissolution and dissolu-tion/permeation experiments for predicting systemic exposure following oral administration of the BCS class II drug clar-ithromycin. Eur. J. Pharm. Sci. 2017, 101, 211–219. [Google Scholar] [CrossRef]
- Lindenberg, M.; Kopp, S.; Dressman, J.B. Classification of orally administered drugs on the World Health Organization Model list of Essential Medicines according to the biopharmaceutics classification system. Eur. J. Pharm. Biopharm. 2004, 58, 265–278. [Google Scholar] [CrossRef]
- Liu, D.; Li, L.Z.; Rostami-Hodjegan, A.; Bois, F.Y.; Jamei, M. Considerations and Caveats when Applying Global Sensitivity Analysis Methods to Physiologically Based Pharmacokinetic Models. AAPS J. 2020, 22, 1–13. [Google Scholar] [CrossRef]
- Miranda, C.; Perez-Rodriguez, Z.; Hernández-Armengol, R.; Quiñones-García, Y.; Betancourt-Purón, T.; Cabrera-Pérez, M. Biowaiver or Bioequivalence: Ambiguity in Sildenafil Citrate BCS Classification. AAPS PharmSciTech 2018, 19, 1693–1698. [Google Scholar] [CrossRef]
- Nader, A.M.; Quinney, S.K.; Fadda, H.M.; Foster, D.R. Effect of Gastric Fluid Volume on the In Vitro Dissolution and In Vivo Absorption of BCS Class II Drugs: A Case Study with Nifedipine. AAPS J. 2016, 18, 981–988. [Google Scholar] [CrossRef]
- Wu, C.-Y.; Benet, L.Z. Predicting Drug Disposition via Application of BCS: Transport/Absorption/ Elimination Interplay and Development of a Biopharmaceutics Drug Disposition Classification System. Pharm. Res. 2005, 22, 11–23. [Google Scholar] [CrossRef] [PubMed]
- Drug Development and Drug Interactions: Table of Substrates, Inhibitors and Inducers. Available online: https://www.fda.gov/drugs/drug-interactions-labeling/drug-development-and-drug-interactions-table-substrates-inhibitors-and-inducers (accessed on 3 October 2020).
- De Castro, W.V.; Mertens-Talcott, S.; Rubner, A.; Butterweck, V.; Derendorf, H. Variation of Flavonoids and Furanocoumarins in Grapefruit Juices: A Potential Source of Variability in Grapefruit Juice−Drug Interaction Studies. J. Agric. Food Chem. 2006, 54, 249–255. [Google Scholar] [CrossRef] [PubMed]
- Hanley, M.J.; Cancalon, P.; Widmer, W.W.; Greenblatt, D.J. The effect of grapefruit juice on drug disposition. Expert Opin. Drug Metab. Toxicol. 2011, 7, 267–286. [Google Scholar] [CrossRef]
- Edwards, D.J.; Fitzsimmons, M.E.; Schuetz, E.G.; Yasuda, K.; Ducharme, M.P.; Warbasse, L.H.; Woster, P.M.; Schuetz, J.D.; Watkins, P. 6′,7′-dihydroxybergamottin in grapefruit juice and seville orange juice: Effects on cyclosporine disposition, en-terocyte CYP3A4, and P-glycoprotein. Clin. Pharmacol. Ther. 1999, 65, 237–244. [Google Scholar] [CrossRef]
- Penzak, S.R.; Acosta, E.P.; Turner, M.; Edwards, D.J.; Hon, Y.Y.; Desai, H.D.; Jann, M.W. Effect of Seville orange juice and grapefruit juice on indinavir pharmacokinetics. J. Clin. Pharmacol. 2002, 42, 1165–1170. [Google Scholar] [CrossRef] [PubMed]
- Offman, E.; Freeman, D.J.; Dresser, G.K.; Muñoz, C.; Bend, J.R.; Bailey, D.G. Red wine–cisapride interaction: Comparison with grapefruit juice. Clin. Pharmacol. Ther. 2001, 70, 17–23. [Google Scholar] [CrossRef]
- Piver, B.; Berthou, F.; Dreano, Y.; Lucas, D. Inhibition of CYP3A, CYP1A and CYP2E1 activities by resveratrol and other non volatile red wine components. Toxicol. Lett. 2001, 125, 83–91. [Google Scholar] [CrossRef]
- Berginc, K.; Kristl, A. The Mechanisms Responsible for Garlic—Drug Interactions and their In Vivo Relevance. Curr. Drug Metab. 2013, 14, 90–101. [Google Scholar] [CrossRef] [PubMed]
- Berginc, K.; Milisav, I.; Kristl, A. Garlic Flavonoids and Organosulfur Compounds: Impact on the Hepatic Pharmacokinetics of Saquinavir and Darunavir. Drug Metab. Pharmacokinet. 2010, 25, 521–530. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.; Sy-Cordero, A.; Graf, T.N.; Brantley, S.J.; Paine, M.F.; Oberlies, N.H. Isolation and Identification of Intestinal CYP3A Inhibitors from Cranberry (Vaccinium macrocarpon) Using Human Intestinal Microsomes. Planta Medica 2010, 77, 265–270. [Google Scholar] [CrossRef] [PubMed]
- Srovnalova, A.; Svecarova, M.; Zapletalova, M.K.; Anzenbacher, P.; Bachleda, P.; Anzenbacherova, E.; Dvorak, Z. Effects of Anthocyanidins and Anthocyanins on the Expression and Catalytic Activities of CYP2A6, CYP2B6, CYP2C9, and CYP3A4 in Primary Human Hepatocytes and Human Liver Microsomes. J. Agric. Food Chem. 2014, 62, 789–797. [Google Scholar] [CrossRef] [PubMed]
- Rahimi, R.; Abdollahi, M. An update on the ability of St. John’s wort to affect the metabolism of other drugs. Expert Opin. Drug Metab. Toxicol. 2012, 8, 691–708. [Google Scholar] [CrossRef]
- Goey, A.K.; Mooiman, K.D.; Beijnen, J.H.; Schellens, J.H.; Meijerman, I. Relevance of in vitro and clinical data for predicting CYP3A4-mediated herb–drug interactions in cancer patients. Cancer Treat. Rev. 2013, 39, 773–783. [Google Scholar] [CrossRef]
- Yale, S.H.; Glurich, I. Analysis of the inhibitory potential of ginkgo biloba, echinacea purpurea, and serenoa repens on the metabolic activity of cytochrome P450 3A4, 2D6, AND 2C9. J. Altern. Complement. Med. 2005, 11, 433–439. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.-F.; Deng, Y.; Bi, H.-C.; Zhao, L.-Z.; Wang, X.-D.; Chen, J.; Ou, Z.-M.; Ding, L.; Xu, L.-J.; Guan, S.; et al. Induction of cytochrome P450 3A by the Ginkgo biloba extract and bilobalides in human and rat primary hepatocytes. Drug Metab. Lett. 2008, 2, 60–66. [Google Scholar] [CrossRef]
- Gurley, B.J.; Gardner, S.F.; Hubbard, M.A.; Williams, D.K.; Gentry, W.B.; Khan, I.A.; Shah, A. In vivo effects of goldenseal, kava kava, black cohosh, and valerian on human cytochrome p450ia2, 2d6, 2e1, and 3a4/5 phenotypes. Clin. Pharmacol. Ther. 2005, 77, 415–426. [Google Scholar] [CrossRef]
- Gurley, B.J.; Swain, A.; Barone, G.W.; Williams, D.K.; Breen, P.; Yates, C.R.; Stuart, L.B.; Hubbard, M.A.; Tong, Y.; Che-boyina, S. Effect of goldenseal (Hydrastis canadensis) and kava kava (Piper methysticum) supplementation on digoxin phar-macokinetics in humans. Drug Metab. Dispos. 2007, 35, 240–245. [Google Scholar] [CrossRef]
- Boušová, I.; Matoušková, P.; Bártíková, H.; Szotáková, B.; Hanusova, V.; Tománková, V.; Anzenbacherová, E.; Liskova, B.; Anzenbacher, P.; Skálová, L. Influence of diet supplementation with green tea extract on drug-metabolizing enzymes in a mouse model of monosodium glutamate-induced obesity. Eur. J. Nutr. 2015, 55, 361–371. [Google Scholar] [CrossRef]
- Darweesh, R.S.; El-Elimat, T.; Zayed, A.; Khamis, T.N.; Babaresh, W.M.; Arafat, T.; Al Sharie, A.H. The effect of grape seed and green tea extracts on the pharmacokinetics of imatinib and its main metabolite, N-desmethyl imatinib, in rats. BMC Pharmacol. Toxicol. 2020, 21, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Silberstein, S.D. The pharmacology of ergotamine and dihydroergotamine. Headache 1997, 37, S15–S25. [Google Scholar] [PubMed]
- Liaudet, L.; Buclin, T.; Jaccard, C.; Eckert, P. Drug points: Severe ergotism associated with interaction between ritonavir and ergotamine. BMJ 1999, 318, 771. [Google Scholar] [CrossRef] [PubMed]
- Bu, H.Z. A Literature Review of Enzyme Kinetic Parameters for CYP3A4-Mediated Metabolic Reactions of 113 Drugs in Human Liver Microsomes: Structure- Kinetics Relationship Assessment. Curr. Drug Metab. 2006, 7, 231–249. [Google Scholar] [CrossRef]
- Amidon, G.L.; Lennernäs, H.; Shah, V.P.; Crison, J.R. A theoretical basis for a biopharmaceutic drug classification: The correlation of in vitro drug product dissolution and in vivo bioavailability. Pharm. Res. 1995, 12, 413–420. [Google Scholar] [CrossRef]
- Benet, L.Z. The role of BCS (biopharmaceutics classification system) and BDDCS (biopharmaceutics drug disposition classi-fication system) in drug development. J. Pharm. Sci. 2013, 102, 34–42. [Google Scholar] [CrossRef]
- Fujita, K.-I. Food-drug interactions via human cytochrome P450 3A (CYP3A). Drug Metab. Drug Interact. 2004, 20, 195–217. [Google Scholar] [CrossRef] [PubMed]
- Won, C.S.; Oberlies, N.; Paine, M.F. Mechanisms underlying food–drug interactions: Inhibition of intestinal metabolism and transport. Pharmacol. Ther. 2012, 136, 186–201. [Google Scholar] [CrossRef]
- Bailey, D.; Spence, J.D.; Munoz, C.; Arnold, J. Interaction of citrus juices with felodipine and nifedipine. Lancet 1991, 337, 268–269. [Google Scholar] [CrossRef]
- Bailey, D.G.; Arnold, J.M.O.; Munoz, C.; Spence, J.D. Grapefruit juice–felodipine interaction: Mechanism, predictability, and effect of naringin. Clin. Pharmacol. Ther. 1993, 53, 637–642. [Google Scholar] [CrossRef]
- Ducharme, M.P.; Warbasse, L.H.; Edwards, D.J. Disposition of intravenous and oral cyclosporine after administration with grapefruit juice. Clin. Pharmacol. Ther. 1995, 57, 485–491. [Google Scholar] [CrossRef]
- Kupferschmidt, H.H.; Ha, H.R.; Ziegler, W.H.; Meier, P.J.; Krähenbühl, S. Interaction between grapefruit juice and midazolam in humans. Clin. Pharmacol. Ther. 1995, 58, 20–28. [Google Scholar] [CrossRef]
- Lown, K.S.; Bailey, D.G.; Fontana, R.J.; Janardan, S.K.; Adair, C.H.; A Fortlage, L.; Brown, M.B.; Guo, W.; Watkins, P.B. Grapefruit juice increases felodipine oral availability in humans by decreasing intestinal CYP3A protein expression. J. Clin. Investig. 1997, 99, 2545–2553. [Google Scholar] [CrossRef]
- Greenblatt, D.J.; Von Moltke, L.L.; Harmatz, J.S.; Chen, G.; Weemhoff, J.L.; Jen, C.; Kelley, C.J.; LeDuc, B.W.; Zinny, M.A. Time course of recovery of cytochrome p450 3A function after single doses of grapefruit juice. Clin. Pharmacol. Ther. 2003, 74, 121–129. [Google Scholar] [CrossRef]
- Basheer, L.; Kerem, Z. Interactions between CYP3A4 and Dietary Polyphenols. Oxidative Med. Cell. Longev. 2015, 2015, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Hippius, H. St john’s wort (‘hypericum perforatum’)—A herbal antidepressant. Curr. Med. Res. Opin. 1998, 14, 171–184. [Google Scholar] [CrossRef]
- Chrubasik-Hausmann, S.; Vlachojannis, J.; McLachlan, A.J. Understanding drug interactions with St john’s wort (hypericum perforatum l.): Impact of hyperforin content. J. Pharmacy. Pharmacol. 2019, 71, 129–138. [Google Scholar] [CrossRef]
- Madabushi, R.; Frank, B.; Drewelow, B.; Derendorf, H.; Butterweck, V. Hyperforin in St. John’s wort drug interactions. Eur. J. Clin. Pharmacol. 2006, 62, 225–233. [Google Scholar] [CrossRef]
- Mitchell, M.J.; Billingsley, M.M.; Haley, R.M.; Wechsler, M.E.; Peppas, N.A.; Langer, R. Engineering precision nanoparticles for drug delivery. Nat. Rev. Drug Discov. 2020, 20, 101–124. [Google Scholar] [CrossRef]
- Müller, R.; Runge, S.; Ravelli, V.; Mehnert, W.; Thünemann, A.; Souto, E. Oral bioavailability of cyclosporine: Solid lipid nanoparticles (SLN®) versus drug nanocrystals. Int. J. Pharm. 2006, 317, 82–89. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Chen, J.; Zhang, Y.; Shen, Q.; Pan, W. Characterization and evaluation of nanostructured lipid carrier as a vehicle for oral delivery of etoposide. Eur. J. Pharm. Sci. 2011, 43, 174–179. [Google Scholar] [CrossRef]
- Gambhire, V.M.; Gambhire, M.S.; Ranpise, N.S. Solid Lipid Nanoparticles of Dronedarone Hydrochloride for Oral Delivery: Optimization, In Vivo Pharmacokinetics and Uptake Studies. Pharm. Nanotechnol. 2019, 7, 375–388. [Google Scholar] [CrossRef] [PubMed]
- Makwana, V.; Jain, R.; Patel, K.; Nivsarkar, M.; Joshi, A. Solid lipid nanoparticles (SLN) of Efavirenz as lymph targeting drug delivery system: Elucidation of mechanism of uptake using chylomicron flow blocking approach. Int. J. Pharm. 2015, 495, 439–446. [Google Scholar] [CrossRef] [PubMed]
- Elmowafy, M.; Ibrahim, H.M.; Ahmed, M.A.; Shalaby, K.; Salama, A.; Hefesha, H. Atorvastatin-loaded nanostructured lipid carriers (NLCs): Strategy to overcome oral delivery drawbacks. Drug Deliv. 2017, 24, 932–941. [Google Scholar] [CrossRef] [PubMed]
- Bhalekar, M.R.; Upadhaya, P.; Madgulkar, A.R.; Kshirsagar, S.J.; Dube, A.; Bartakke, U.S. In-vivo bioavailability and lymphatic uptake evaluation of lipid nanoparticulates of darunavir. Drug Deliv. 2015, 23, 2581–2586. [Google Scholar] [CrossRef]
- Patel, M.; Mundada, V.; Sawant, K. Enhanced intestinal absorption of asenapine maleate by fabricating solid lipid nanoparticles using TPGS: Elucidation of transport mechanism, permeability across Caco-2 cell line and in vivo pharmacokinetic studies. Artif. Cells Nanomedicine Biotechnol. 2019, 47, 144–153. [Google Scholar] [CrossRef]
- Sharma, M.; Gupta, N.; Gupta, S. Implications of designing clarithromycin loaded solid lipid nanoparticles on their pharma-cokinetics, antibacterial activity and safety. RSC Advances 2016, 6, 76621–76631. [Google Scholar] [CrossRef]
- Obinu, A.; Burrai, G.; Cavalli, R.; Galleri, G.; Migheli, R.; Antuofermo, E.; Rassu, G.; Gavini, E.; Giunchedi, P. Transmucosal Solid Lipid Nanoparticles to Improve Genistein Absorption via Intestinal Lymphatic Transport. Pharmaceutics 2021, 13, 267. [Google Scholar] [CrossRef]
- Yao, M.; McClements, D.J.; Zhao, F.; Craig, R.W.; Xiao, H. Controlling the gastrointestinal fate of nutraceutical and pharmaceutical-enriched lipid nanoparticles: From mixed micelles to chylomicrons. NanoImpact 2017, 5, 13–21. [Google Scholar] [CrossRef]
- Gao, F.; Zhang, Z.; Bu, H.; Huang, Y.; Gao, Z.; Shen, J.; Zhao, C.; Li, Y. Nanoemulsion improves the oral absorption of can-desartan cilexetil in rats: Performance and mechanism. J. Control. Release 2011, 149, 168–174. [Google Scholar] [CrossRef]
- Yuan, H.; Chen, C.-Y.; Chai, G.; Du, Y.-Z.; Hu, F.-Q. Improved Transport and Absorption through Gastrointestinal Tract by PEGylated Solid Lipid Nanoparticles. Mol. Pharm. 2013, 10, 1865–1873. [Google Scholar] [CrossRef] [PubMed]
- Han, H.-K.; Shin, H.-J.; Ha, D.H. Improved oral bioavailability of alendronate via the mucoadhesive liposomal delivery system. Eur. J. Pharm. Sci. 2012, 46, 500–507. [Google Scholar] [CrossRef] [PubMed]
- Pooja, D.; Kulhari, H.; Kuncha, M.; Rachamalla, S.S.; Adams, D.; Bansal, V.; Sistla, R. Improving Efficacy, Oral Bioavailability, and Delivery of Paclitaxel Using Protein-Grafted Solid Lipid Nanoparticles. Mol. Pharm. 2016, 13, 3903–3912. [Google Scholar] [CrossRef]
- Shi, L.-L.; Xie, H.; Lu, J.; Cao, Y.; Liu, J.-Y.; Zhang, X.-X.; Zhang, H.; Cui, J.-H.; Cao, Q.-R. Positively Charged Surface-Modified Solid Lipid Nanoparticles Promote the Intestinal Transport of Docetaxel through Multifunctional Mechanisms in Rats. Mol. Pharm. 2016, 13, 2667–2676. [Google Scholar] [CrossRef] [PubMed]
- Baek, J.S.; Cho, C.W. Surface modification of solid lipid nanoparticles for oral delivery of curcumin: Improvement of bioa-vailability through enhanced cellular uptake, and lymphatic uptake. Eur. J. Pharm. Biopharm. 2017, 117, 132–140. [Google Scholar] [CrossRef] [PubMed]
- Hosny, K.M.; Ahmed, O.A.A.; Al-Abdali, R.T. Enteric-coated alendronate sodium nanoliposomes: A novel formula to overcome barriers for the treatment of osteoporosis. Expert Opin. Drug Deliv. 2013, 10, 741–746. [Google Scholar] [CrossRef]
- Rajpoot, K.; Jain, S.K. Oral delivery of ph-responsive alginate microbeads incorporating folic acid-grafted solid lipid nano-particles exhibits enhanced targeting effect against colorectal cancer: A dual-targeted approach. Int. J. Biol. Macromol. 2020, 151, 830–844. [Google Scholar] [CrossRef]
- Guo, X.; Zhang, J.; Cai, Q.; Fan, S.; Xu, Q.; Zang, J.; Yang, H.; Yu, W.; Li, Z.; Zhang, Z. Acetic acid transporter-mediated, oral, multifunctional polymer liposomes for oral delivery of docetaxel. Colloids Surf. B Biointerfaces 2020, 198, 111499. [Google Scholar] [CrossRef]
- Kim, K.S.; Youn, Y.S.; Bae, Y.H. Immune-triggered cancer treatment by intestinal lymphatic delivery of docetaxel-loaded nanoparticle. J. Control. Release 2019, 311-312, 85–95. [Google Scholar] [CrossRef]
- Liu, Y.; Jiang, Z.F.; Hou, X.F.; Xie, X.M.; Shi, J.P.; Shen, J.Y.; He, Y.Z.; Wang, Z.; Feng, N.P. Functional lipid polymeric nanoparticles for oral drug delivery: Rapid mucus penetration and improved cell entry and cellular transport. Nanomed. Nanotechnol. Biol. Med. 2019, 21, 102075. [Google Scholar] [CrossRef]
- Murakami, T. Absorption sites of orally administered drugs in the small intestine. Expert Opin. Drug Discov. 2017, 12, 1219–1232. [Google Scholar] [CrossRef]
- Atuma, C.; Strugala, V.; Allen, A.; Holm, L. The adherent gastrointestinal mucus gel layer: Thickness and physical state in vivo. Am. J. Physiol. Liver Physiol. 2001, 280, G922–G929. [Google Scholar] [CrossRef]
- Ahadian, S.; Finbloom, J.A.; Mofidfar, M.; Diltemiz, S.E.; Nasrollahi, F.; Davoodi, E.; Hosseini, V.; Mylonaki, I.; Sangabathuni, S.; Montazerian, H.; et al. Micro and nanoscale technologies in oral drug delivery. Adv. Drug Deliv. Rev. 2020, 157, 37–62. [Google Scholar] [CrossRef]
- Boddupalli, B.M.; Mohammed, Z.N.K.; Nath, R.A.; Banji, D. Mucoadhesive drug delivery system: An overview. J. Adv. Pharm. Technol. Res. 2010, 1, 381. [Google Scholar] [CrossRef]
- Chen, H.; Langer, R. Oral particulate delivery: Status and future trends. Adv. Drug Deliv. Rev. 1998, 34, 339–350. [Google Scholar] [CrossRef]
- Olbrich, C.; Kayser, O.; Müller, R.H. Enzymatic Degradation of Dynasan 114 SLN—Effect of Surfactants and Particle Size. J. Nanoparticle Res. 2002, 4, 121–129. [Google Scholar] [CrossRef]
- Olbrich, C.; Müller, R.H. Enzymatic degradation of SLN—Effect of surfactant and surfactant mixtures. Int. J. Pharm. 1999, 180, 31–39. [Google Scholar] [CrossRef]
- Bernkop-Schnurch, A.; Schmitz, T. Pre-systemic metabolism of orally administered peptide drugs and strategies to overcome it. Curr. Drug Metab. 2007, 8, 509–517. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T. Regulation of the intestinal barrier by nutrients: The role of tight junctions. Anim. Sci. J. 2020, 91, e13357. [Google Scholar] [CrossRef] [PubMed]
- Bernkop-Schnürch, A.; Kast, C.E.; Guggi, D. Permeation enhancing polymers in oral delivery of hydrophilic macromolecules: Thiomer/GSH systems. J. Control. Release 2003, 93, 95–103. [Google Scholar] [CrossRef]
- Patel, K.; Patil, A.; Mehta, M.; Gota, V.; Vavia, P. Oral delivery of paclitaxel nanocrystal (PNC) with a dual P-gp-CYP3A4 inhibitor: Preparation, characterization and antitumor activity. Int. J. Phar. 2014, 472, 214–223. [Google Scholar] [CrossRef]
- Martin, P.; Giardiello, M.; McDonald, T.O.; Smith, D.; Siccardi, M.; Rannard, S.P.; Owen, A. Augmented Inhibition of CYP3A4 in Human Primary Hepatocytes by Ritonavir Solid Drug Nanoparticles. Mol. Pharm. 2015, 12, 3556–3568. [Google Scholar] [CrossRef] [PubMed]
- Tompkins, L.; Lynch, C.; Haidar, S.; Polli, J.; Wang, H. Effects of Commonly Used Excipients on the Expression of CYP3A4 in Colon and Liver Cells. Pharm. Res. 2010, 27, 1703–1712. [Google Scholar] [CrossRef] [PubMed]
- Trevaskis, N.L.; Charman, W.N.; Porter, C.J.H. Lipid-based delivery systems and intestinal lymphatic drug transport: A mechanistic update. Adv. Drug Deliv. Rev. 2008, 60, 702–716. [Google Scholar] [CrossRef]
- Charman, W.N.; Porter, C. Lipophilic prodrugs designed for intestinal lymphatic transport. Adv. Drug Deliv. Rev. 1996, 19, 149–169. [Google Scholar] [CrossRef]
- Wasan, K.M. The Role of Lymphatic Transport in Enhancing Oral Protein and Peptide Drug Delivery. Drug Dev. Ind. Pharm. 2002, 28, 1047–1058. [Google Scholar] [CrossRef]
- Muranishi, S. Delivery System Design for Improvement of Intestinal Absorption of Peptide Drugs. Yakugaku Zasshi 1997, 117, 394–414. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zhang, R.X.; Li, L.Y.; Li, J.; Xu, Z.; Abbasi, A.Z.; Lin, L.; Amini, M.A.; Weng, W.Y.; Sun, Y.; Rauth, A.M.; et al. Coor-dinating biointeraction and bioreaction of a nanocarrier material and an anticancer drug to overcome membrane rigidity and target mitochondria in multidrug-resistant cancer cells. Adv. Funct. Mater. 2017, 27, 1700804–1700816. [Google Scholar] [CrossRef]
- Kucharzik, T.; Lügering, N.; Rautenberg, K.; Schmidt, M.A.; Stoll, R.; Domschke, W. Role of M Cells in Intestinal Barrier Function. Ann. N. Y. Acad. Sci. 2006, 915, 171–183. [Google Scholar] [CrossRef] [PubMed]
- Lopes, M.A.; Abrahim, B.A.; Cabral, L.M.; Rodrigues, C.R.; Seiça, R.; Veiga, F.; Ribeiro, A.J. Intestinal absorption of insulin nanoparticles: Contribution of M cells. Nanomed. Nanotechnol. Biol. Med. 2014, 10, 1139–1151. [Google Scholar] [CrossRef]
- Fasano, A. Innovative strategies for the oral delivery of drugs and peptides. Trends Biotechnol. 1998, 16, 152–157. [Google Scholar] [CrossRef]
- Davitt, C.J.H.; Lavelle, E.C. Delivery strategies to enhance oral vaccination against enteric infections. Adv. Drug Deliv. Rev. 2015, 91, 52–69. [Google Scholar] [CrossRef] [PubMed]
- Rieux, A.D.; Fievez, V.; Garinot, M.; Schneider, Y.-J.; Préat, V. Nanoparticles as potential oral delivery systems of proteins and vaccines: A mechanistic approach. J. Control. Release 2006, 116, 1–27. [Google Scholar] [CrossRef] [PubMed]
- Salman, H.H.; Gamazo, C.; Campanero, M.A.; Irache, J.M. Salmonella-like bioadhesive nanoparticles. J. Control. Release 2005, 106, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Hussain, N.; Florence, A. O 3 Invasin-induced oral uptake of nanospheres: Utilising bacterial mechanisms of epithelial cell entry. J. Control. Release 1996, 41, S3–S4. [Google Scholar] [CrossRef]
- Maderuelo, C.; Lanao, J.M.; Zarzuelo, A. Enteric coating of oral solid dosage forms as a tool to improve drug bioavailability. Eur. J. Pharm. Sci. 2019, 138, 105019. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Chang, H.H.R.; Shalviri, A.; Li, J.; Lugtu-Pe, J.A.; Kane, A.; Wu, X.Y. Investigation of a new pH-responsive na-noparticulate pore former for controlled release enteric coating with improved processability and stability. Eur.J. Pharm. Biopharm. 2017, 120, 116–125. [Google Scholar] [CrossRef]
- Lugtu-Pe, J.A.; Ghaffari, A.; Chen, K.; Kane, A.; Wu, X.Y. Development of controlled release amorphous solid dispersions (CRASD) using polyvinyl acetate-based release retarding materials: Effect of dosage form design. Eur. J. Pharm. Sci. 2018, 124, 319–327. [Google Scholar] [CrossRef]
- Jelvehgari, M.; Zakeri-Milani, P.; Siahi-Shadbad, M.R.; Loveymi, B.D.; Nokhodchi, A.; Azari, Z.; Valizadeh, H. Development of ph-sensitive insulin nanoparticles using EUDRAGIT l100-55 and chitosan with different molecular weights. AAPS PharmSciTech 2010, 11, 1237–1242. [Google Scholar] [CrossRef]
- Leroux, J.-C.; Cozens, R.; Roesel, J.L.; Galli, B.; Kubel, F.; Doelker, E.; Gurny, R. Pharmacokinetics of a Novel HIV-1 Protease Inhibitor Incorporated into Biodegradable or Enteric Nanoparticles following Intravenous and Oral Administration to Mice. J. Pharm. Sci. 1995, 84, 1387–1391. [Google Scholar] [CrossRef]
- Manthena, V.V.; Catherine, M.A.; Mohammad, U.; Charles, J.R.; Hao, S.; John, L.; Katherine, S.F.; Ayman, F.E.-K. Targeting intestinal transporters for optimizing oral drug absorption. Curr. Drug Metab. 2010, 11, 730–742. [Google Scholar]
- Vitamin B12. Available online: https://ods.od.nih.gov/factsheets/VitaminB12-Consumer/ (accessed on 1 July 2021).
- Gupta, Y.; Kohli, D.V.; Jain, S.K. Vitamin B12-mediated transport: A potential tool for tumor targeting of antineoplastic drugs and imaging agents. Crit. Rev. Ther. Drug Carr. Syst. 2008, 25, 347–379. [Google Scholar] [CrossRef] [PubMed]
- Alsenz, J.; Russell-Jones, G.J.; Westwood, S.; Levet-Trafit, B.; De Smidt, P.C. Oral absorption of peptides through the cobalamin (vitamin B12) pathway in the rat intestine. Pharm. Res. 2000, 17, 825–832. [Google Scholar] [CrossRef]
- Bose, S.; Seetharam, S.; Dahms, N.M.; Seetharam, B. Bipolar Functional Expression of Transcobalamin II Receptor in Human Intestinal Epithelial Caco-2 Cells. J. Biol. Chem. 1997, 272, 3538–3543. [Google Scholar] [CrossRef] [PubMed]
- Reker, D.; Blum, S.M.; Steiger, C.; Anger, K.E.; Sommer, J.M.; Fanikos, J.; Traverso, G. “Inactive” ingredients in oral medica-tions. Sci. Transl. Med. 2019, 11, eaau6753–eaau6767. [Google Scholar] [CrossRef] [PubMed]
- Veeravalli, V.; Cheruvu, H.S.; Srivastava, P.; Madgula, L.M.V. Three-dimensional aspects of formulation excipients in drug discovery: A critical assessment on orphan excipients, matrix effects and drug interactions. J. Pharm. Anal. 2020, 10, 522–531. [Google Scholar] [CrossRef]
- Vasconcelos, T.; Marques, S.; Sarmento, B. The biopharmaceutical classification system of excipients. Ther. Deliv. 2017, 8, 65–78. [Google Scholar] [CrossRef]
- El-Sayed, R.; Bhattacharya, K.; Gu, Z.; Yang, Z.; Weber, J.K.; Li, H.; Leifer, K.; Zhao, Y.; Toprak, M.S.; Zhou, R.; et al. Single-Walled Carbon Nanotubes Inhibit the Cytochrome P450 Enzyme, CYP3A4. Sci. Rep. 2016, 6, 21316–21328. [Google Scholar] [CrossRef]
- Chmielewska, A.; Kozłowska, M.; Rachwał, D.; Wnukowski, P.; Amarowicz, R.; Nebesny, E.; Rosicka-Kaczmarek, J. Cano-la/rapeseed protein—Nutritional value, functionality and food application: A review. Crit. Rev. Food Sci. Nutr. 2020, 1–21. [Google Scholar] [CrossRef]
- Wang, Z.; Zhang, R.X.; Zhang, C.; Dai, C.; Ju, X.; He, R. Fabrication of Stable and Self-Assembling Rapeseed Protein Nanogel for Hydrophobic Curcumin Delivery. J. Agric. Food Chem. 2018, 67, 887–894. [Google Scholar] [CrossRef]
- Wang, Z.; Zhang, R.X.; Zhang, T.; He, C.; He, R.; Ju, X.; Wu, X.Y. In situ proapoptotic peptide-generating rapeseed pro-tein-based nanocomplexes synergize chemotherapy for cathepsin-B overexpressing breast cancer. ACS Appl. Mater. Interfaces 2018, 10, 41056–41069. [Google Scholar] [CrossRef] [PubMed]
- Lovelace, B., Jr. Pfizer’s New At-Home Pill to Treat COVID Could Be Available by End of the Year, CEO Hopes. CNBC 2021. Available online: https://www.cnbc.com/2021/04/27/pfizer-at-home-covid-pill-could-be-available-by-year-end-ceo-albert-bourla-says.html (accessed on 15 July 2021).
- Ganesan, P.; Ramalingam, P.; Karthivashan, G.; Ko, Y.T.; Choi, D.-K. Recent developments in solid lipid nanoparticle and surface-modified solid lipid nanoparticle delivery systems for oral delivery of phyto-bioactive compounds in various chronic diseases. Int. J. Nanomed. 2018, 13, 1569–1583. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Interaction with CYP3A4 a | Drug Examples | Drug Class | Indications | Formulations b | BDDCS Class c | BCS Class d | Log P e |
|---|---|---|---|---|---|---|---|
| Substrates | Alprazolam | Benzodiazepine | Anxiety disorders and panic disorder | Tablet, concentrated liquid | Class 1 | Class 2 | 2.12 |
| Atorvastatin | HMG-CoA reductase inhibitor | Hypercholesterolemia | Tablet | Class 2 | Class 2 | 6.36 | |
| Carbamazepine | Anticonvulsant | Seizures, bipolar disorder, trigeminal neuralgia, diabetic neuropathy | Tablet, capsule, suspension | Class 2 | Class 2 | 2.45 | |
| Cyclosporine | Immuno-suppressant | Prevention organ rejection severe psoriasis, severe rheumatoid arthritis | Capsule, solution | Class 2 | Class 2 | 1.4 | |
| Dexamethasone | Steroid derivative | Different inflammatory conditions: allergic disorders, psoriasis, rheumatoid arthritis, ulcerative colitis | Tablet, solution | Class 1 | Class 1/Class 3 | 1.83 | |
| Ethinyl estradiol | Hormone derivative | Contraceptive, menopausal symptoms | Tablet | Class 1 | Class 1 | 3.67 | |
| Felodipine | Calcium channel blocker | Hypertension | Tablet | Class 2 | Class 2 | 3.86 | |
| Indinavir | HIV protease inhibitor | Cocktails for HIV infections | Capsule | Class 2 | Class 4 | 3.49 | |
| Itraconazole | Azole antifungal | Infections caused by fungus, including the lungs, mouth or throat, fingernails. | Capsule, tablet, solution | Class 2 | Class 2 | 5.66 | |
| Lovastatin | HMG-CoA reductase inhibitor | Hypercholesterolemia | Tablet | Class 2 | Class 2 | 4.26 | |
| Ritonavir | HIV protease inhibitor | Cocktails for HIV infections | Capsule, tablet, solution | Class 2 | Class 4 | 6.27 | |
| Saquinavir | HIV protease inhibitor | Cocktails for HIV infections | Capsule, tablet | Class 2 | Class 4 | 4.7 | |
| Sildenafil | cGMP Phosphodiesterase-5 inhibitor | Erectile dysfunction, pulmonary arterial hypertension | Tablet, capsule, suspension | Class 1 | Class 2 | 2.75 | |
| Simvastatin | HMG-CoA reductase inhibitor | Hypercholesterolemia | Tablet, suspension | Class 2 | Class 2 | 4.68 | |
| Tadalafil | cGMP Phosphodiesterase-5 inhibitor | Erectile dysfunction, enlarged prostate | Tablet | Class 2 | Class 2 | 1.42 | |
| Triazolam | Benzodiazepine | Insomnia | Tablet | Class 1 | n/a | 2.42 | |
| Vardenafil | cGMP Phosphodiesterase-5 inhibitor | Erectile dysfunction, pulmonary arterial hypertension | Tablet | Class 1 | Class 2 | 2.79 | |
| Verapamil | Calcium channel blocker | Hypertension, angina, cardiac arrhythmias | Tablet | Class 1 | Class 1 | 3.79 at pH 9.0; 2.15 at pH 7.0 | |
| Inhibitors | Clarithromycin | Macrolide antibiotic | Bacterial infections: stomach ulcers caused by Helicobacter pylori | Tablet, suspension | Class 3 | Class 2 | 3.16 |
| Erythromycin | Macrolide antibiotic | Different types of bacterial infections | Capsule, tablet, liquid | Class 3 | Class 3 | 3.06 | |
| Fluconazole | Azole antifungal | Fungus infections, cryptococcal meningitis | Powder, tablet | Class 3 | Class 1 | 0.5 | |
| Ketoconazole | Azole antifungal | Fungus infections | Tablet | Class 2 | Class 2 | 4.34 | |
| Midazolam | Benzodiazepine | Anesthesia, anxiety, panic disorder, seizures | Syrup | Class 1 | Class 1 | 4.33 | |
| Nicardipine | Calcium channel blocker | Hypertension, angina, cardiac arrhythmias | Capsule | Class 1 | Class 2 | 3.82 | |
| Nifedipine | Calcium channel blocker | Hypertension, angina, cardiac arrhythmias | Tablet | Class 2 | Class 2 | 2.20 | |
| Tacrolimus | Immuno-suppressant | Prevention of graft rejection following solid organ or bone marrow transplantation | Capsule, granule, tablet | Class 2 | Class 2 | 3.03 | |
| Inducers | Phenobarbital f | Anticonvulsant | Seizures, sedation, insomnia | Elixir, tablet | Class 1 | Class 1 | 1.47 |
| Phenytoin | Anticonvulsant | Seizures, arrhythmia | Capsule, tablet, suspension | Class 2 | Class 2 | 2.47 |
| Effect on CYP3A4 Activity | Diet & Herbal | Main Use | Modulating Constituent a | References |
|---|---|---|---|---|
| Inhibition | Grapefruit | Fruit, juice | Bergaptol, flavonoids (naringin, naringenin, kaempferol and quercetin), furanocoumarins (bergamottin and DHB), paradisin C | [73,74] |
| Inhibition | Seville orange | Fruit, juice, marmalade | DHB, bergamottin, | [75,76] |
| Inhibition | Red wine | drinks | Polyphenolic (trans-resveratrol), red wine solids (flavonoids and other polyphenols), gallic acid | [77,78] |
| Inhibition | Garlic | Food, flavoring agent, supplement | Flavonoids (tangeretin, nobiletin, rutin, quercetin), garlic sulfur containing compounds (DADS, DAS, AMS) | [79,80] |
| Inhibition | Cranberry | Fruit, juice, supplement | Triterpenes (maslinic acid, corosolic acid, and ursolic acid), anthocyanidins, anthocyanins | [81,82] |
| Inhibition/Induction | St. John’s wort | Herbal/dietary supplement | Hyperforin, hypericin, quercitrin | [83,84] |
| Inhibition/Induction | Ginkgo biloba | Alternative medicine | Bilobalide, ginkgolide A | [85,86] |
| Inhibition | Goldenseal | Botanical supplement | Individual isoquinoline alkaloids (berberine, hydrastine) | [87,88] |
| Inhibition | Green tea | Drinks, dietary supplement | Green tea catechins, EC, EGC, ECG, EGCG | [89,90] |
| Induction | Ergot alkaloids | Medicine | Ergotamine | [91,92] |
| LNS a | Delivery Mechanism | Nanoformulations b | Drug Payload | BDDCS Class c | Study Models | Main Effects b | Reference |
|---|---|---|---|---|---|---|---|
| Lipid NPs | Mucoadhesive | SLN | Cyclosporin A | Class 2 | Young pig |
| [109] |
| Mucoadhesive | VP16-NLC | Etoposide | Class 3 | Rat intestinal membrane, Healthy rat |
| [110] | |
| Clathrin-mediated endocytosis | DRD-SLN | Dronedarone hydrochloride | Class 2 | Healthy rat |
| [111] | |
| Lymphatic transport via chylomicrons | EFV-SLN | Efavirenz | Class 2 | Chylomicron blocking rat model, Mesenteric lymph duct cannulated rat model |
| [112] | |
| Lymphatic transport via chylomicrons | AT-NLC | Atorvastatin | Class 2 | Both High-fat diet treated and health rats |
| [113] | |
| Portal vein and lymphatic pathway transport | Darunavir-SLN | Darunavir | Class 2 | Everted rat intestine, Chylomicron blocking rat model, Healthy rats |
| [114] | |
| Portal vein and lymphatic pathway transport | AM-SLNs | Asenapine maleate | Class 1 | Caco-2 monolayer, Chylomicron blocking rat model |
| [115] | |
| Lymphatic absorption | CLA-SLN | Clarithromycin | Class 3 | Healthy rat |
| [116] | |
| Lymphatic absorption | GEN-loaded SLN | Genistein | CYP3A4 inhibitor | In vitro characterization of chylomicrons Caco-2 cells, Ex vivo porcine duodenum |
| [117] | |
| Lymphatic transport via chylomicrons | micelles | 5-demethylnobiletin | N/A | Caco-2 monolayer |
| [118] | |
| Portal vein and lymphatic pathway transport | CCN | Candesartan cilexetil | Class 4 | Caco-2 monolayer, in situ single-pass intestine perfusion, ligated intestinal loop model, Healthy rats |
| [119] | |
| PLN | Mucus penetration | pSLN | Doxorubicin | Class 1 | Caco-2/HT29 co-culture, Everted rat intestine, Intestine loops model, Healthy rat |
| [120] |
| Mucoadhesive | Chitosan coated liposome | Alendronate | Class 3 | Caco-2 monolayer, Healthy rat |
| [121] | |
| Enterocyte adhesive via WGA-lectin binding | LPSN | Paclitaxel | Class 2 | A549 cells, Healthy rats |
| [122] | |
| M-cell phagocytosis, TJ opening, and caveola-mediated endocytosis | HACC-DTX-SLN | Docetaxel | Class 2 | Caco-2 monolayer, FAE monolayer, Healthy rat |
| [123] | |
| Lymphatic uptake | NCC-SLN | Curcumin | CYP3A4 inhibitor | Healthy rat |
| [124] | |
| pH responsive drug release (i.e., pH 1.2 and pH 7.4) | EuC-NLS | Alendronate sodium | Class 3 | Healthy rabbit |
| [125] | |
| pH-responsive drug release, (i.e., pH>7.0) | IRSLNF3 | Irinotecan hydrochloride trihydrate | Class 1 | Healthy mice, HT-29 bearing mice |
| [126] | |
| Targeting MCT1 transport | DTX-ACSL-Lip | Docetaxel | Class 2 | 4T1 and Caco-2 cells, Healthy rats |
| [127] | |
| Targeting ASBT in the distal ileum | DSLN-CSG | Docetaxel | Class 2 | Lymph fistula rat model, Healthy rats and tumor bearing mice |
| [128] | |
| Targeting VB12 mediated endocytosis | H/VC-LPN | Curcumin | CYP3A4 inhibitor | Caco-2/HT29-MTX co-culture, Healthy mice & rats |
| [129] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, R.X.; Dong, K.; Wang, Z.; Miao, R.; Lu, W.; Wu, X.Y. Nanoparticulate Drug Delivery Strategies to Address Intestinal Cytochrome P450 CYP3A4 Metabolism towards Personalized Medicine. Pharmaceutics 2021, 13, 1261. https://doi.org/10.3390/pharmaceutics13081261
Zhang RX, Dong K, Wang Z, Miao R, Lu W, Wu XY. Nanoparticulate Drug Delivery Strategies to Address Intestinal Cytochrome P450 CYP3A4 Metabolism towards Personalized Medicine. Pharmaceutics. 2021; 13(8):1261. https://doi.org/10.3390/pharmaceutics13081261
Chicago/Turabian StyleZhang, Rui Xue, Ken Dong, Zhigao Wang, Ruimin Miao, Weijia Lu, and Xiao Yu Wu. 2021. "Nanoparticulate Drug Delivery Strategies to Address Intestinal Cytochrome P450 CYP3A4 Metabolism towards Personalized Medicine" Pharmaceutics 13, no. 8: 1261. https://doi.org/10.3390/pharmaceutics13081261
APA StyleZhang, R. X., Dong, K., Wang, Z., Miao, R., Lu, W., & Wu, X. Y. (2021). Nanoparticulate Drug Delivery Strategies to Address Intestinal Cytochrome P450 CYP3A4 Metabolism towards Personalized Medicine. Pharmaceutics, 13(8), 1261. https://doi.org/10.3390/pharmaceutics13081261