
The Neuropeptide Cortistatin Alleviates Neuropathic Pain in Experimental Models of Peripheral Nerve Injury

 , , , , and
, , , , and 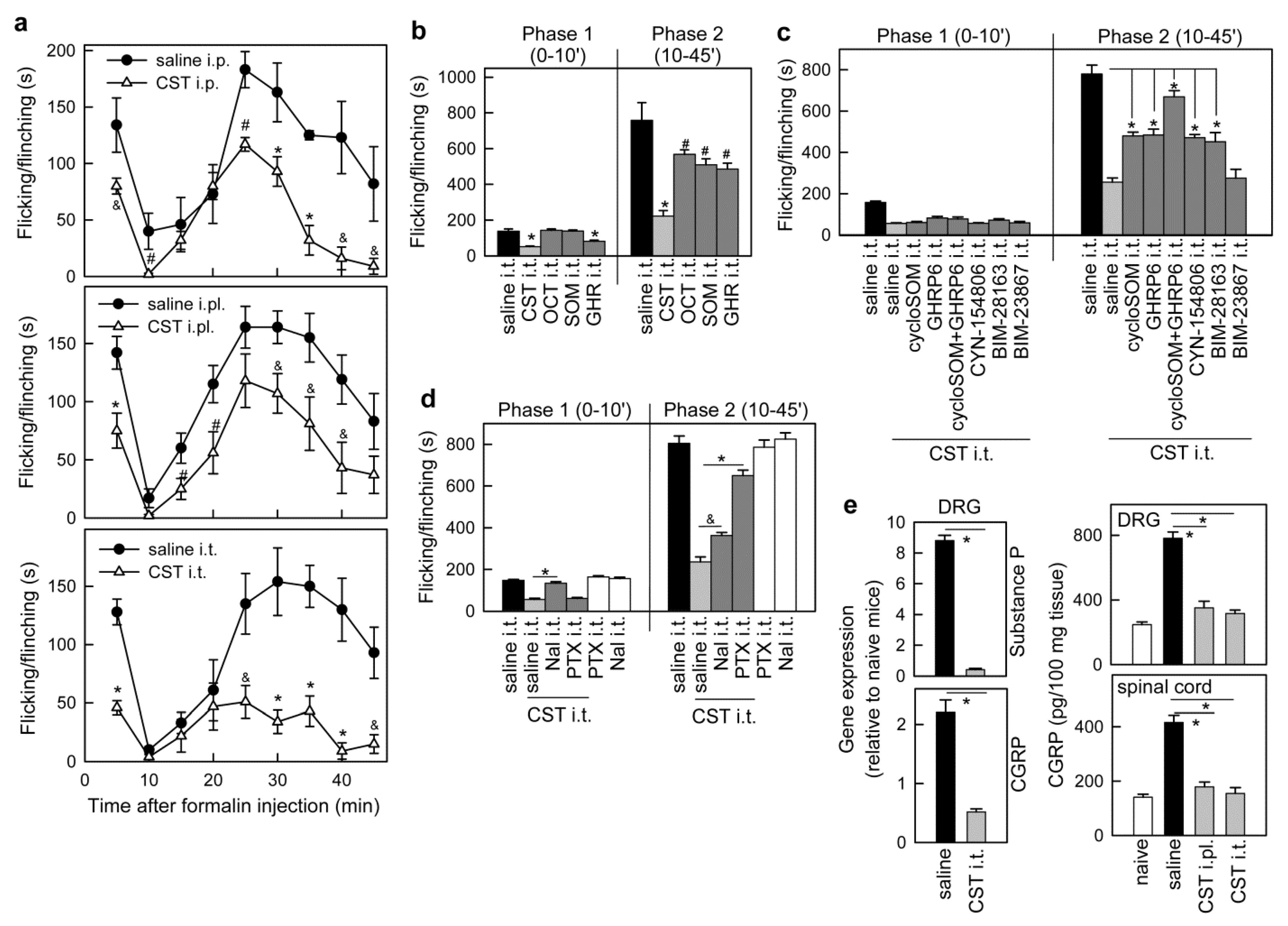{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals and Ethic Statement
2.2. Reagents
2.3. Induction and Treatment of Acute Pain Evoked with Formalin
2.4. Induction and Treatment of Chronic Constriction Injury (CCI) of Sciatic Nerve
2.5. Induction and Treatment of Spared Nerve Injury (SNI)
2.6. Induction and Treatment for the Sciatic Nerve Transection (SNT) Model
2.7. Induction and Treatment of Neuropathic Pain in Diabetic Mice
2.8. Analysis of Functional Tests in the SNT Model
- -
- Determination of the clinical score: to assess the functional nerve recovery following SNT, walk tracking videos were recorded (Sony HDR-XR260VE) for each mouse one day before and at different times (7, 17, and 42 days) after nerve lesion, and they were evaluated by two independent blinded examiners to obtain four parameters affecting locomotion: gait, toe spreading, ankle dorsiflexion, and foot step [32,33]. Each of these parameters were individually scored on a 0–2 scale as follows, giving a maximum possible score of 8 per animal: Gait: 0—no motor deficits, 1—mild loss of coordination (manifests in an uneven gait or waddling), 2—walking deficits characterized by lateral disequilibrium (weak support with the lesion); toe spreading: 0—normal toes separation, 1—extended toes close together, 2—curved toes tightly close; ankle dorsiflexion (based on the lateral foot-base angle measurements, see below): 0—ankle angle > 90°, 1—ankle angle of 45–90°, 2—ankle angle < 45°; foot step: 0—normal foot dorsiflexion in the steps, 1—partial loss of foot dorsiflexion, 2—total absence of foot dorsiflexion and rigidity (ipsilateral steps are characterized by “flat” foot).
- -
- Measurement of the LFBA: left- and right-side views of one walking trial for all mice were captured once one day before and at different time-points (7, 17, and 42 days) after surgery. The video sequences were examined using VLC 3.0.12 multimedia player platform (available at https://www.videolan.org/vlc/, accessed on 24 July 2020), in order to select adequate frames, in which the animals could be seen in defined phases of the step cycle. The LFBA was measured using ImageJ software, as previously described [33]. Briefly, side-view frames showing the ipsilateral foot at the toe-off position with maximal plantar flexion and the contralateral foot in the initial stance phase were evaluated, in which the angle is formed by a line parallel to the mid- and hind foot (i.e., excluding the toes) and the horizontal line. Four to six frames were evaluated for each mouse and the mean of these values was considered as LFBA for each time-point.
2.9. Measurement of Nocifensive Responses to Peripheral Nerve Injury
- -
- Thermal nociceptive responses were determined using Hargreaves’s radiant heat apparatus (IITC Life Sciences). Briefly, mice were placed in Plexiglas boxes placed on a glass floor maintained at 30 °C and allowed to adjust for at least one hour. After acclimation, mice received an automatic heat source (50 W/10 V) onto the plantar surface of the hind paw and the paw withdrawal latency (PWL) was recorded. Each mouse was tested three times (with one-minute intervals between consecutive measurements) and the latencies were averaged for each animal. We adjusted the basal PWL to 9–12 s and set a cut-off of 20 s (defined as complete analgesia) to prevent tissue damage.
- -
- Mechanical pressure hypersensitivity in the hind paw was determined by using a Randall–Selitto Pressure Analgesiometer (IITC Life Sciences). Briefly, mice were restrained as such where they were able to flex their legs. A constantly increasing pressure was applied to the plantar surface of both the ipsilateral and contralateral hind paws through the tip of the paw pressure analgesiometer. Readings (in g) were obtained when the mice attempted to withdraw the paw, and the results were expressed as the decrease (in percentage) in threshold response to pressure stimulation between contralateral and ipsilateral hind paws. The cut-off force was set at 200 g to prevent further injury to the paws.
- -
- Mechanical/tactile allodynia was determined by quantifying the withdrawal threshold of the hind paw in response to stimulation with flexible von Frey filaments (range 0.02–3.0 g; IITC Life Sciences). Mice were placed in Plexiglas boxes on a stainless-steel mesh floor and allowed to habituate for at least 30 min. We then applied a series of calibrated von Frey hairs (six times each on the basis of the Dixon’s up-and-down method) perpendicularly to the lateral and medial plantar surface of the ipsilateral and contralateral hind paw with sufficient force to bend the filament for 4–5 s. Brisk withdrawal or paw flinching was considered positive responses. In the absence of a response, a filament with the next greatest force was applied. In the presence of a response, a filament with the next lowest force was applied. We recorded the 50% withdrawal threshold (i.e., force of the von Frey hair to which an animal reacts in 50% of the presentations).
- -
- Cold allodynia was determined via acetone test, as previously described [34]. A drop of acetone solution was delicately dropped onto the lateral plantar surface of the paw, using a blunt needle connected to a syringe without touching the skin. Acetone was applied three times to the ipsilateral hind paw at intervals of 30 s, and the duration of biting or licking of the hind paw was recorded. We reported the cumulative time of biting/licking in all three measurements with an arbitrary minimal value of 0.5 s and a maximum of 10 s in each of the three trials.
2.10. Histomorphometric Analysis
2.11. Measurement of Neuropeptide, Neurotrophin, and Cytokine Contents
2.12. Determination of Gene Expression
2.13. Statistical Analysis
3. Results
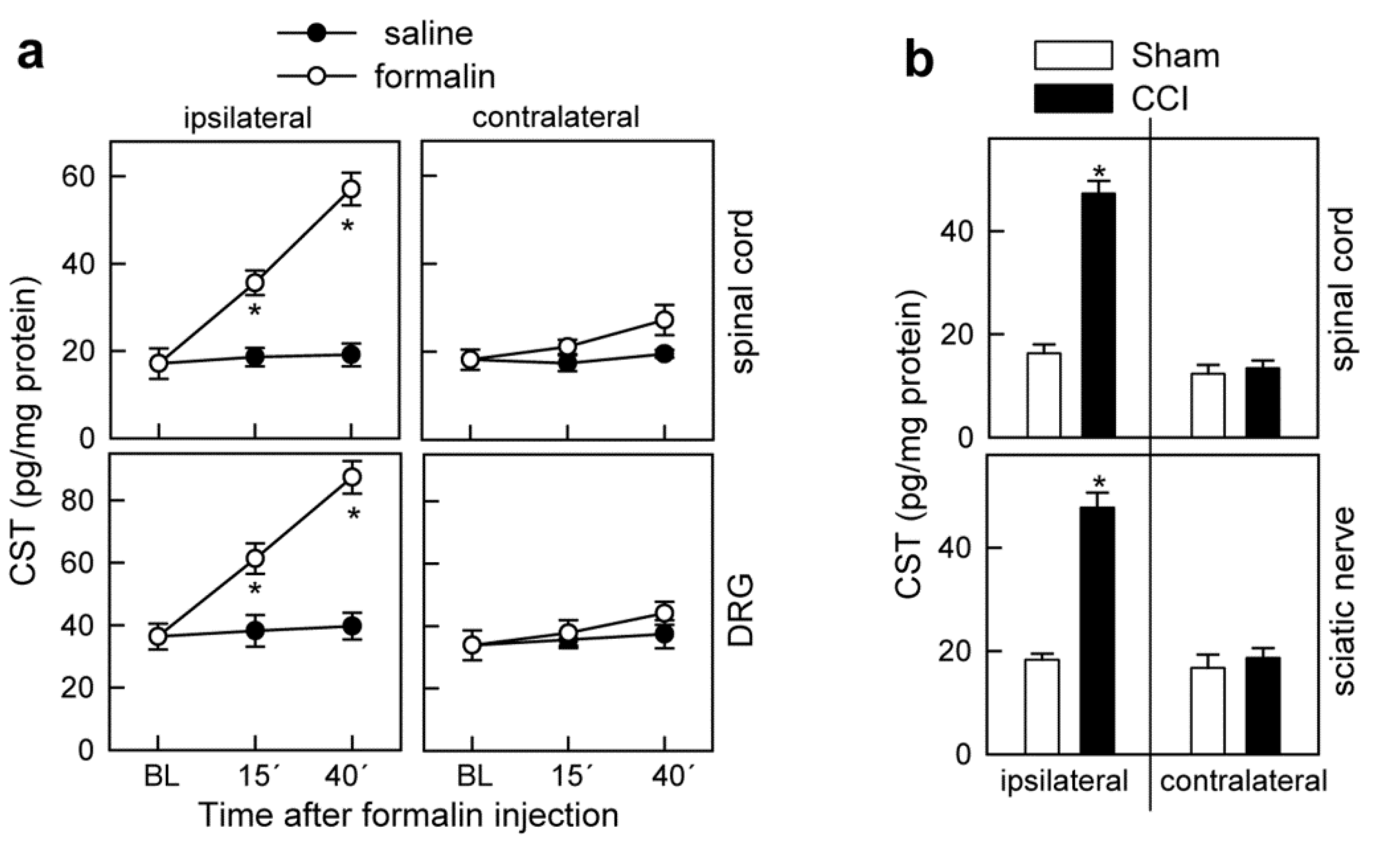
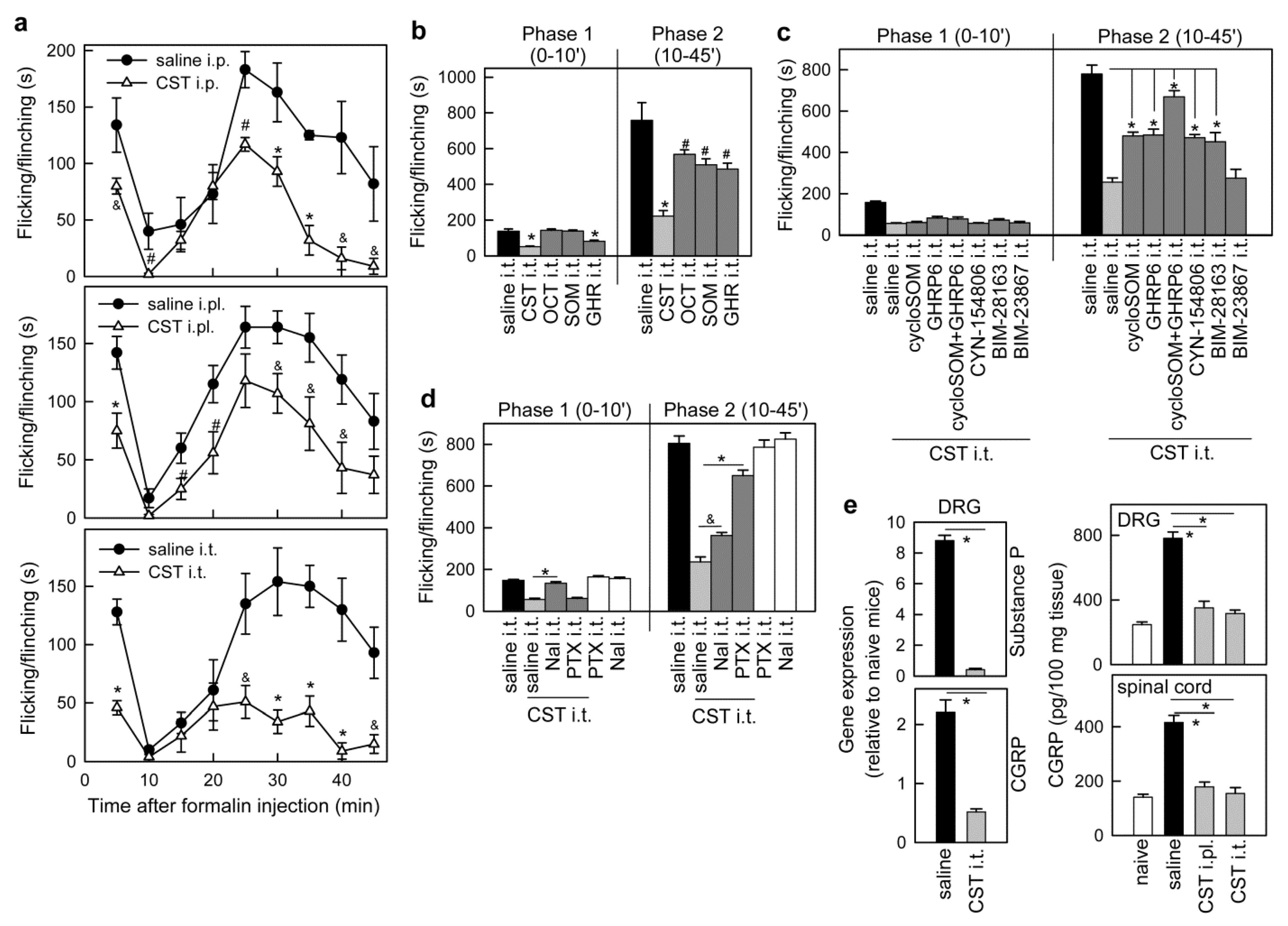
3.1. Cortistatin Alleviates Acute Pain Evoked by Formalin
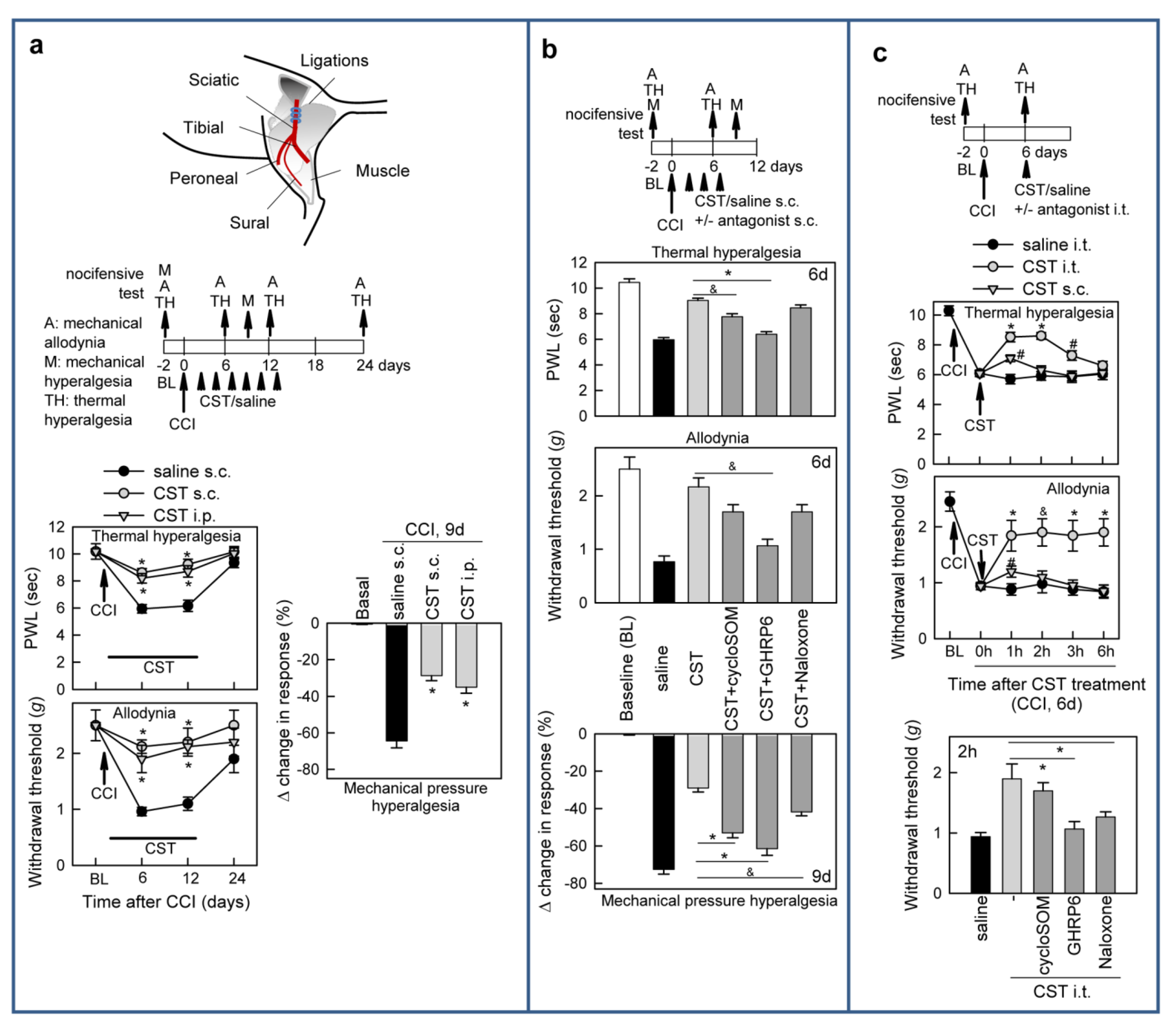
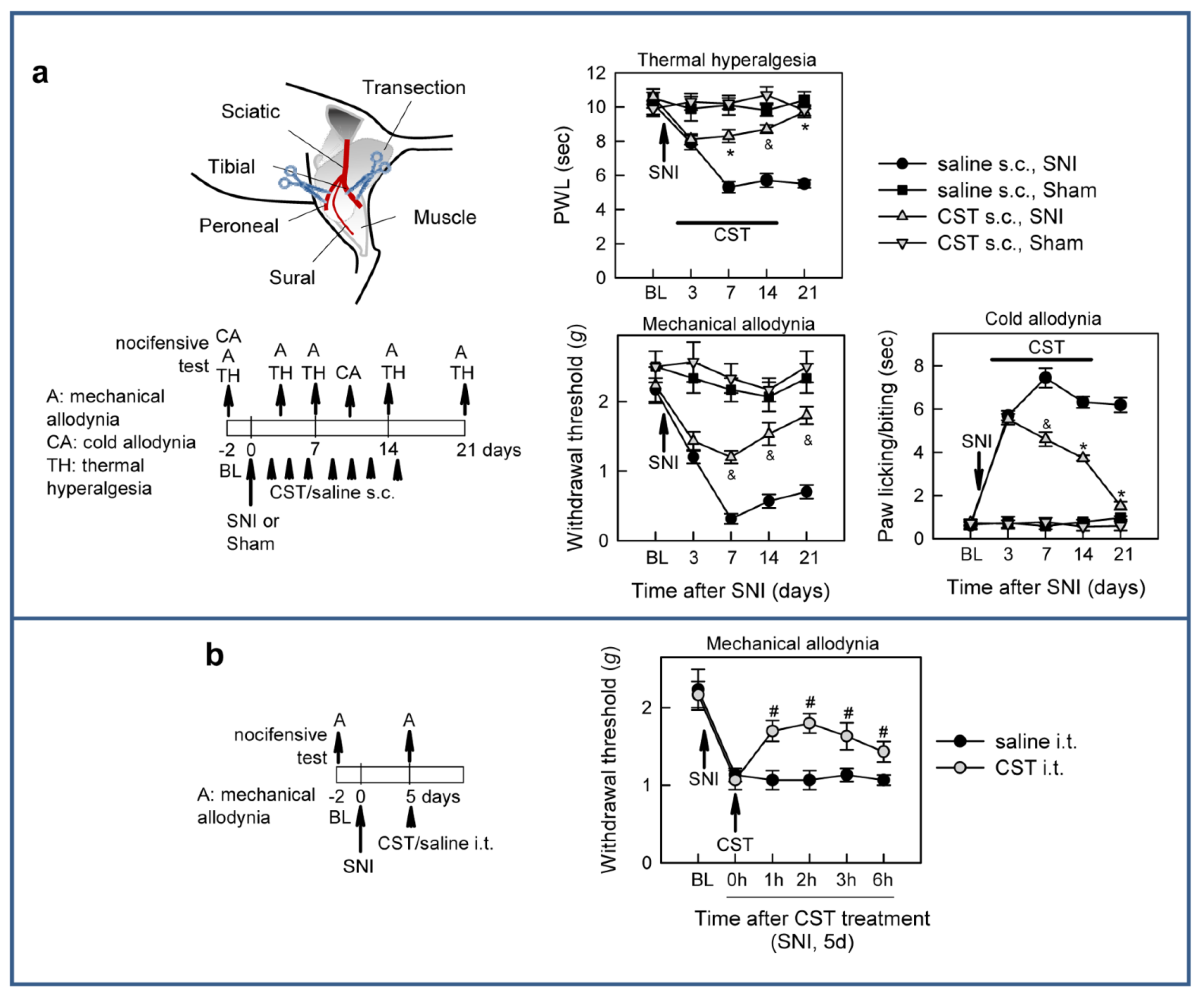
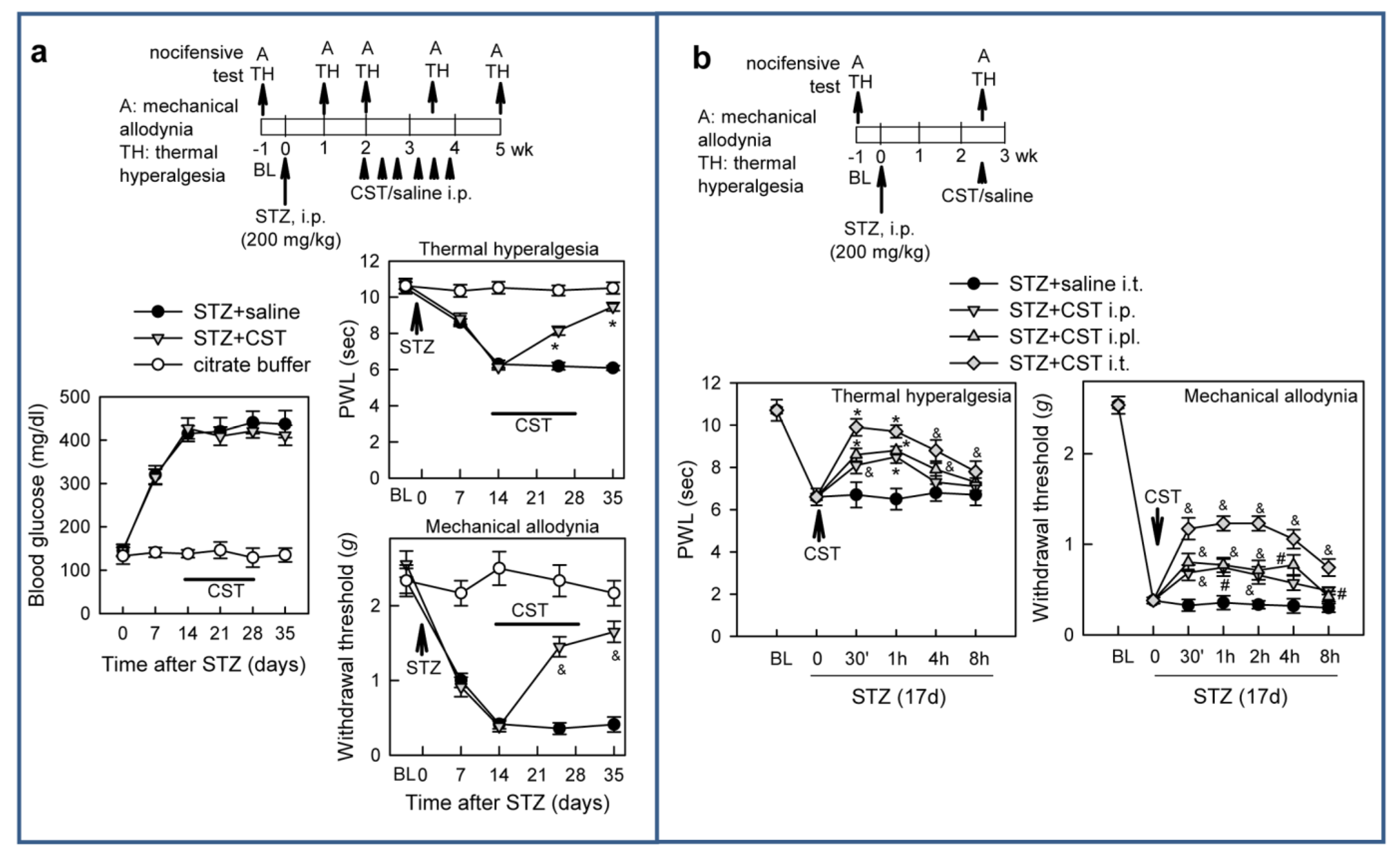
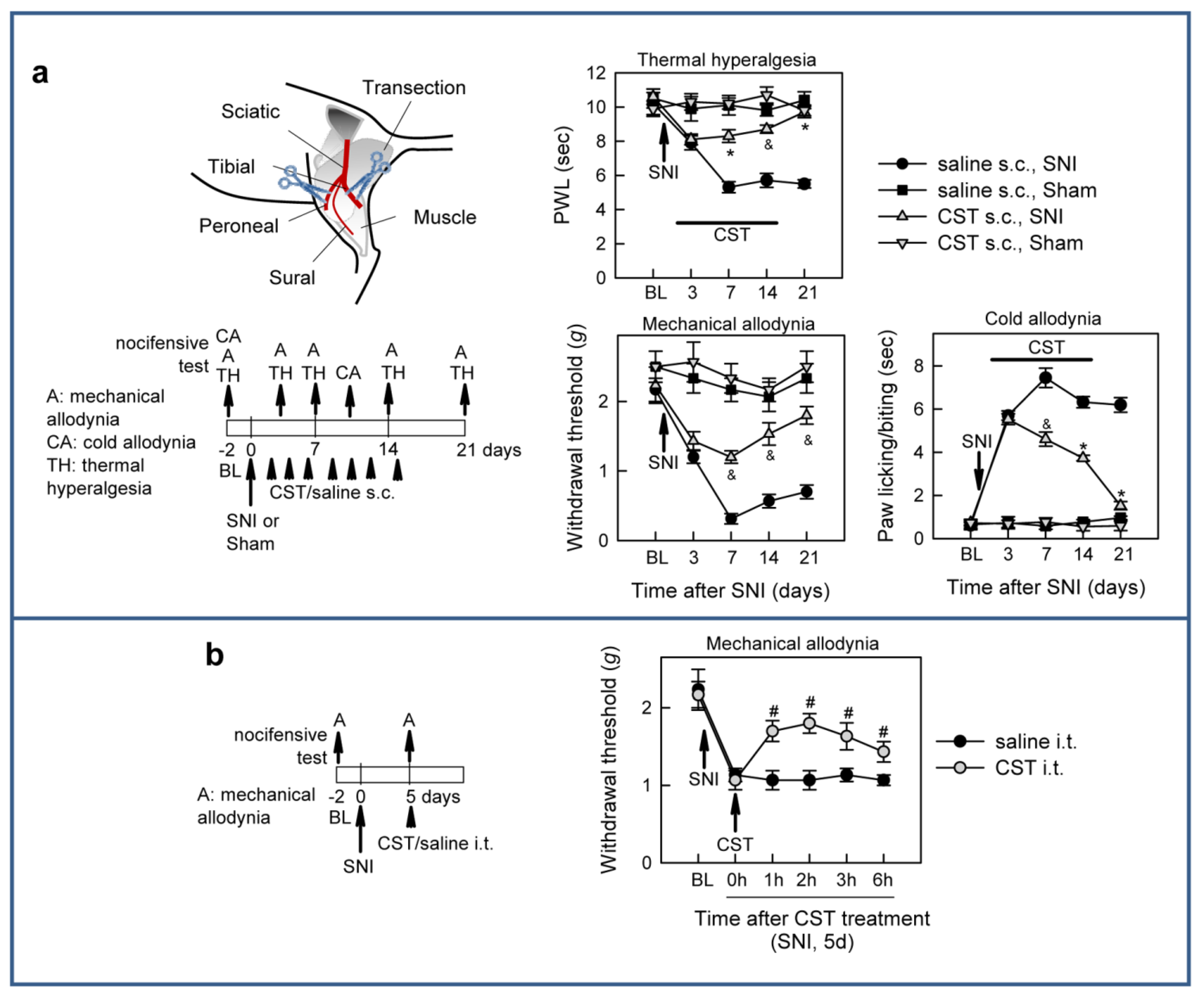
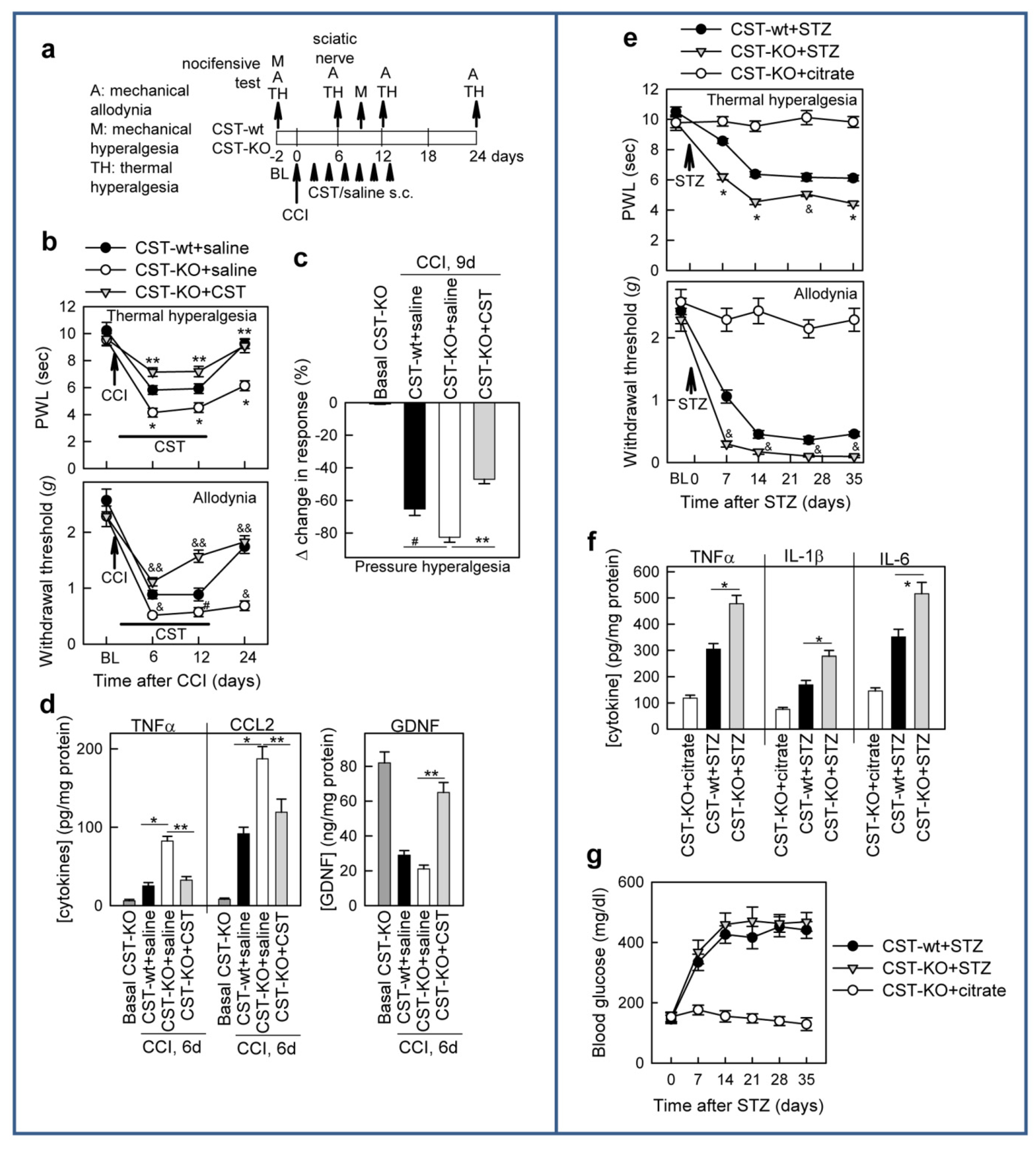
3.2. Cortistatin Attenuates Neuropathic Pain Caused by Peripheral Nerve Injury
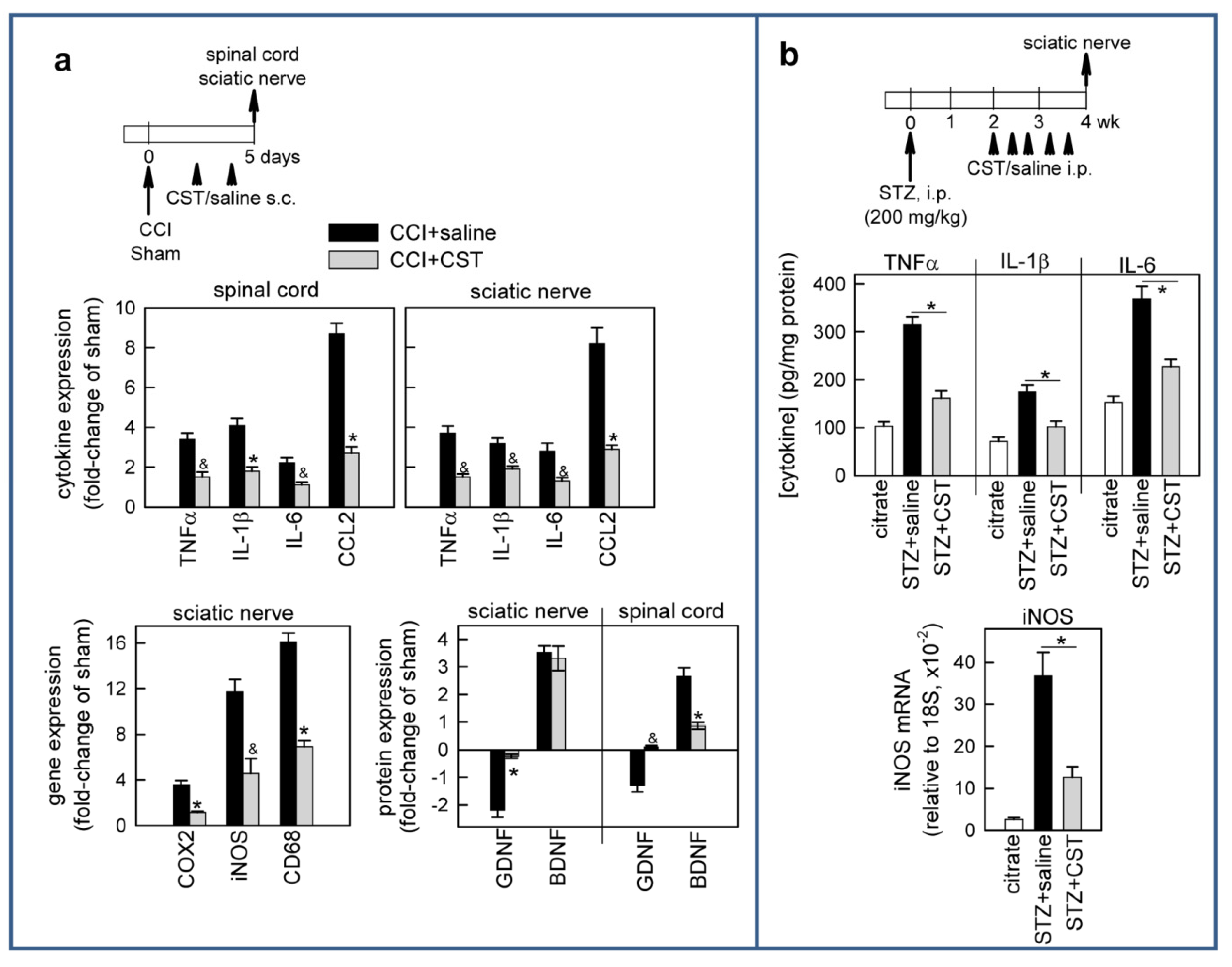
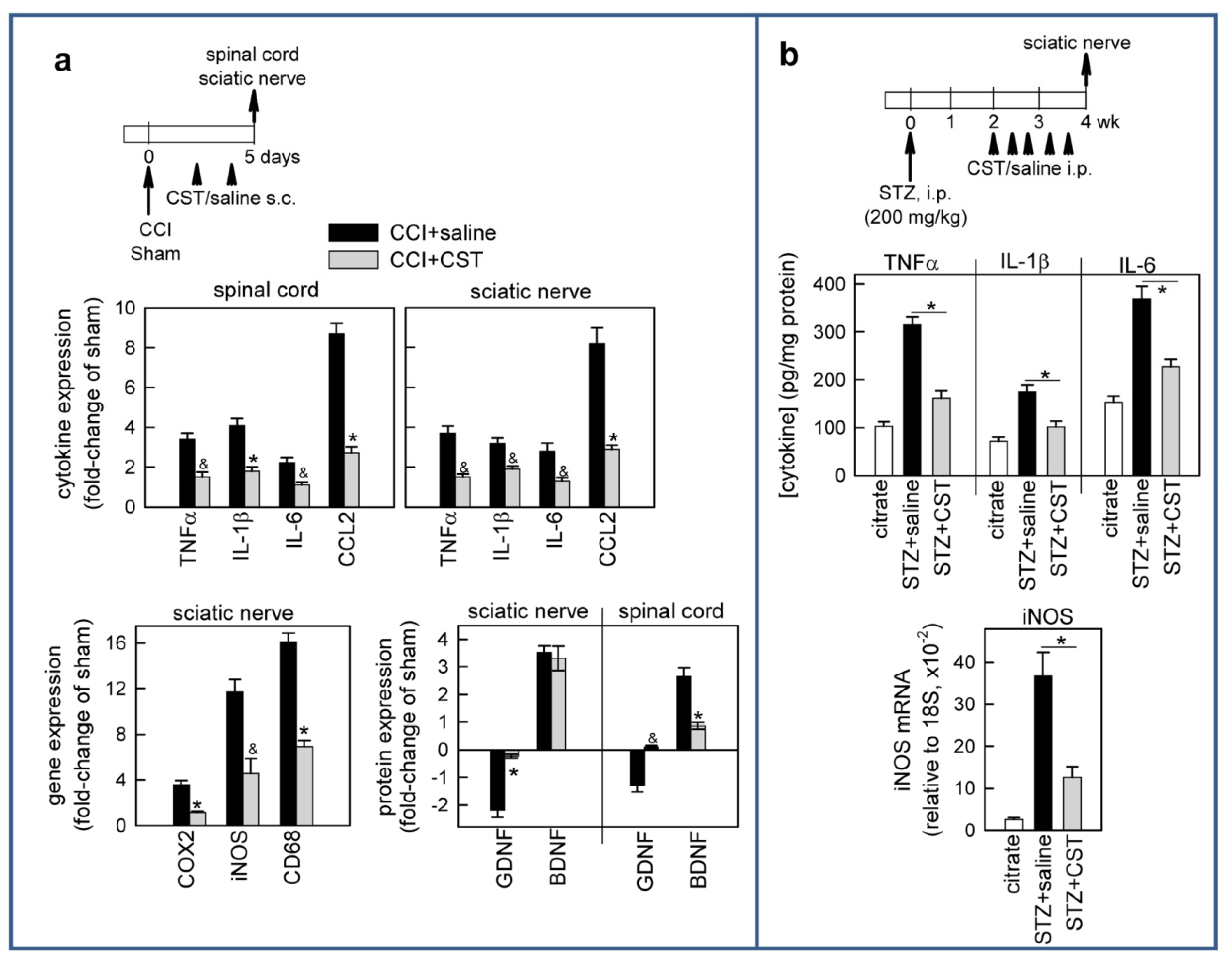
3.3. Cortistatin Regulates Inflammatory and Neurotrophic Mediators of Neuropathic Pain
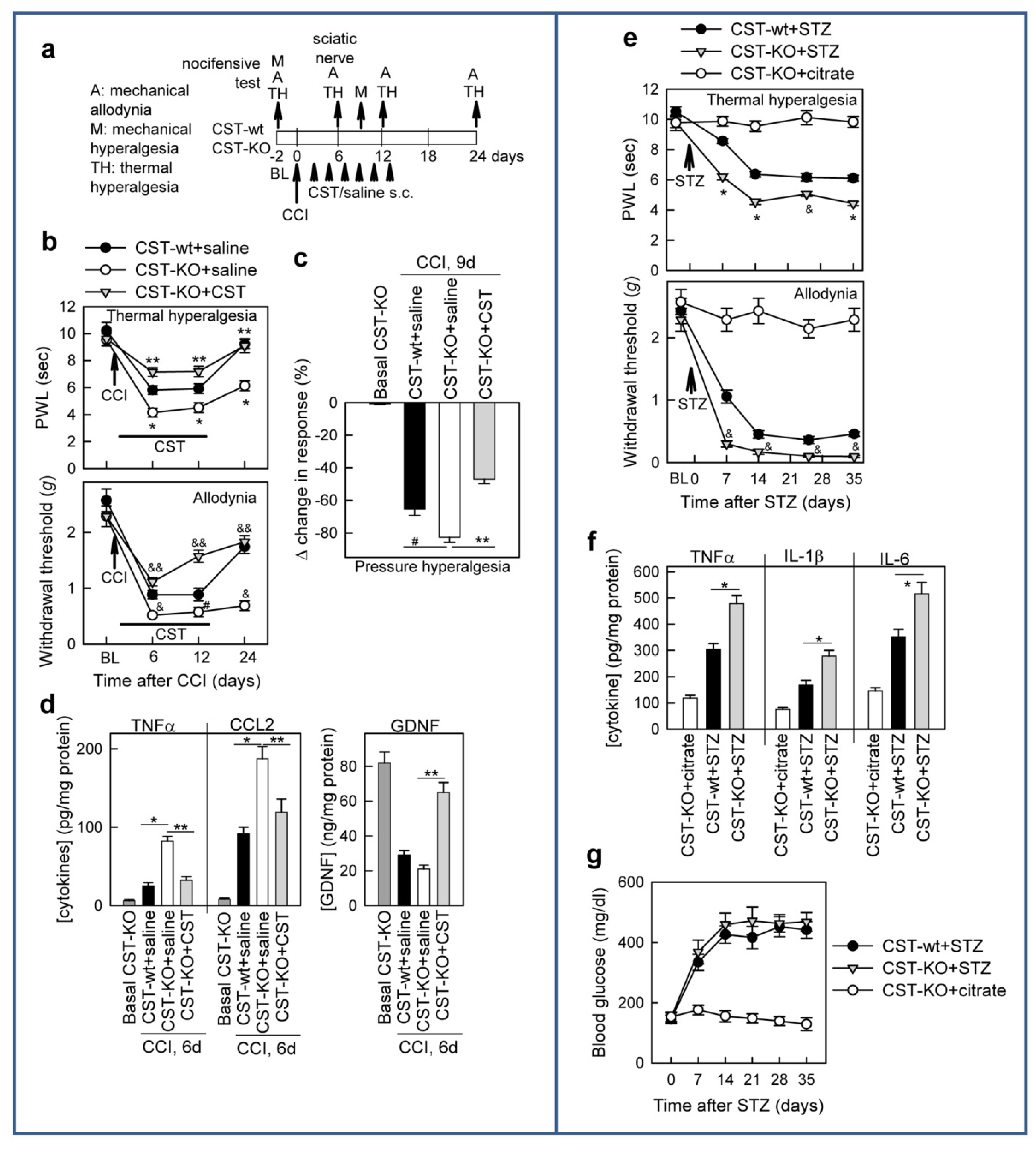
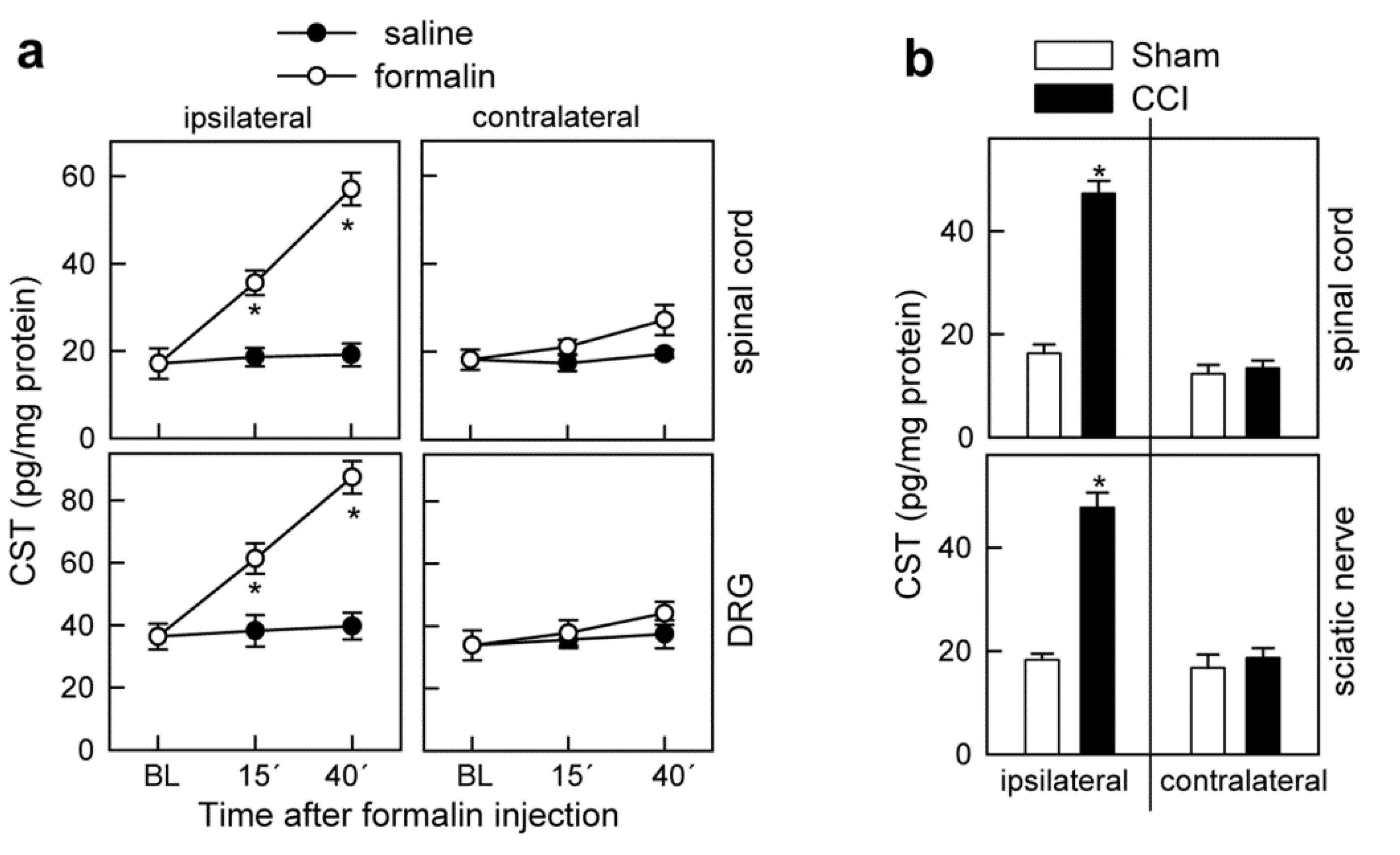
3.4. Cortistatin Plays an Endogenous Regulatory Role in the Development of Neuropathic Pain
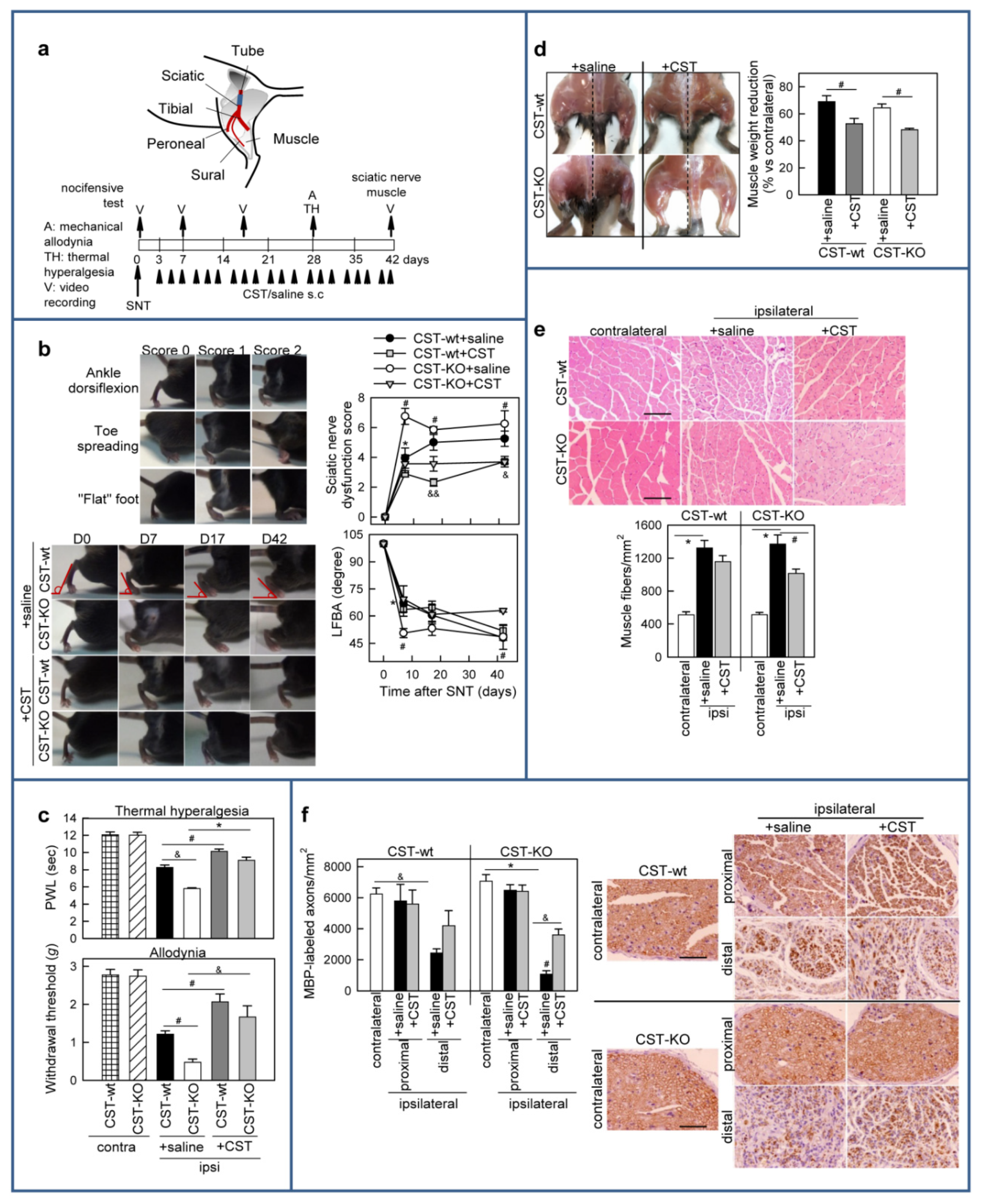
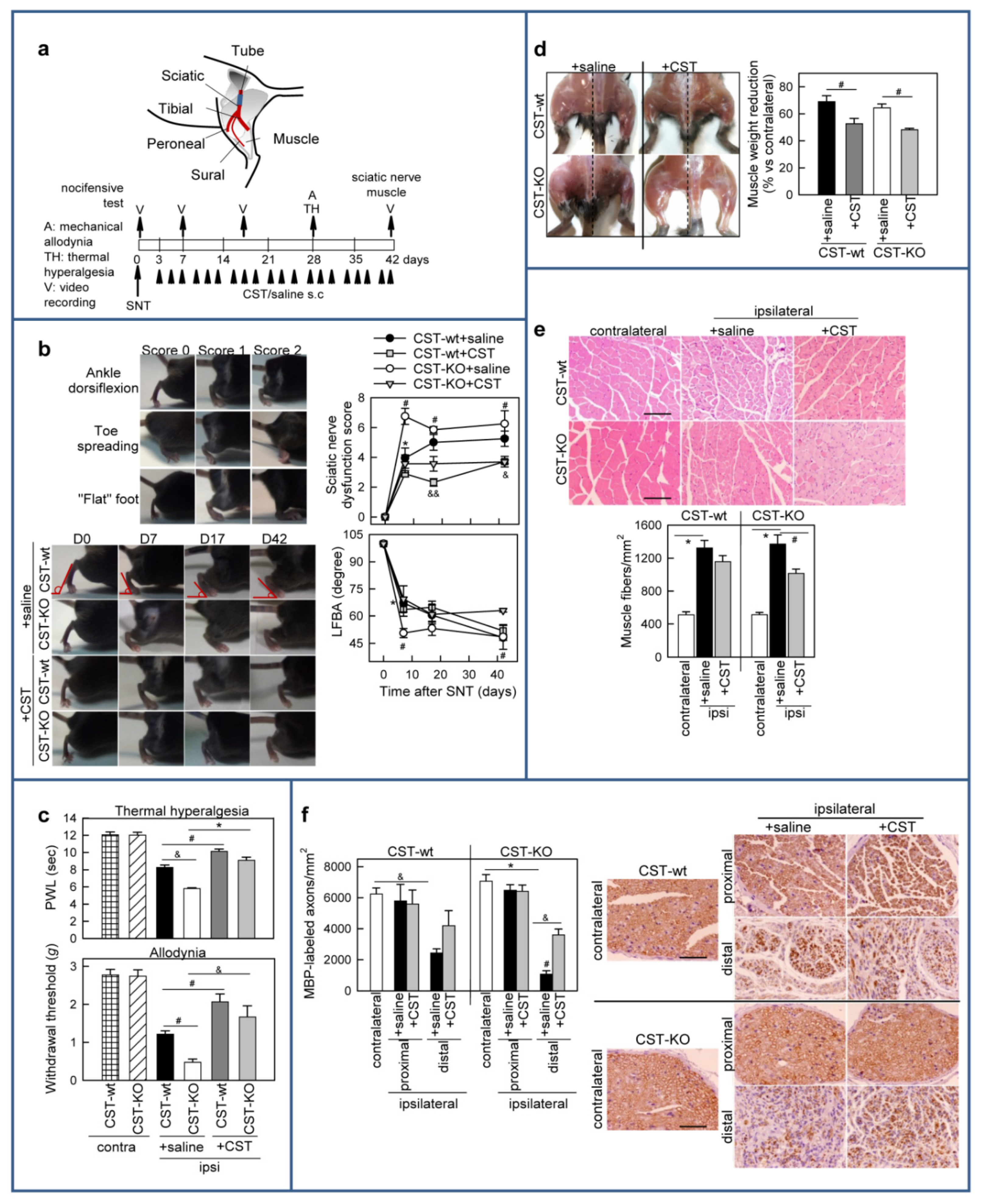
3.5. Cortistatin Promotes Functional Recovery after Sciatic Nerve Transection (SNT)
3.6. Treatment with Cortistatin Attenuates Denervation-Induced Muscle Atrophy and Influences Nerve Regeneration after Sciatic Nerve Transection
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Van Hecke, O.; Austin, S.K.; Khan, R.A.; Smith, B.H.; Torrance, N. Neuropathic pain in the general population: A systematic review of epidemiological studies. Pain 2014, 155, 654–662. [Google Scholar] [CrossRef]
- Colloca, L.; Ludman, T.; Bouhassira, D.; Baron, R.; Dickenson, A.H.; Yarnitsky, D.; Freeman, R.; Truini, A.; Attal, N.; Finnerup, N.B.; et al. Neuropathic pain. Nat. Rev. Dis. Primers 2017, 3, 17002. [Google Scholar] [CrossRef] [Green Version]
- Jensen, T.S.; Finnerup, N.B. Allodynia and hyperalgesia in neuropathic pain: Clinical manifestations and mechanisms. Lancet Neurol. 2014, 13, 924–935. [Google Scholar] [CrossRef]
- Selvarajah, D.; Kar, D.; Khunti, K.; Davies, M.J.; Scott, A.R.; Walker, J.; Tesfaye, S. Diabetic peripheral neuropathy: Advances in diagnosis and strategies for screening and early intervention. Lancet Diabetes Endocrinol. 2019, 7, 938–948. [Google Scholar] [CrossRef] [Green Version]
- Finnerup, N.B.; Attal, N.; Haroutounian, S.; McNicol, E.; Baron, R.; Dworkin, R.H.; Gilron, I.; Haanpää, M.; Hansson, P.; Jensen, T.S.; et al. Pharmacotherapy for neuropathic pain in adults: A systematic review and meta-analysis. Lancet Neurol. 2015, 14, 162–173. [Google Scholar] [CrossRef] [Green Version]
- Austin, P.J.; Moalem-Taylor, G. The neuro-immune balance in neuropathic pain: Involvement of inflammatory immune cells, immune-like glial cells and cytokines. J. Neuroimmunol. 2010, 229, 26–50. [Google Scholar] [CrossRef] [PubMed]
- Rotshenker, S. Wallerian degeneration: The innate-immune response to traumatic nerve injury. J. Neuroinflammation 2011, 8, 109. [Google Scholar] [CrossRef] [Green Version]
- Hucho, T.; Levine, J.D. Signaling pathways in sensitization: Toward a nociceptor cell biology. Neuron 2007, 55, 365–376. [Google Scholar] [CrossRef] [Green Version]
- Todd, A.J. Neuronal circuitry for pain processing in the dorsal horn. Nat. Rev. Neurosci. 2010, 11, 823–836. [Google Scholar] [CrossRef] [Green Version]
- Pan, H.L.; Wu, Z.Z.; Zhou, H.Y.; Chen, S.R.; Zhang, H.M.; Li, D.P. Modulation of pain transmission by G-protein-coupled receptors. Pharmacol. Ther. 2008, 117, 141–161. [Google Scholar] [CrossRef] [Green Version]
- De Lecea, L.; Criado, J.R.; Prospero-Garcia, O.; Gautvik, K.M.; Schweitzer, P.; Danielson, P.E.; Dunlop, C.L.; Siggins, G.R.; Henriksen, S.J.; Sutcliffe, J.G. A cortical neuropeptide with neuronal depressant and sleep-modulating properties. Nature 1996, 381, 242–245. [Google Scholar] [CrossRef] [PubMed]
- Gahete, M.D.; Duran-Prado, M.; Luque, R.M.; Martinez-Fuentes, A.J.; Vazquez-Martinez, R.; Malagon, M.M.; Castaño, J.P. Are somatostatin and cortistatin two siblings in regulating endocrine secretions? In vitro work ahead. Mol. Cell. Endocrinol. 2008, 286, 128–134. [Google Scholar] [CrossRef]
- Deghenghi, R.; Papotti, M.; Ghigo, E.; Muccioli, G. Cortistatin, but not somatostatin, binds to growth hormone secretagogue (GHS) receptors of human pituitary gland. J. Endocrinol. Investig. 2001, 24, RC1–RC3. [Google Scholar] [CrossRef]
- Robas, N.; Mead, E.; Fidock, M. MrgX2 is a high potency cortistatin receptor expressed in dorsal root ganglion. J. Biol. Chem. 2003, 278, 44400–44404. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez-Rey, E.; Chorny, A.; Delgado, M. Regulation of immune tolerance by anti- inflammatory neuropeptides. Nat. Rev. Immunol. 2007, 7, 52–63. [Google Scholar] [CrossRef]
- Delgado-Maroto, V.; Benitez, R.; Forte-Lago, I.; Morell, M.; Maganto-Garcia, E.; Souza- Moreira, L.; O’Valle, F.; Duran-Prado, M.; Lichtman, A.H.; Gonzalez-Rey, E.; et al. Cortistatin reduces atherosclerosis in hyperlipidemic ApoE-deficient mice and the formation of foam cells. Sci. Rep. 2017, 7, 46444. [Google Scholar] [CrossRef] [Green Version]
- Delgado-Maroto, V.; Falo, C.P.; Forte-Lago, I.; Adan, N.; Morell, M.; Maganto-Garcia, E.; Robledo, G.; O’Valle, F.; Lichtman, A.H.; Gonzalez-Rey, E.; et al. The neuropeptide cortistatin attenuates experimental autoimmune myocarditis via inhibition of cardiomyogenic T cell-driven inflammatory responses. Br. J. Pharmacol. 2017, 174, 267–280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duran-Prado, M.; Morell, M.; Delgado-Maroto, V.; Castaño, J.P.; Aneiros-Fernandez, J.; de Lecea, L.; Culler, M.D.; Hernandez-Cortes, P.; O’Valle, F.; Delgado, M. Cortistatin inhibits migration and proliferation of human vascular smooth muscle cells and decreases neointimal formation on carotid artery ligation. Circ. Res. 2013, 112, 1444–1455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mendez-Diaz, M.; Guevara-Martinez, M.; Alquicira, C.R.; Guzman-Vasquez, K.; Prospero- Garcia, O. Cortistatin, a modulatory peptide of sleep and memory, induces analgesia in rats. Neurosci. Lett. 2004, 354, 242–244. [Google Scholar] [CrossRef] [PubMed]
- Markovics, A.; Szoke, É.; Sándor, K.; Börzsei, R.; Bagoly, T.; Kemény, Á.; Elekes, K.; Pintér, E.; Szolcsányi, J.; Helyes, Z. Comparison of the anti-inflammatory and antinociceptive effects of cortistatin-14 and somatostatin-14 in distinct in vitro and in vivo model systems. J. Mol. Neurosci. 2012, 46, 40–50. [Google Scholar] [CrossRef] [PubMed]
- Morell, M.; Souza-Moreira, L.; Caro, M.; O’Valle, F.; Forte-Lago, I.; de Lecea, L.; Gonzalez-Rey, E.; Delgado, M. Analgesic effect of the neuropeptide cortistatin in murine models of arthritic inflammatory pain. Arthritis Rheum. 2013, 65, 1390–1401. [Google Scholar] [CrossRef] [Green Version]
- Morell, M.; Camprubí-Robles, M.; Culler, M.D.; de Lecea, L.; Delgado, M. Cortistatin attenuates inflammatory pain via spinal and peripheral actions. Neurobiol. Dis. 2014, 63, 141–154. [Google Scholar] [CrossRef]
- Colleoni, M.; Sacerdote, P. Murine models of human neuropathic pain. Biochim. Biophys. Acta 2010, 1802, 924–933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Percie du Sert, N.; Hurst, V.; Ahluwalia, A.; Alam, S.; Avey, M.T.; Baker, M.; Browne, W.J.; Clark, A.; Cuthill, I.C.; Dirnagl, U.; et al. The ARRIVE guidelines 2.0: Updated guidelines for reporting animal research. Br. J. Pharmacol. 2020, 177, 3617–3624. [Google Scholar] [CrossRef]
- Cordoba-Chacon, J.; Gahete, M.D.; Pozo-Salas, A.I.; Martínez-Fuentes, A.J.; de Lecea, L.; Gracia-Navarro, F.; Kineman, R.D.; Castaño, J.P.; Luque, R.M. Cortistatin is not a somatostatin analogue but stimulates prolactin release and inhibits GH and ACTH in a gender-dependent fashion: Potential role of ghrelin. Endocrinology 2011, 152, 4800–4812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tulipano, G.; Soldi, D.; Bagnasco, M.; Culler, M.D.; Taylor, J.E.; Cocchi, D.; Giustina, A. Characterization of newselective somatostatin receptor subtype-2 (sst2) antagonists, BIM-23627 and BIM-23454. Effects of BIM-23627 on GH release in anesthetizedmale rats after short-term high-dose dexamethasone treatment. Endocrinology 2002, 143, 1218–1224. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Rey, E.; Chorny, A.; Robledo, G.; Delgado, M. Cortistatin, a new antiinflammatory peptide with therapeutic effect on lethal endotoxemia. J. Exp. Med. 2006, 203, 563–571. [Google Scholar] [CrossRef]
- Takeda, M.; Kadoi, J.; Takahashi, M.; Nasu, M.; Matsumoto, S. Somatostatin inhibits the excitability of rat small-diameter trigeminal ganglion neurons that innervate nasal mucosa and project to the upper cervical dorsal horn via activation of somatostatin 2a receptor. Neuroscience 2007, 148, 744–756. [Google Scholar] [CrossRef]
- Jerlhag, E.; Egecioglu, E.; Dickson, S.L.; Engel, J.A. Glutamatergic regulation of ghrelin-induced activation of the mesolimbic dopamine system. Addict. Biol. 2010, 16, 82–91. [Google Scholar] [CrossRef] [Green Version]
- Inhof, A.K.; Glück, L.; Gajda, M.; Lupp, A.; Bräuer, R.; Schaible, H.G.; Schulz, S. Differential antiinflammatory and antinociceptive effects of the somatostatin analogs octreotide and pasireotide in a mouse model of immune-mediated arthritis. Arthritis Rheum. 2011, 63, 2352–2362. [Google Scholar] [CrossRef]
- Kehoe, S.; Zhang, X.F.; Boyd, D. FDA approved guidance conduits and wraps for peripheral nerve injury: A review of materials and efficacy. Injury 2012, 43, 553–572. [Google Scholar] [CrossRef] [PubMed]
- Griffin, J.W.; Pan, B.; Polley, M.A.; Hoffman, P.N.; Farah, M.H. Measuring nerve regeneration in the mouse. Exp. Neurol. 2010, 223, 60–71. [Google Scholar] [CrossRef] [PubMed]
- Fey, A.; Schachner, M.; Irintchev, A. A novel motion analysis approach reveals late recovery in C57BL/6 mice and deficits in NCAM-deficient mice after sciatic nerve crush. J. Neurotrauma 2010, 27, 815–828. [Google Scholar] [CrossRef] [PubMed]
- Nieto, F.R.; Entrena, J.M.; Cendan, C.M.; Del Pozo, E.; Vela, J.M.; Baeyens, J.M. Tetrodotoxin inhibits the development and expression of neuropathic pain induced by paclitaxel in mice. Pain 2008, 137, 520–531. [Google Scholar] [CrossRef]
- Anderson, P.; Souza-Moreira, L.; Morell, M.; Caro, M.; O’Valle, F.; Gonzalez-Rey, E.; Delgado, M. Adipose-derived mesenchymal stromal cells induce immunomodulatory macrophages which protect from experimental colitis and sepsis. Gut 2013, 62, 1131–1141. [Google Scholar] [CrossRef]
- Coderre, T.J.; Fundytus, M.E.; McKenna, J.E.; Dalal, S.; Melzack, R. The formalin test: A validation of the weighted-scores method of behavioural pain rating. Pain 1993, 54, 43–50. [Google Scholar] [CrossRef]
- Segond von Banchet, G.; Schindler, M.; Hervieu, G.J.; Beckmann, B.; Emson, P.C.; Heppelmann, B. Distribution of somatostatin receptor subtypes in rat lumbar spinal cord examined with gold-labelled somatostatin and anti-receptor antibodies. Brain Res. 1999, 816, 254–257. [Google Scholar] [CrossRef]
- Bar, K.J.; Schurigt, U.; Scholze, A.; Segond Von Banchet, G.; Stopfel, N.; Bräuer, R.; Halbhuber, K.J.; Schaible, H.G. The expression and localization of somatostatin receptors in dorsal root ganglion neurons of normal and monoarthritic rats. Neuroscience 2004, 127, 197–206. [Google Scholar] [CrossRef]
- Vergnano, A.M.; Ferrini, F.; Salio, C.; Lossi, L.; Baratta, M.; Merighi, A. The gastrointestinal homrone ghrelin modulates inhibitory neurotransmission in deep laminae of mouse spinal cord dorsal horn. Endocrinology 2008, 149, 2306–2312. [Google Scholar] [CrossRef] [Green Version]
- Pinter, E.; Helyes, Z.; Szolcsanyi, J. Inhibitory effect of somatostatin on inflammation and nociception. Pharmacol. Ther. 2006, 112, 440–456. [Google Scholar] [CrossRef]
- Dezaki, K.; Damdindorj, B.; Sone, H.; Dyachok, O.; Tengholm, A.; Gylfe, E.; Kurashina, T.; Yoshida, M.; Kakei, M.; Yada, T. Ghrelin attenuates cAMP-PKA signaling to evoke insulinostatic cascade in islet beta-cells. Diabetes 2011, 6, 2315–2324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bennett, G.J.; Xie, Y.K. A peripheral mononeuropathy in rat that produces disorders of pain sensation like those seen in man. Pain 1988, 33, 87–107. [Google Scholar] [CrossRef]
- Decosterd, I.; Woolf, C.J. Spared nerve injury: An animal model of persistent peripheral neuropathic. Pain 2000, 87, 149–158. [Google Scholar] [CrossRef]
- Sullivan, K.A.; Lentz, S.I.; Roberts, J.L., Jr.; Feldman, E.L. Criteria for creating and assessing mouse models of diabetic neuropathy. Curr. Drug Targets 2008, 9, 3–13. [Google Scholar] [PubMed] [Green Version]
- Davies, A.J.; Rinaldi, S.; Costigan, M.; Oh, S.B. Cytotoxic immunity in peripheral nerve injury and pain. Front. Neurosci. 2020, 14, 142. [Google Scholar] [CrossRef]
- Ji, R.R.; Xu, Z.Z.; Gao, Y.J. Emerging targets in neuroinflammation-driven chronic pain. Nat. Rev. Drug Discov. 2014, 13, 533–548. [Google Scholar] [CrossRef] [Green Version]
- Ji, R.R.; Chamessian, A.; Zhang, Y.Q. Pain regulation by non-neuronal cells and inflammation. Science 2016, 354, 572–577. [Google Scholar] [CrossRef] [Green Version]
- Myers, R.R.; Campana, W.M.; Shubayev, V.I. The role of neuroinflammation in neuropathic pain: Mechanisms and therapeutic targets. Drug Discov. Today 2006, 11, 8–20. [Google Scholar] [CrossRef]
- Pinho-Ribeiro, F.A.; Verri, W.A., Jr.; Chiu, I.M. Nociceptor sensory neuron-immune interactions in pain and inflammation. Trends Immunol. 2017, 38, 5–19. [Google Scholar] [CrossRef] [Green Version]
- Scholz, J.; Woolf, C.J. The neuropathic pain triad: Neurons, immune cells and glia. Nat. Neurosci. 2007, 10, 1361–1368. [Google Scholar] [CrossRef]
- Jessen, K.R.; Mirsky, R.; Lloyd, A.C. Schwann Cells: Development and Role in Nerve Repair. Cold Spring Harb. Perspect. Biol. 2015, 7, a020487. [Google Scholar] [CrossRef]
- Goulart, C.O.; Martinez, A.M.B. Tubular conduits, cell-based therapy and exercise to improve peripheral nerve regeneration. Neural Regen. Res. 2015, 10, 565–567. [Google Scholar] [PubMed]
- Shen, Y.; Zhang, R.; Xu, L.; Wan, Q.; Zhu, J.; Gu, J.; Huang, Z.; Ma, W.; Shen, M.; Ding, F.; et al. Microarray Analysis of Gene Expression Provides New Insights into Denervation-Induced Skeletal Muscle Atrophy. Front. Physiol. 2019, 10, 1298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finnerup, N.B.; Haroutounian, S.; Baron, R.; Dworkin, R.H.; Gilron, I.; Haanpaa, M.; Jensen, T.S.; Kamerman, P.R.; McNicol, E.; Moore, A.; et al. Neuropathic pain clinical trials: Factors associated with decreases in estimated drug efficacy. Pain 2018, 159, 2339–2346. [Google Scholar] [CrossRef] [PubMed]
- Aley, K.O.; Levine, J.D. Different peripheral mechanisms mediate enhanced in metabolic/toxic and traumatic painful peripheral neuropathies in the rat. Neuroscience 2002, 111, 389–397. [Google Scholar] [CrossRef]
- Finnerup, N.B.; Haroutounian, S.; Kamerman, P.; Baron, R.; Bennett, D.L.H.; Bouhassira, D.; Cruccu, G.; Freeman, R.; Hansson, P.; Nurmikko, T.; et al. Neuropathic pain: An updated grading system for research and clinical practice. Pain 2016, 157, 1599–1606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, G.; Zhang, Y.Q.; Qadri, Y.J.; Serhan, C.N.; Ji, R.R. Microglia in pain: Detrimental and protective roles in pathogenesis and resolution of pain. Neuron 2018, 100, 1292–1311. [Google Scholar] [CrossRef] [Green Version]
- Chen, P.; Piao, X.; Bonaldo, P. Role of macrophages in Wallerian degeneration and axonal regeneration after peripheral nerve injury. Acta Neuropathol. 2015, 130, 605–618. [Google Scholar] [CrossRef] [PubMed]
- Jiang, B.C.; Liu, T.; Gao, Y.J. Chemokines in chronic pain: Cellular and molecular mechanisms and therapeutic potential. Pharmacol Ther. 2020, 212, 107581. [Google Scholar] [CrossRef]
- Barriga, M.; Benitez, R.; Ferraz-Paula, V.; Caro, M.; Robledo, G.; O´Valle, F.; Campos-Salinas, J.; Delgado, M. Protective role of cortistatin in pulmonary inflammation and fibrosis. Br. J. Pharmacol. 2021, in press. [Google Scholar]
- Gonzalez-Rey, E.; Varela, N.; Sheibanie, A.F.; Chorny, A.; Ganea, D.; Delgado, M. Cortistatin, an antiinflammatory peptide with therapeutic action in inflammatory bowel disease. Proc. Natl. Acad. Sci. USA 2006, 103, 4228–4233. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez-Rey, E.; Chorny, A.; Del Moral, R.G.; Varela, N.; Delgado, M. Therapeutic effect of cortistatin on experimental arthritis by downregulating inflammatory and Th1 responses. Ann. Rheum. Dis. 2007, 66, 582–588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiu, C.; Li, J.; Luo, D.; Chen, X.; Qu, R.; Liu, T.; Li, F.; Liu, Y. Cortistatin protects against inflammatory airway diseases through curbing CCL2 and antagonizing NF-κB signaling pathway. Biochem. Biophys. Res. Commun. 2020, 531, 595–601. [Google Scholar] [CrossRef]
- Souza-Moreira, L.; Morell, M.; Delgado-Maroto, V.; Pedreño, M.; Martinez-Escudero, L.; Caro, M.; O’Valle, F.; Luque, R.; Gallo, M.; de Lecea, L.; et al. Paradoxical effect of cortistatin treatment and its deficiency on experimental autoimmune encephalomyelitis. J. Immunol. 2013, 191, 2144–2154. [Google Scholar] [CrossRef]
- Niederberger, E.; Geisslinger, G. The IKK-NF-jB pathway: A source for novel molecular drug targets in pain therapy. FASEB J. 2008, 22, 3433–3442. [Google Scholar] [CrossRef]
- Sommer, C.; Lindenlaub, T.; Teuteberg, P.; Schäfers, M.; Hartung, T.; Toyka, K.V. Anti- TNF-neutralizing antibodies reduce pain-related behavior in two different mouse models of painful mononeuropathy. Brain Res. 2001, 913, 86–89. [Google Scholar] [CrossRef]
- Boucher, T.J.; Okuse, K.; Bennett, D.L.; Munson, J.B.; Wood, J.N.; McMahon, S.B. Potent analgesic effects of GDNF in neuropathic pain states. Science 2000, 290, 124–127. [Google Scholar] [CrossRef] [PubMed]
- Wei, Z.; Fei, Y.; Su, W.; Chen, G. Emerging role of schwann cells in neuropathic pain: Receptors, glial mediators and myelination. Front. Cell. Neurosci. 2019, 13, 116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sikandar, S.; Minett, M.S.; Millet, Q.; Santana-Varela, S.; Lau, J.; Wood, J.N.; Zhao, J. Brain-derived neurotrophic factor derived from sensory neurons plays a critical role in chronic pain. Brain 2018, 141, 1028–1039. [Google Scholar] [CrossRef]
- Kotliarova, A.A.; Sidorova, Y.A. Glial cell line-derived neurotrophic factor family ligands, players at the interface of neuroinflammation and neuroprotection: Focus onto the glia. Front. Cell. Neurosci. 2021, 15, 223. [Google Scholar] [CrossRef]
- Chen, Z.L.; Yu, W.M.; Strickland, S. Peripheral regeneration. Annu. Rev. Neurosci. 2007, 30, 209–233. [Google Scholar] [CrossRef]
- Radtke, C.; Vogt, P.M. Peripheral nerve regeneration: A current perspective. Eplasty 2009, 9, e47. [Google Scholar]
- Dubový, P.; Jančálek, R.; Kubek, T. Chapter Seven—Role of Inflammation and Cytokines in Peripheral Nerve Regeneration. Int. Rev. Neurobiol. 2013, 108, 173–206. [Google Scholar]
- Gaudet, A.D.; Popovich, P.G.; Ramer, M.S. Wallerian degeneration: Gaining perspective on inflammatory events after peripheral nerve injury. J. Neuroinflammation 2011, 8, 110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mietto, B.S.; Mostacada, K.; Martinez, A.M. Neurotrauma and inflammation: CNS and PNS responses. Mediat. Inflamm. 2015, 2015, 251204. [Google Scholar] [CrossRef]
- Zhang, J.M.; Donnelly, D.F.; Song, X.J.; Lamotte, R.H. Axotomy increases the excitability of dorsal root ganglion cells with unmyelinated axons. J. Neurophysiol. 1997, 78, 2790–2794. [Google Scholar] [CrossRef] [PubMed]
- Boyd, J.G.; Gordon, T. Glial cell line-derived neurotrophic factor and brain-derived neurotrophic factor sustain the axonal regeneration of chronically axotomized motoneurons in vivo. Exp. Neurol. 2003, 183, 610–619. [Google Scholar] [CrossRef]
- Deister, C.; Schmidt, C.E. Optimizing neurotrophic factor combinations for neurite outgrowth. J. Neural Eng. 2006, 3, 172–179. [Google Scholar] [CrossRef]
- Gardell, L.R.; Wang, R.; Ehrenfels, C.; Ossipov, M.H.; Rossomando, A.J.; Miller, S.; Buckley, C.; Cai, A.K.; Tse, A.; Foley, S.F.; et al. Multiple actions of systemic artemin in experimental neuropathy. Nat. Med. 2003, 9, 1383–1389. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Rossomando, A.; Sah, D.W.Y.; Ossipov, M.H.; King, T.; Porreca, F. Artemin induced functional recovery and reinnervation after partial nerve injury. Pain 2014, 155, 476–484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Backonja, M.; Williams, L.; Miao, X.; Katz, N.; Chen, C. Safety and efficacy of neublastin in painful lumbosacral radiculopathy: A randomized, double-blinded, placebo-controlled phase 2 trial using Bayesian adaptive design (the SPRINT trial). Pain 2017, 158, 1802–1812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahato, A.K.; Sidorova, Y.A. Glial cell line-derived neurotrophic factors (GFLs) and small molecules targeting RET receptor for the treatment of pain and Parkinson’s disease. Cell Tissue Res. 2020, 382, 147–160. [Google Scholar] [CrossRef]
- Sidorova, Y.A.; Bespalov, M.M.; Wong, A.W.; Kambur, O.; Jokinen, V.; Lilius, T.O.; Suleymanova, I.; Karelson, G.; Rauhala, P.V.; Karelson, M.; et al. A Novel small molecule GDNF receptor RET agonist, BT13, promotes neurite growth from sensory neurons in vitro and attenuates experimental neuropathy in the rat. Front. Pharmacol. 2017, 8, 365. [Google Scholar] [CrossRef] [PubMed]
- Viisanen, H.; Nuotio, U.; Kambur, O.; Mahato, A.K.; Jokinen, V.; Lilius, T.; Li, W.; Santos, H.A.; Karelson, M.; Rauhala, P.; et al. Novel RET agonist for the treatment of experimental neuropathies. Mol. Pain 2020, 16, 1744806920950866. [Google Scholar] [CrossRef] [PubMed]
- Clements, M.P.; Byrne, E.; Camarillo Guerrero, L.F.; Cattin, A.L.; Zakka, L.; Ashraf, A.; Burden, J.J.; Khadayate, S.; Lloyd, A.C.; Marguerat, S.; et al. The Wound Microenvironment Reprograms Schwann Cells to Invasive Mesenchymal-like Cells to Drive Peripheral Nerve Regeneration. Neuron 2017, 96, 98–114.e7. [Google Scholar] [CrossRef] [Green Version]
- Gerber, D.; Pereira, J.A.; Gerber, J.; Tan, G.; Dimitrieva, S.; Yángüez, E.; Suter, U. Transcriptional profiling of mouse peripheral nerves to the single-cell level to build a sciatic nerve ATlas (SNAT). eLife 2021, 10, e58591. [Google Scholar] [CrossRef] [PubMed]
- Cattin, A.L.; Lloyd, A.C. The multicellular complexity of peripheral nerve regeneration. Curr. Opin. Neurobiol. 2016, 39, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Carr, M.J.; Toma, J.S.; Johnston, A.P.W.; Steadman, P.E.; Yuzwa, S.A.; Mahmud, N.; Frankland, P.W.; Kaplan, D.R.; Miller, F.D. Mesenchymal precursor cells in adult nerves contribute to mammalian tissue repair and regeneration. Cell Stem Cell 2019, 24, 240–256. [Google Scholar] [CrossRef] [Green Version]
- Falo, C.P.; Castillo-González, J.; Forte-Lago, I.; Stucchi, A.; O’Valle, F.; Caro, M.; González-Rey, E. Atypical myelin physiology and dynamics in a cortistatin-deficient environment. Glia Porto 2019, 67, E460–E461. [Google Scholar]
- Falo, C.P.; Ferraz-de-Paula, V.; Castillo-González, J.; Forte-Lago, I.; Caro, M.; Serrano, I.; O’Valle, F.; Andrés-Leon, E.; Macklin, W.B.; González-Rey, E. Cortistatin Exerts a Fine-Tuning on the Glial Dynamics Affecting Neuroinflammation and Remyelination in Multiple Sclerosis. Available online: https://digital.csic.es/handle/10261/229034 (accessed on 24 July 2020).
- Bombeiro, A.L.; Santini, J.C.; Thomé, R.; Ferreira, E.R.L.; Nunes, S.L.O.; Moreira, B.M.; Bonet, I.J.M.; Sartori, C.R.; Verinaud, L.; Oliveira, A.L.R. Enhanced Immune Response in Immunodeficient Mice Improves Peripheral Nerve Regeneration Following Axotomy. Front. Cell. Neurosci. 2016, 10, 151. [Google Scholar] [CrossRef] [Green Version]
- Giordano, R.; Picu, A.; Bonelli, L.; Broglio, F.; Prodam, F.; Grottoli, S.; Muccioli, G.; Ghigo, E.; Arvat, E. The activation of somatostatinergic receptors by either somatostatin-14 or cortistatin-17 often inhibits ACTH hypersecretion in patients with Cushing’s disease. Eur. J. Endocrinol. 2007, 157, 393–398. [Google Scholar] [CrossRef]
- Rol, A.; Todorovski, T.; Martin-Malpartida, P.; Escola, A.; González-Rey, E.; Aragon, E.; Verdaguer, X.; Valles-Miret, M.; Farrera-Sinfreu, J.; Puig, E.; et al. Structure-based design of a cortistatin analogue with immunoregulatory activity in models of inflammatory bowel disease. Nat. Commun. 2021, 12, 1869. [Google Scholar] [CrossRef] [PubMed]
- Rosenberger, D.C.; Blechschmidt, V.; Timmerman, H.; Wolff, A.; Treede, R.D. Challenges of neuropathic pain: Focus on diabetic neuropathy. J. Neural Transm. 2020, 127, 589–624. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Falo, C.P.; Benitez, R.; Caro, M.; Morell, M.; Forte-Lago, I.; Hernandez-Cortes, P.; Sanchez-Gonzalez, C.; O’Valle, F.; Delgado, M.; Gonzalez-Rey, E. The Neuropeptide Cortistatin Alleviates Neuropathic Pain in Experimental Models of Peripheral Nerve Injury. Pharmaceutics 2021, 13, 947. https://doi.org/10.3390/pharmaceutics13070947
Falo CP, Benitez R, Caro M, Morell M, Forte-Lago I, Hernandez-Cortes P, Sanchez-Gonzalez C, O’Valle F, Delgado M, Gonzalez-Rey E. The Neuropeptide Cortistatin Alleviates Neuropathic Pain in Experimental Models of Peripheral Nerve Injury. Pharmaceutics. 2021; 13(7):947. https://doi.org/10.3390/pharmaceutics13070947
Chicago/Turabian StyleFalo, Clara P., Raquel Benitez, Marta Caro, Maria Morell, Irene Forte-Lago, Pedro Hernandez-Cortes, Clara Sanchez-Gonzalez, Francisco O’Valle, Mario Delgado, and Elena Gonzalez-Rey. 2021. "The Neuropeptide Cortistatin Alleviates Neuropathic Pain in Experimental Models of Peripheral Nerve Injury" Pharmaceutics 13, no. 7: 947. https://doi.org/10.3390/pharmaceutics13070947
APA StyleFalo, C. P., Benitez, R., Caro, M., Morell, M., Forte-Lago, I., Hernandez-Cortes, P., Sanchez-Gonzalez, C., O’Valle, F., Delgado, M., & Gonzalez-Rey, E. (2021). The Neuropeptide Cortistatin Alleviates Neuropathic Pain in Experimental Models of Peripheral Nerve Injury. Pharmaceutics, 13(7), 947. https://doi.org/10.3390/pharmaceutics13070947