
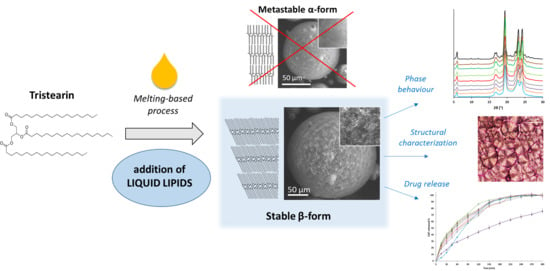
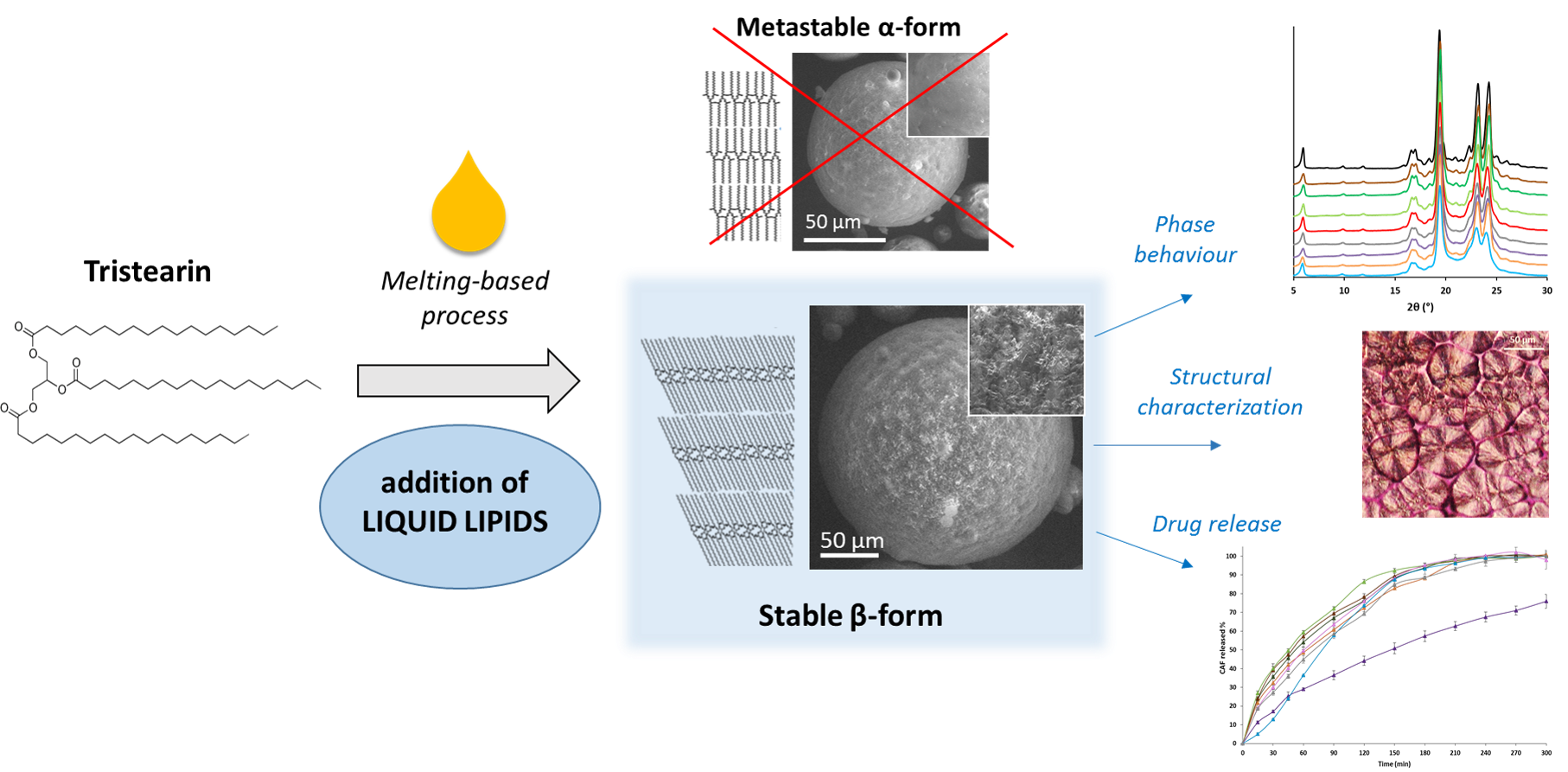
Liquid Lipids Act as Polymorphic Modifiers of Tristearin-Based Formulations Produced by Melting Technologies



Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Preparation of Microparticles (MPs)
2.3. Particle Size
2.4. Differential Scanning Calorimetry (DSC)
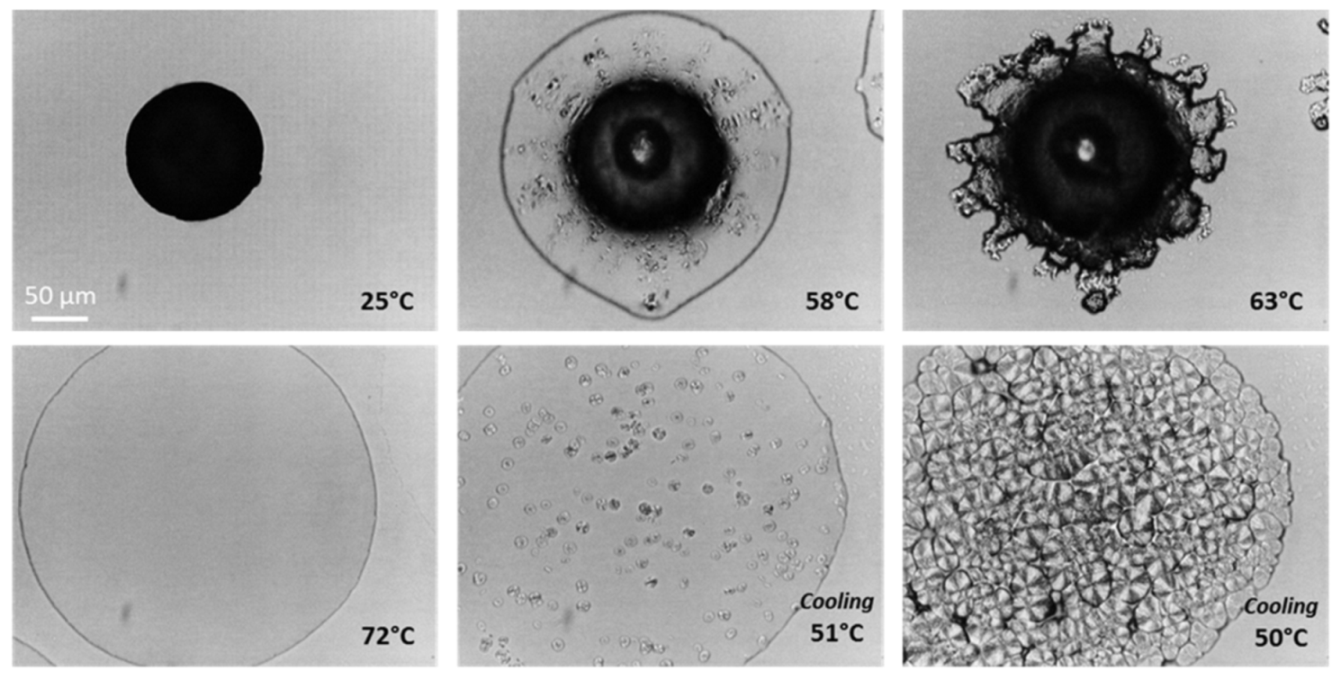
2.5. Hot Stage Microscopy (HSM) and Hot-Stage Polarized Light Microscopy (HS-PLM)
2.6. Powder X-ray Diffraction (PXRD)
2.7. Drug Loading Determination
2.8. Release Studies
2.9. Statistical Analysis
3. Results
3.1. Phase Behavior of Tristearin MPs
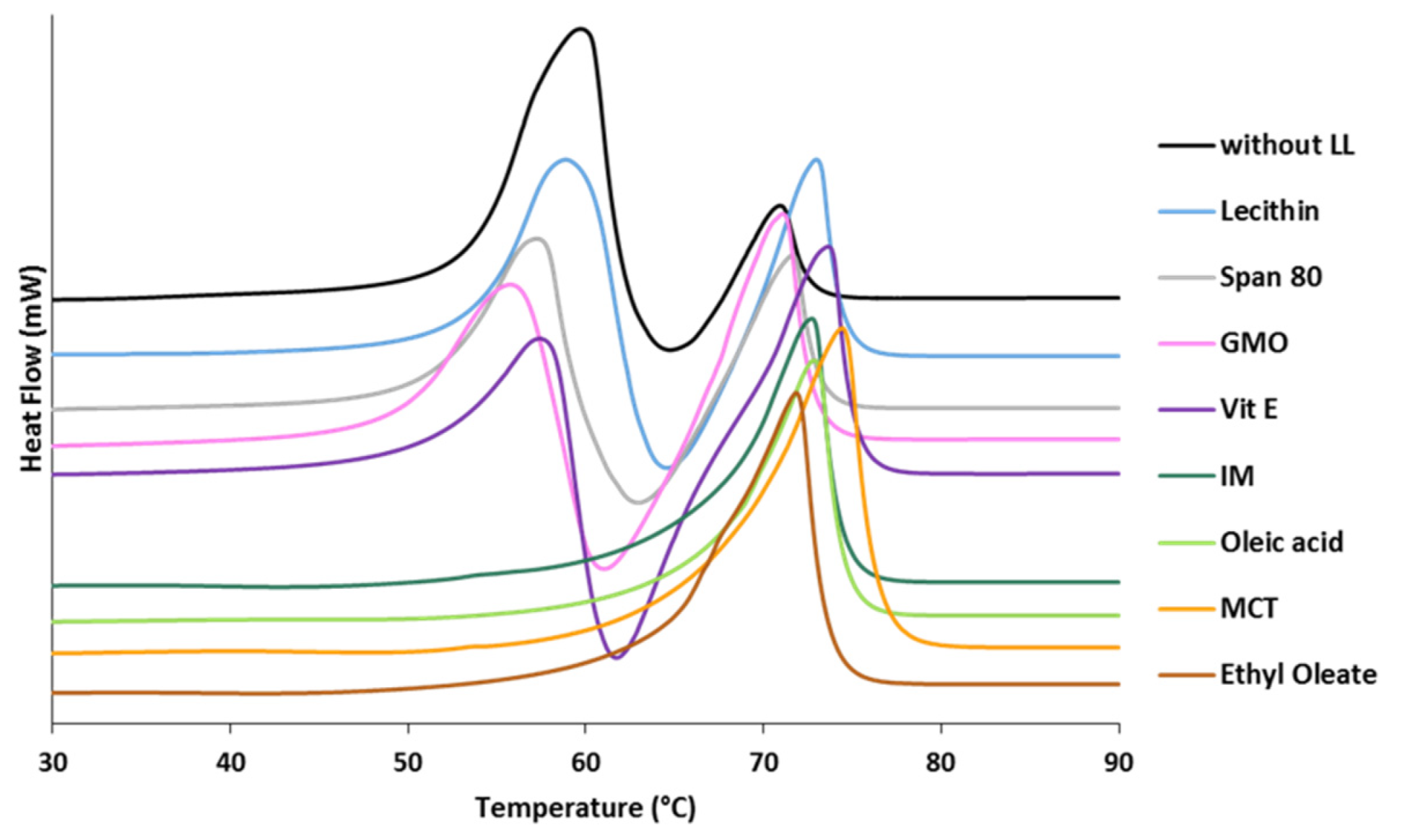
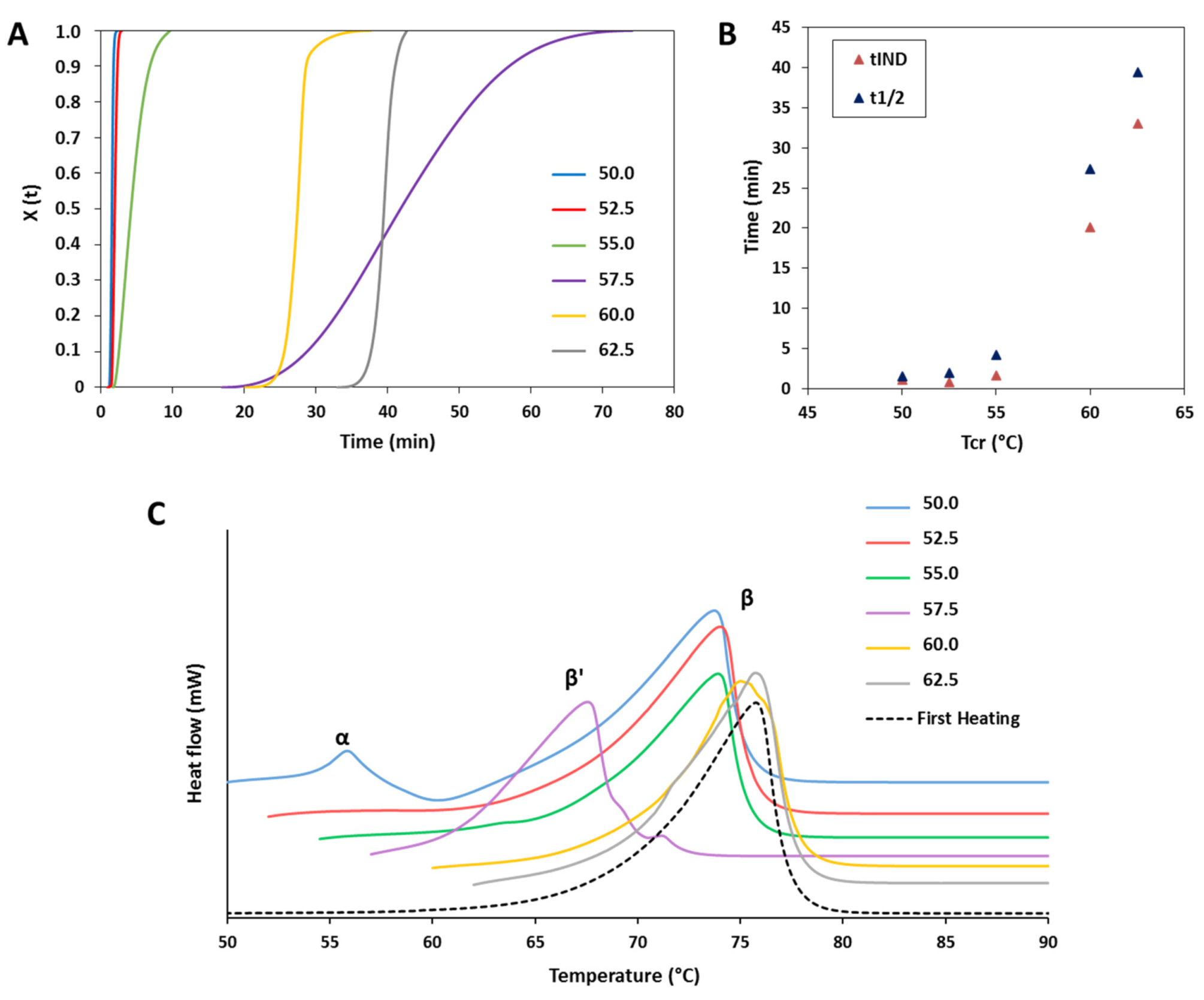
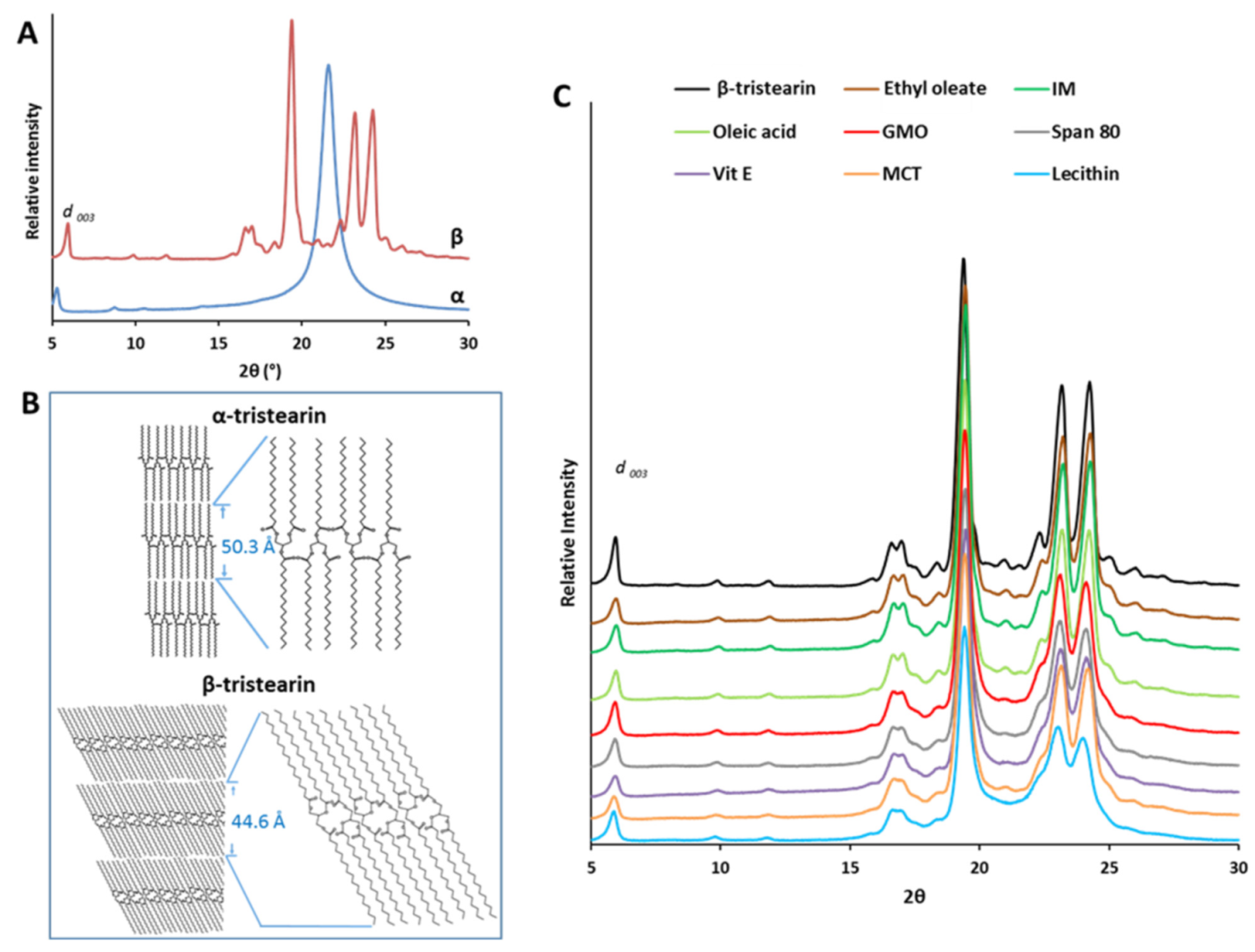
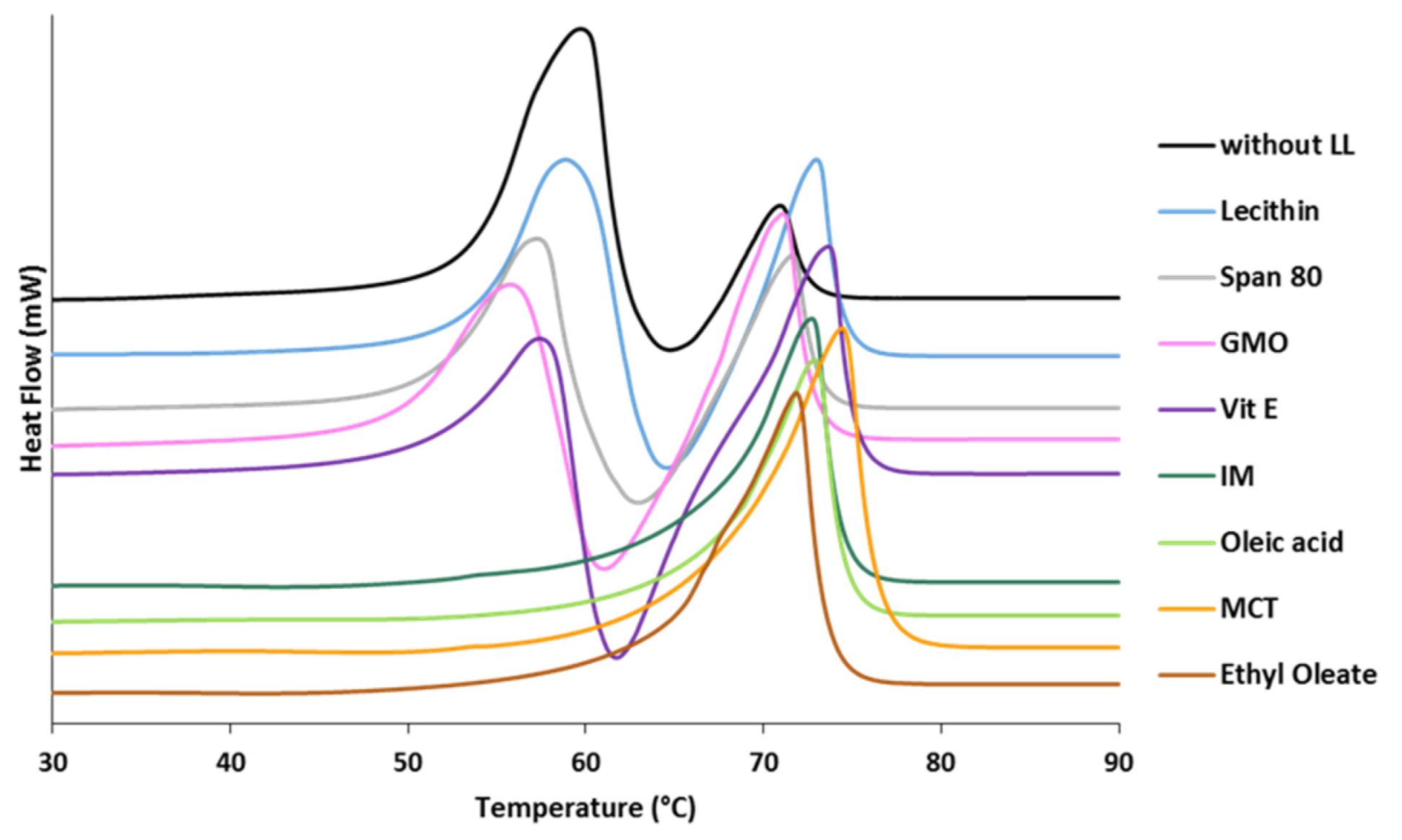
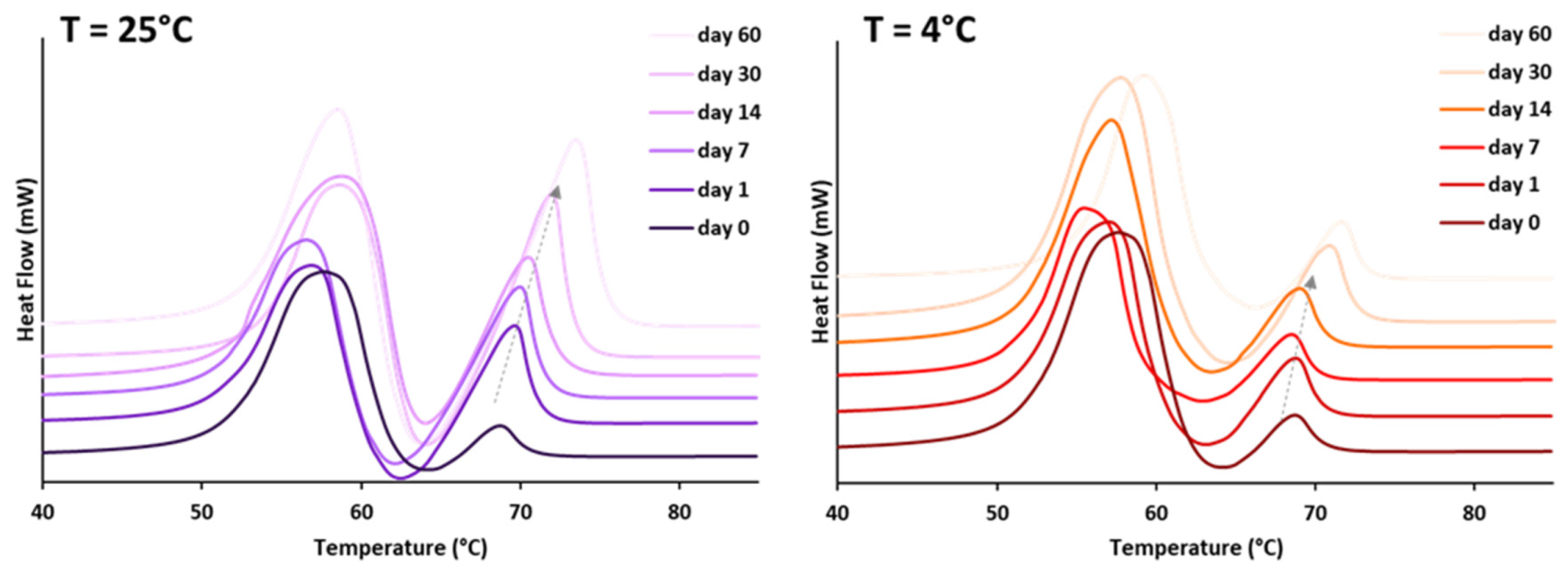
3.1.1. Polymorphic Behavior
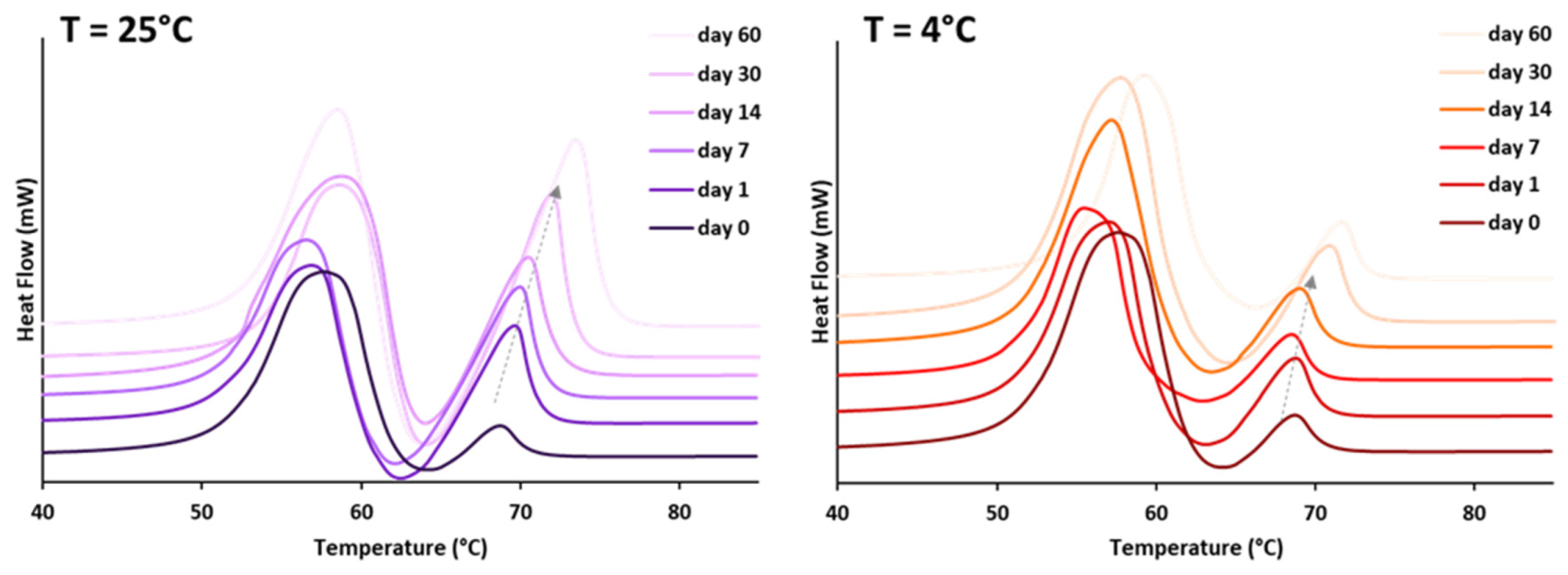
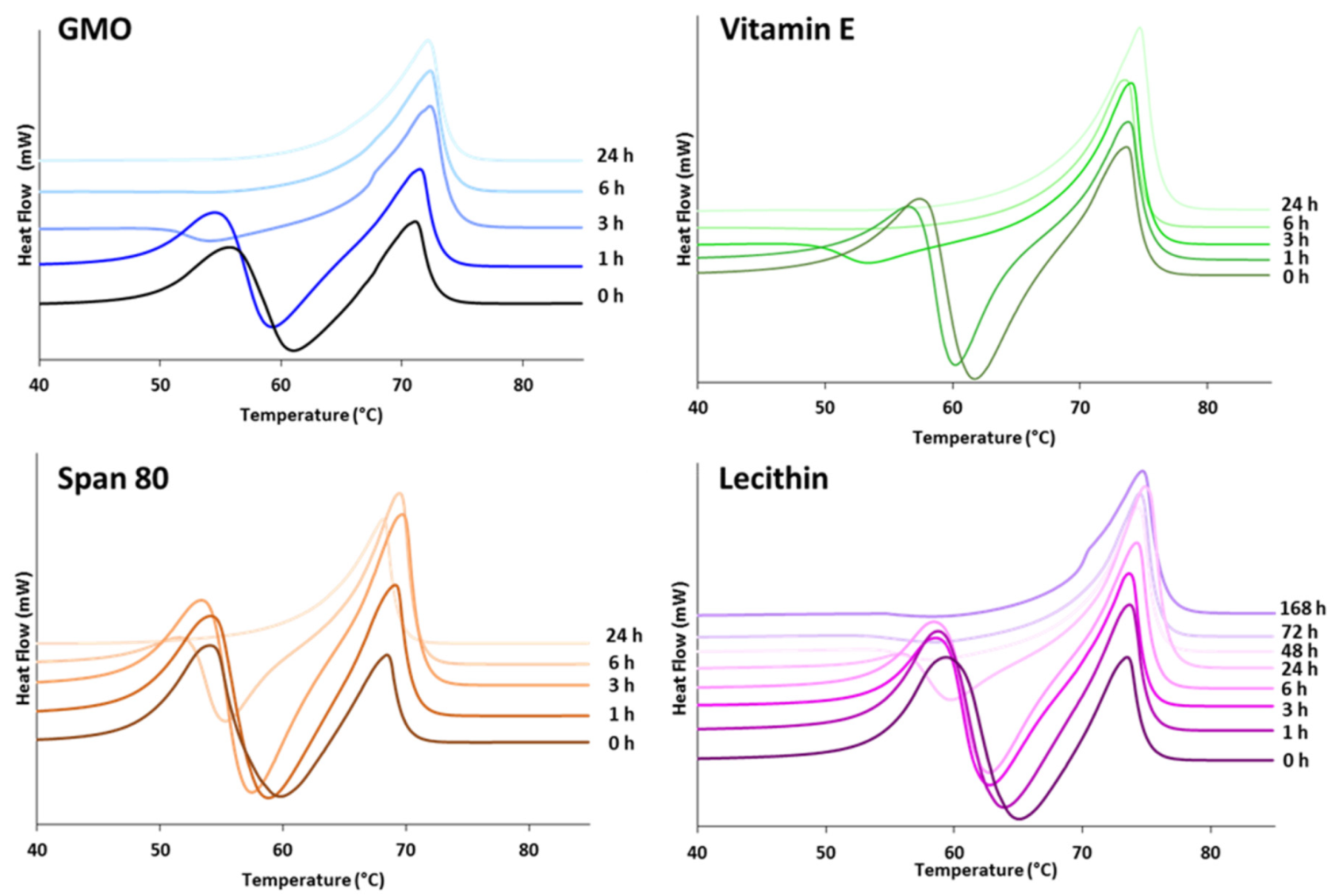
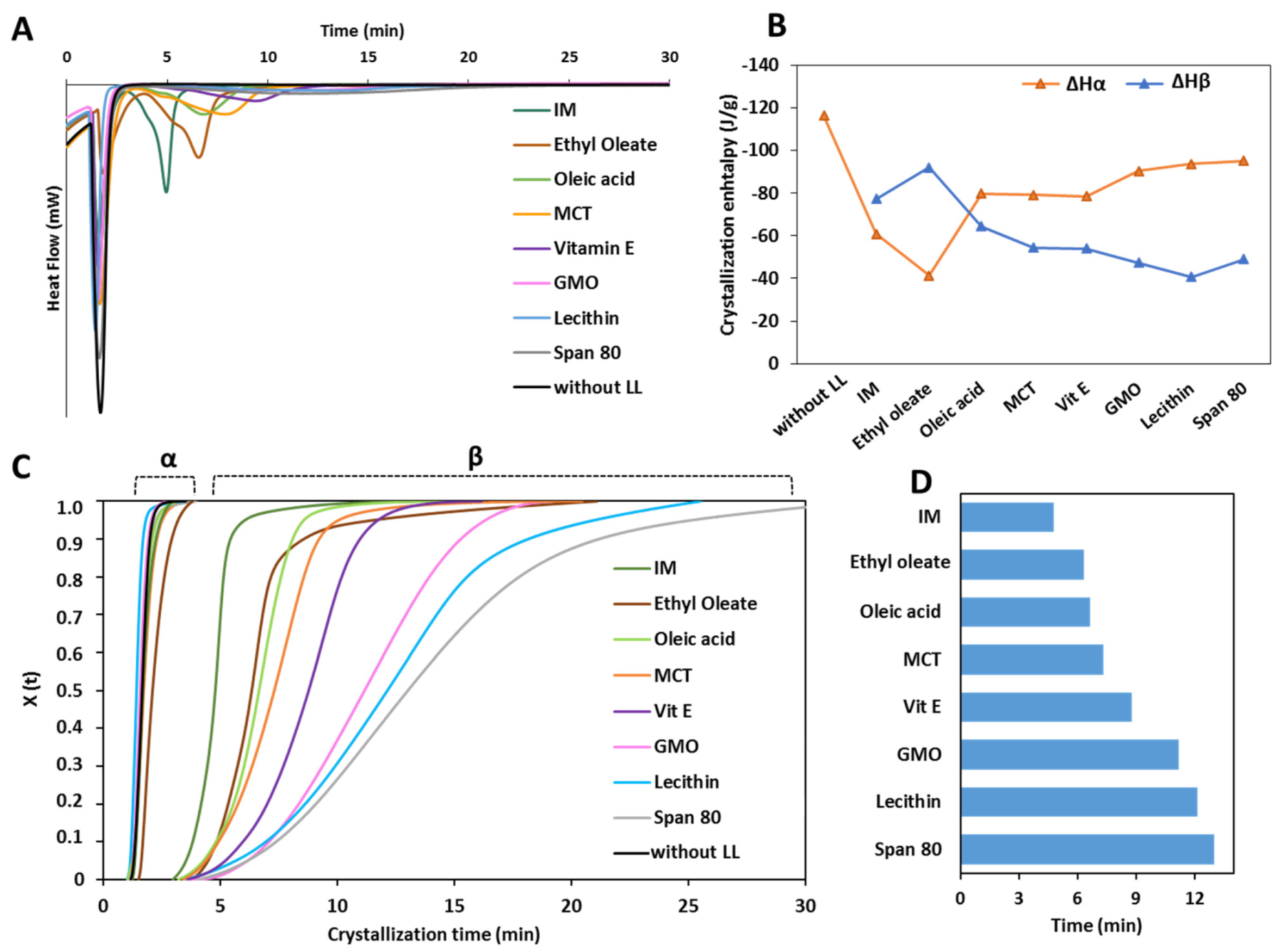
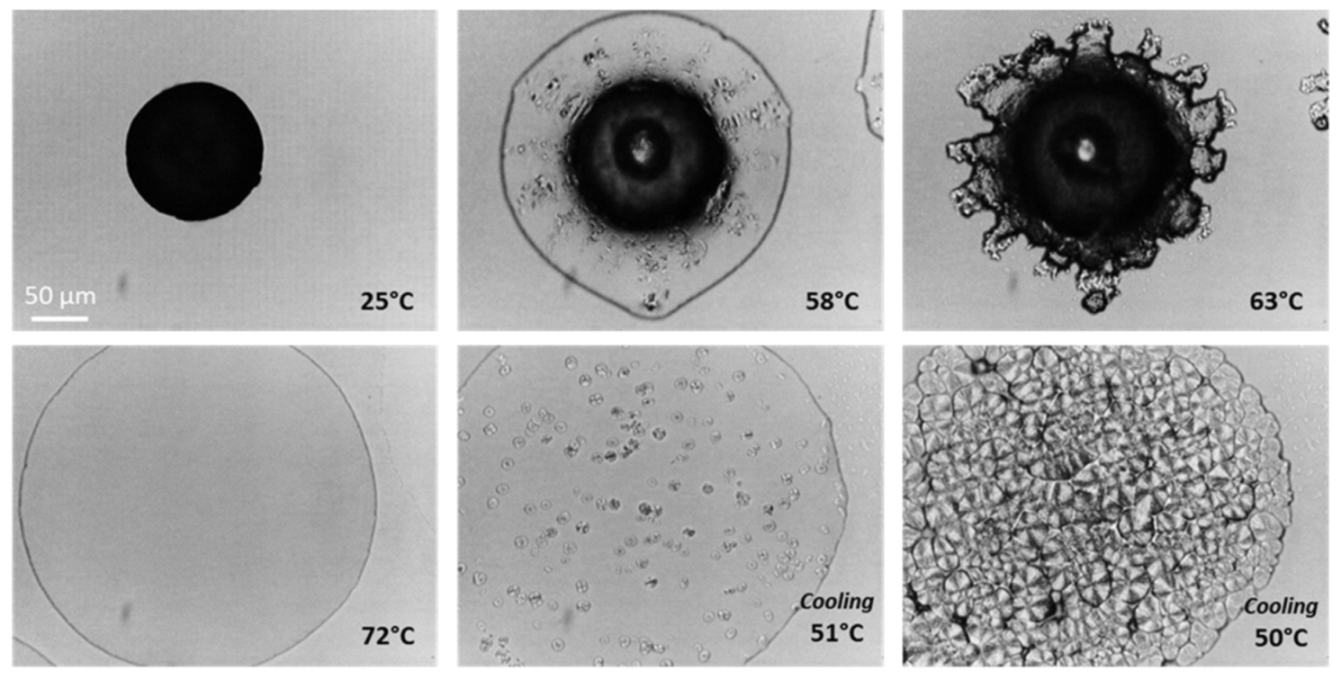
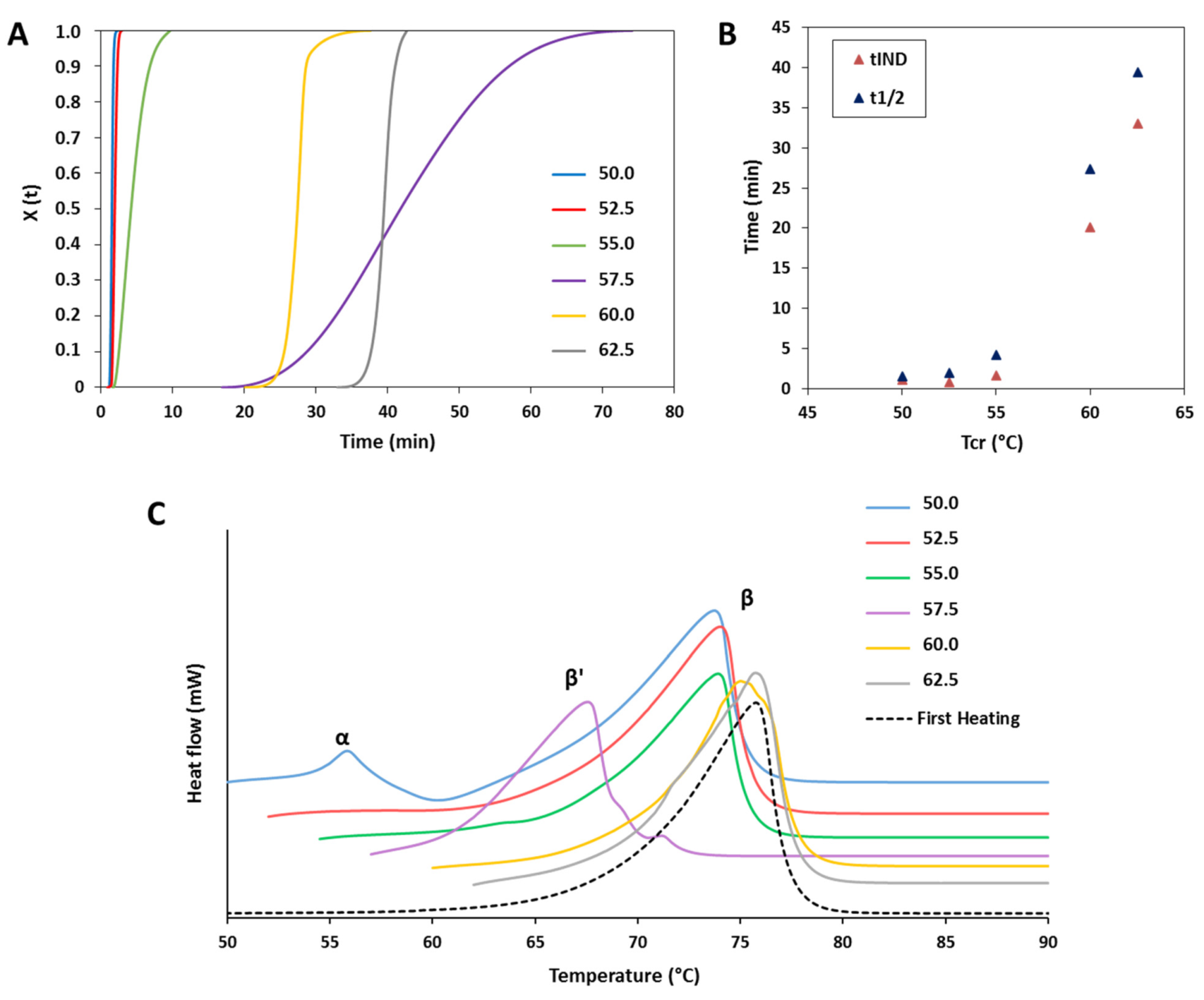
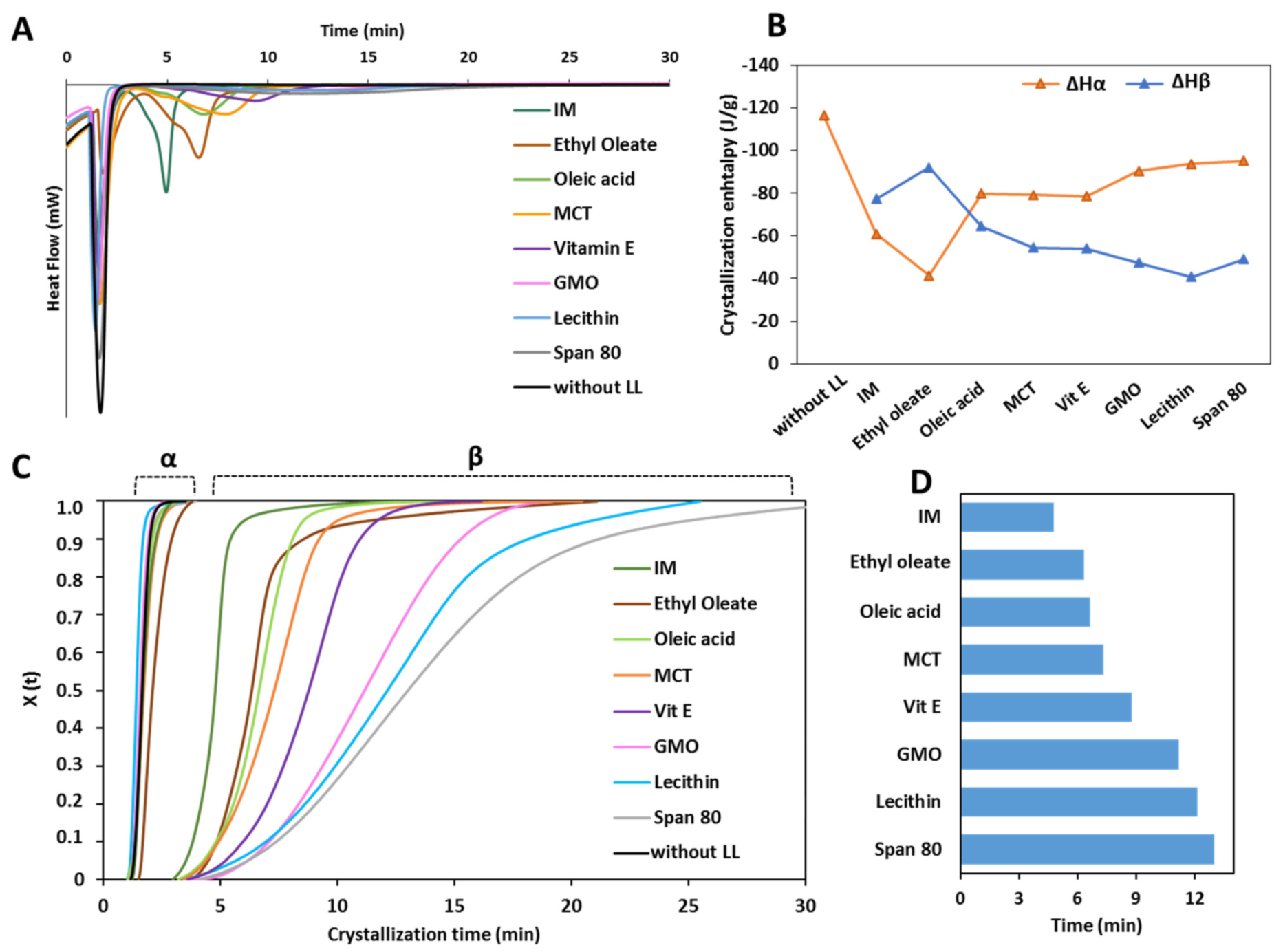
3.1.2. Crystallization Behavior
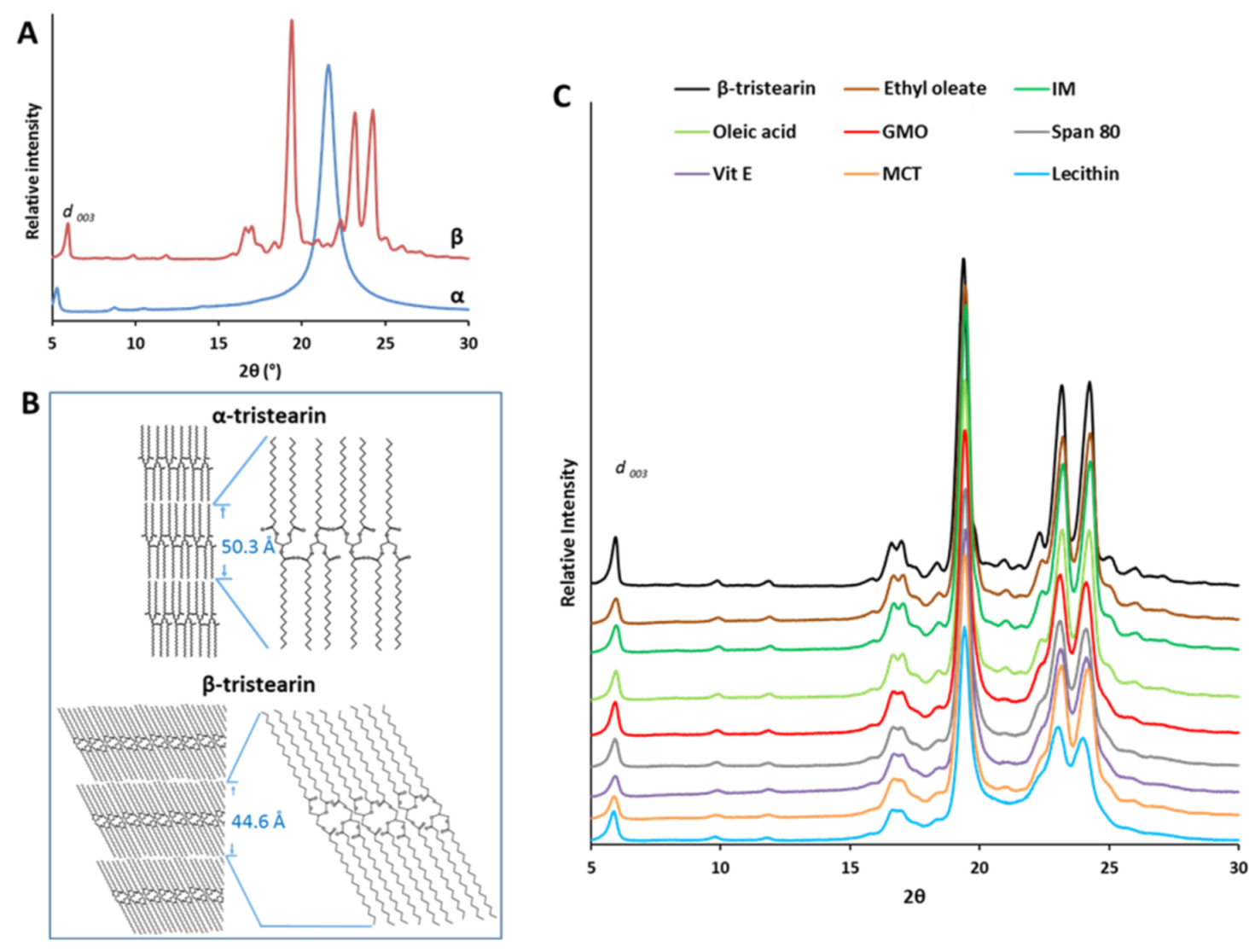
3.2. Structural Characterization of Tristearin MPs
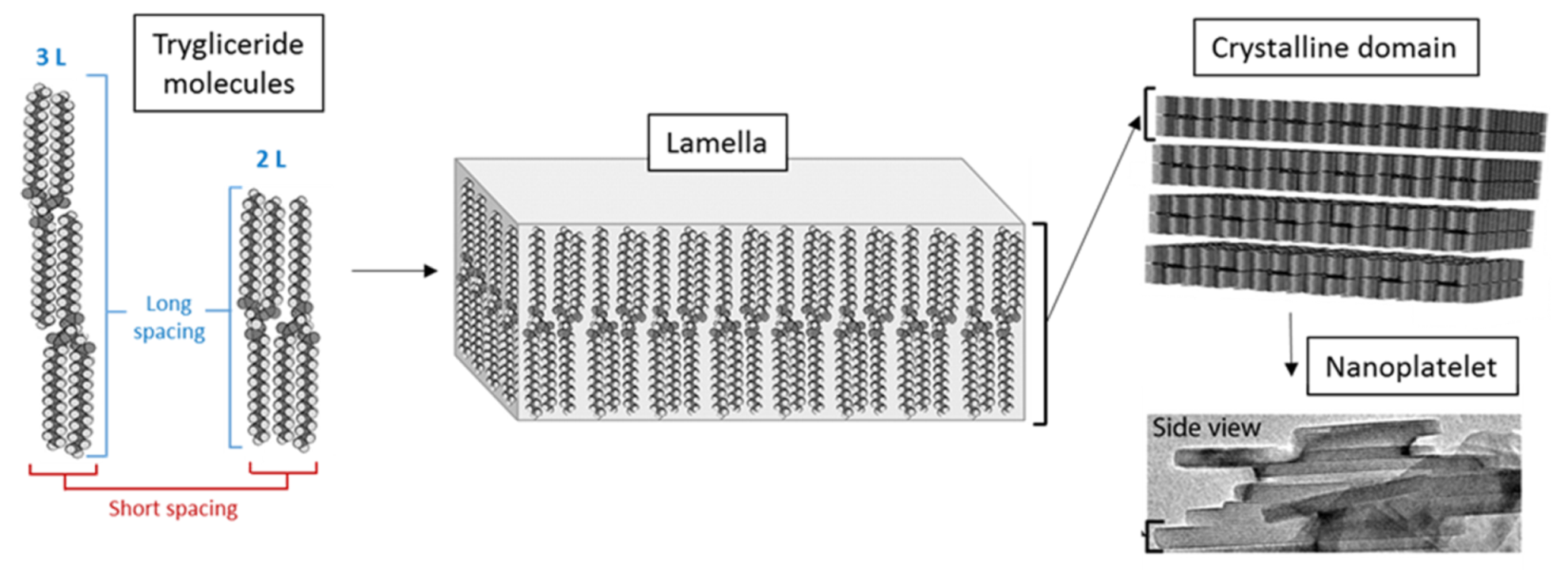
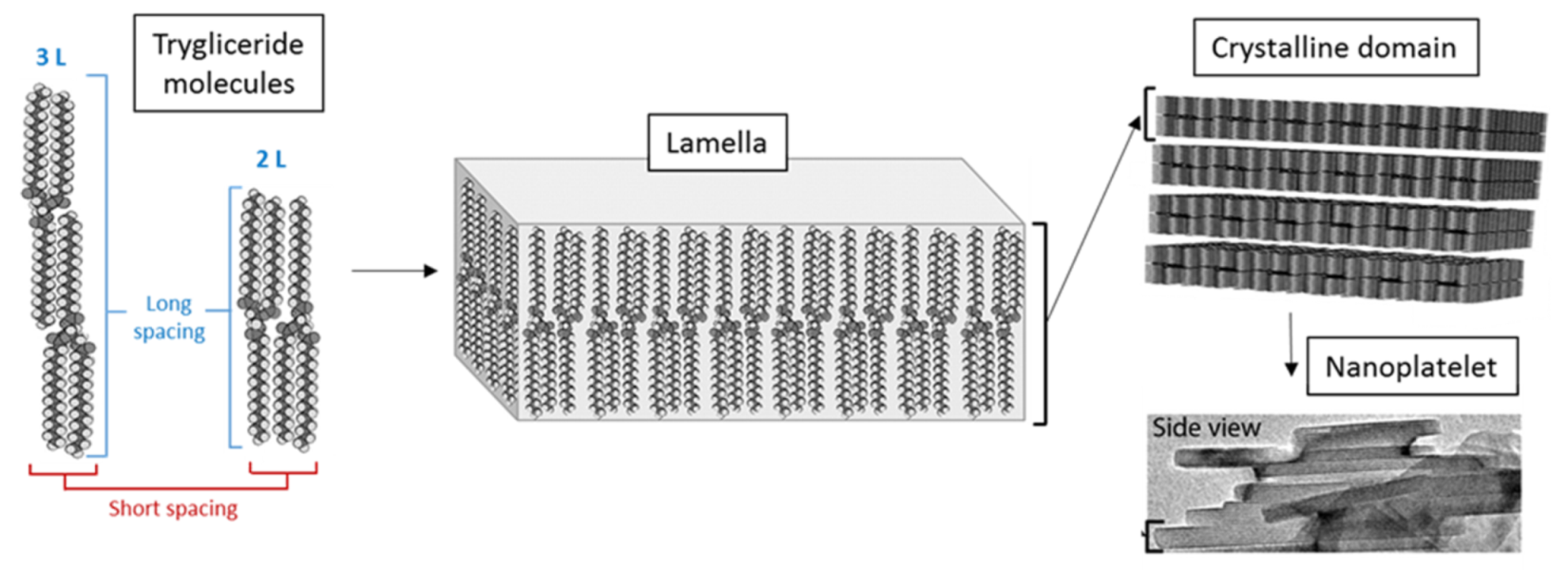
3.2.1. Nanostructural Level
3.2.2. Microstructural Level
3.2.3. Macrostructural Level
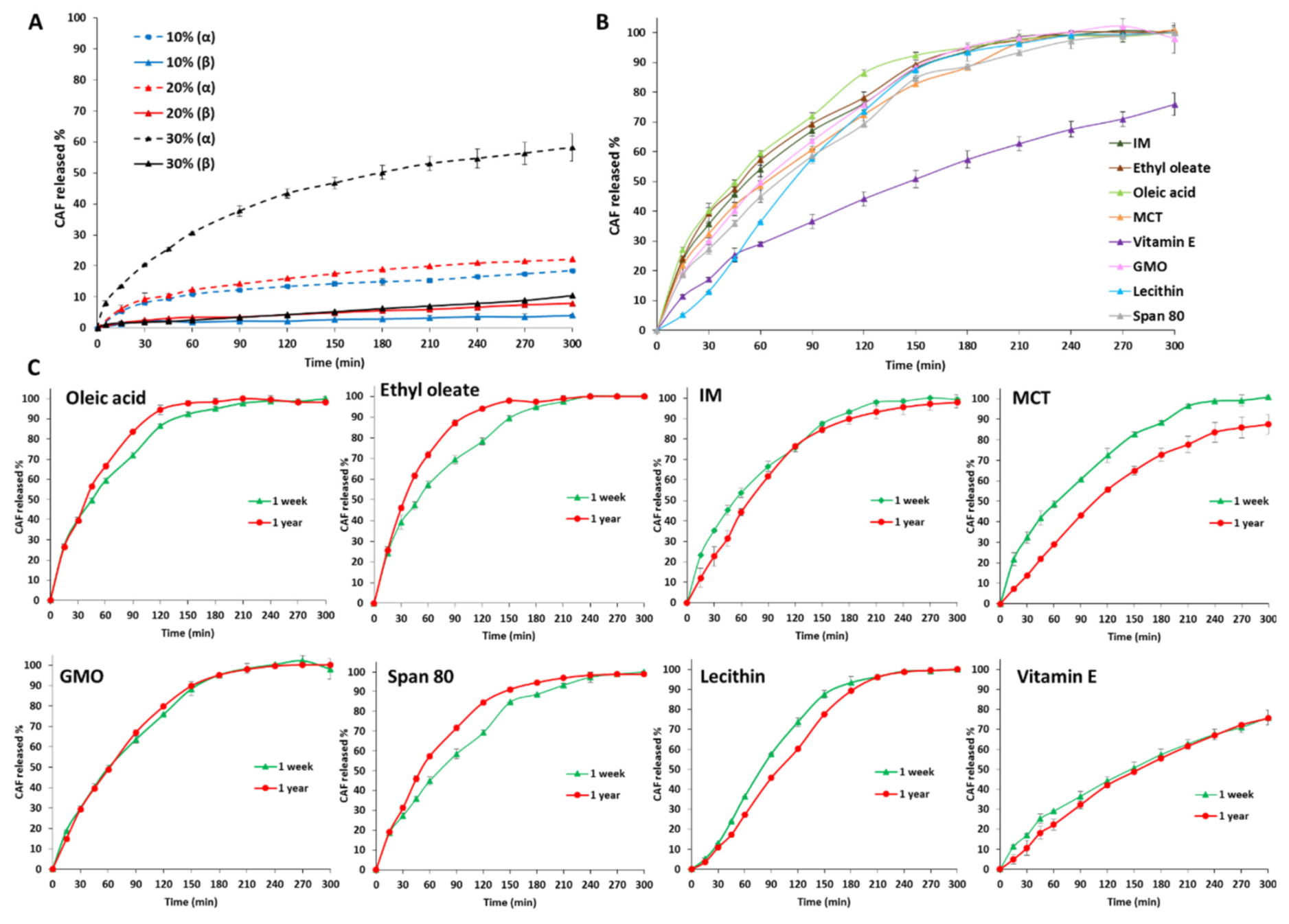
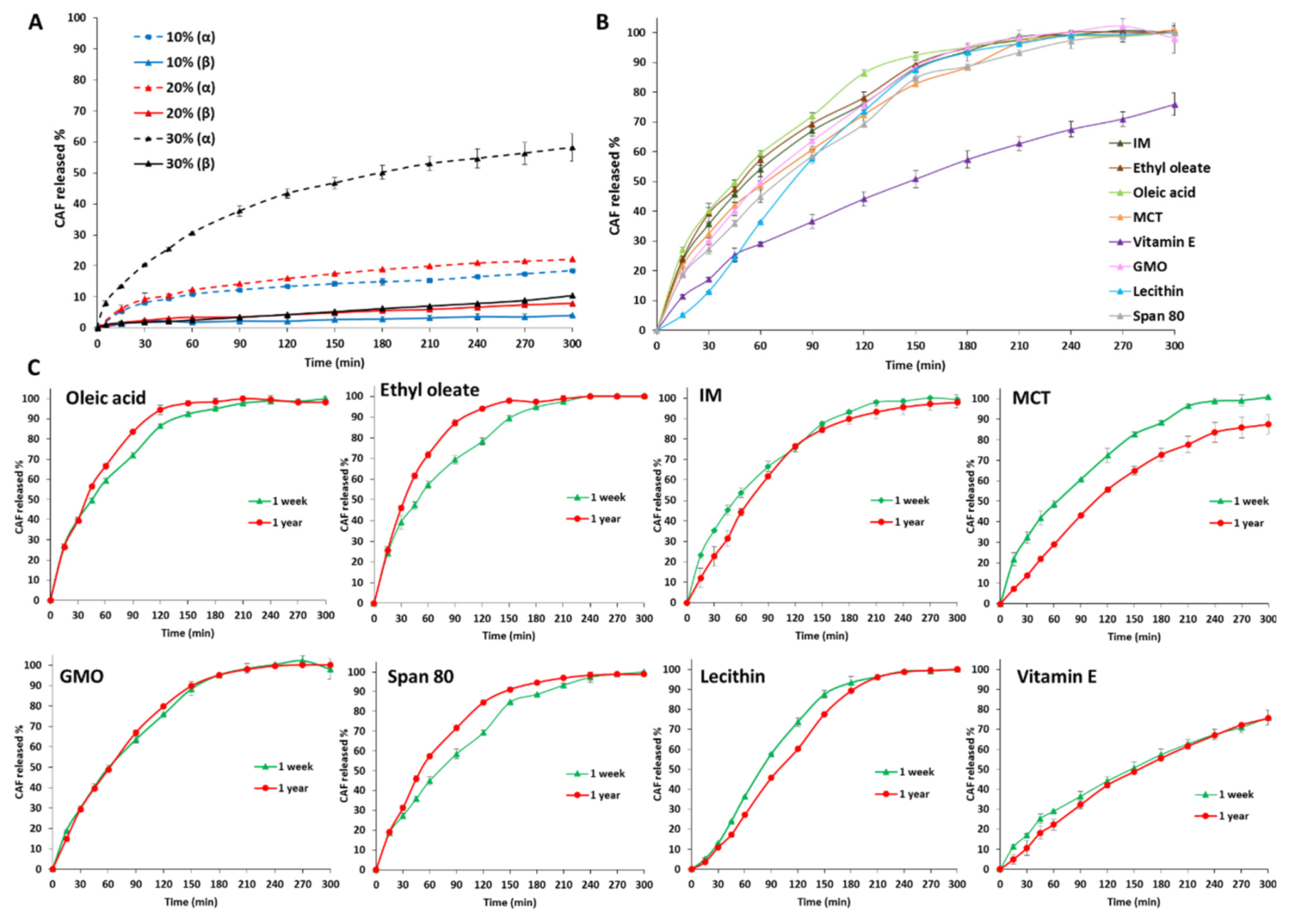
3.3. Drug Loading ad Release Profiles of Tristearin MPs
4. Discussion
- Class I LL (IM, oleic acid, ethyl oleate and MCT) strongly accelerated the polymorphic transition to the stable form, which was completed in few minutes.
- Class II LL (GMO, Vitamin E, span 80 and lecithin) promoted the β phase transition to a lower extent and the time needed to achieve complete polymorphic transition ranged from 6 h to 7 days at room temperature.
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Jannin, V.; Cuppok, Y. Hot-melt coating with lipid excipients. Int. J. Pharm. 2013, 457, 480–487. [Google Scholar] [CrossRef] [PubMed]
- Jannin, V.; Rosiaux, Y.; Doucet, J. Exploring the possible relationship between the drug release of Compritol®—Containing tablets and its polymorph forms using micro X-ray diffraction. J. Control. Release 2015, 197, 158–164. [Google Scholar] [CrossRef] [PubMed]
- Becker, K.; Saurugger, E.-M.; Kienberger, D.; Lopes, D.; Haack, D.; Köberle, M.; Stehr, M.; Lochmann, D.; Zimmer, A.; Salar-Behzadi, S. Advanced stable lipid-based formulations for a patient-centric product design. Int. J. Pharm. 2016, 497, 136–149. [Google Scholar] [CrossRef] [PubMed]
- Gonnet, M.; Lethuaut, L.; Boury, F. New trends in encapsulation of liposoluble vitamins. J. Control. Release 2010, 146, 276–290. [Google Scholar] [CrossRef]
- Helgason, T.; Awad, T.S.; Kristbergsson, K.; Decker, E.A.; McClements, D.J.; Weiss, J. Impact of surfactant properties on oxidative stability of β-carotene encapsulated within solid lipid nanoparticles. J. Agric. Food Chem. 2009, 57, 8033–8040. [Google Scholar] [CrossRef] [PubMed]
- Bertoni, S.; Passerini, N.; Albertini, B. Nanomaterials for oral drug administration. In Nanotechnology for Oral Drug Delivery From Concept to Applications, 1st ed.; Elsevier: Amsterdam, The Netherlands, 2020; pp. 27–76. [Google Scholar]
- Al Khafaji, A.S.; Donovan, M.D. Endocytic uptake of solid lipid nanoparticles by the nasal mucosa. Pharmaceutics 2021, 13, 761. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Pi, J.; Zhang, Y.; Qin, H.; Zhang, B.; Li, N.; Li, Z.; Liu, Z. Enhanced anticancer efficacy of dual drug-loaded self-assembled nanostructured lipid carriers mediated by PH-responsive folic acid and human-derived cell penetrating peptide DNP2. Pharmaceutics 2021, 13, 600. [Google Scholar] [CrossRef] [PubMed]
- Becker, K.; Salar-behzadi, S.; Zimmer, A. Solvent-free melting techniques for the preparation of lipid-based solid oral formulations. Pharm. Res. 2015, 32, 1519–1545. [Google Scholar] [CrossRef] [Green Version]
- Chapman, D. The polymorphism of glycerides. Chem. Rev. 1962, 62, 433–456. [Google Scholar] [CrossRef]
- Olbrich, C.; Kayser, O.; Müller, R.H. Lipase degradation of dynasan 114 and 116 solid lipid nanoparticles (SLN)—Effect of surfactants, storage time and crystallinity. Int. J. Pharm. 2002, 237, 119–128. [Google Scholar] [CrossRef]
- Windbergs, M.; Strachan, C.J.; Kleinebudde, P. Understanding the solid-state behaviour of triglyceride solid lipid extrudates and its influence on dissolution. Eur. J. Pharm. Biopharm. 2009, 71, 80–87. [Google Scholar] [CrossRef]
- Jenning, V.; Schäfer-Korting, M.; Gohla, S. Vitamin A-loaded solid lipid nanoparticles for topical use: Drug release properties. J. Control. Release 2000, 66, 115–126. [Google Scholar] [CrossRef]
- Ribeiro, A.P.B.; Masuchi, M.H.; Miyasaki, E.K.; Domingues, M.A.F.; Stroppa, V.L.Z.; de Oliveira, G.M.; Kieckbusch, T.G. Crystallization modifiers in lipid systems. J. Food Sci. Technol. 2015, 52, 3925–3946. [Google Scholar] [CrossRef] [Green Version]
- Larsson, K. Lipids: Molecular Organization, Physical Functions and Technical Applications; Oily Press: Bridgwater, UK, 1994. [Google Scholar]
- Hoerr, C.W. Morphology of fats, oils, and shortenings. J. Am. Oil Chem. Soc. 1960, 37, 539–546. [Google Scholar] [CrossRef]
- Lopes, D.G.; Koutsamanis, I.; Becker, K.; Scheibelhofer, O.; Laggner, P.; Haack, D.; Stehr, M.; Zimmer, A.; Salar-Behzadi, S. Microphase separation in solid lipid dosage forms as the cause of drug release instability. Int. J. Pharm. 2017, 517, 403–412. [Google Scholar] [CrossRef] [PubMed]
- Khan, N.; Craig, D.Q.M. Role of blooming in determining the storage stability of lipid-based dosage forms. J. Pharm. Sci. 2004, 93, 2962–2971. [Google Scholar] [CrossRef] [PubMed]
- Windbergs, M.; Strachan, C.J.; Kleinebudde, P. Influence of structural variations on drug release from lipid/polyethylene glycol matrices. Eur. J. Pharm. Sci. 2009, 37, 555–562. [Google Scholar] [CrossRef]
- Oh, J.-H.; McCurdy, A.R.; Clark, S.; Swanson, B.G. Stabilizing polymorphic transitions of tristearin using diacylglycerols and sucrose polyesters. J. Am. Oil Chem. Soc. 2005, 82, 13–19. [Google Scholar] [CrossRef]
- Cerdeira, M.; Martini, S.; Candal, R.J.; Herrera, M.L. Polymorphism and growth behavior of low-trans fat blends formulated with and without emulsifiers. J. Am. Oil Chem. Soc. 2006, 83, 489–496. [Google Scholar] [CrossRef]
- Elisabettini, P.; Desmedt, A.; Durant, F. Polymorphism of stabilized and nonstabilized tristearin, pure and in the presence of food emulsifiers. J. Am. Oil Chem. Soc. 1996, 73, 187–192. [Google Scholar] [CrossRef]
- Garti, N.; Wellner, E.; Sarig, S. Crystal structure modifications of tristearin by food emulsifiers. J. Am. Oil Chem. Soc. 1982, 59, 181–185. [Google Scholar] [CrossRef]
- Aronhime, J.S.; Sarig, S.; Garti, N. Mechanistic considerations of polymorphic transformations of tristearin in the presence of emulsifiers. J. Am. Oil Chem. Soc. 1987, 64, 529–533. [Google Scholar] [CrossRef]
- Aronhime, J.S.; Sarig, S.; Garti, N. Dynamic control of polymorphic transformation in triglycerides by surfactants: The button syndrome. J. Am. Oil Chem. Soc. 1988, 65, 1144–1150. [Google Scholar] [CrossRef]
- Helgason, T.; Awad, T.S.; Kristbergsson, K.; Mc Clements, D.J.; Weiss, J. Effect of surfactant surface coverage on formation of solid lipid nanoparticles (SLN). J. Colloid Interface Sci. 2009, 334, 75–81. [Google Scholar] [CrossRef]
- Rosenblatt, K.M.; Bunjes, H. Poly(vinyl alcohol) as emulsifier stabilizes solid triglyceride drug carrier nanoparticles in the α-modification. Mol. Pharm. 2009, 6, 105–120. [Google Scholar] [CrossRef] [PubMed]
- Joseph, S.; Rappolt, M.; Schoenitz, M.; Huzhalska, V.; Augustin, W.; Scholl, S.; Bunjes, H. Stability of the metastable α-polymorph in solid triglyceride drug-carrier nanoparticles. Langmuir 2015, 31, 6663–6674. [Google Scholar] [CrossRef] [PubMed]
- Jennings, J.; Butler, M.F.; McLeod, M.; Csányi, E.; Ryan, A.J.; Mykhaylyk, O.O. Stearyl methacrylate-based polymers as crystal habit modifiers for triacylglycerols. Cryst. Growth Des. 2018, 18, 7094–7105. [Google Scholar] [CrossRef]
- Pattarino, F.; Bettini, R.; Foglio Bonda, A.; Della Bella, A.; Giovannelli, L. Polymorphism and kinetic behavior of binary mixtures of triglycerides. Int. J. Pharm. 2014, 473, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Windbergs, M.; Strachan, C.J.; Kleinebudde, P. Investigating the principles of recrystallization from glyceride melts. AAPS PharmSciTech 2009, 10, 1224–1233. [Google Scholar] [CrossRef] [Green Version]
- Kellens, M.; Reynaers, H. Study of the polymorphism of saturated monoacid triglycerides I: Melting and crystallization behaviour of tristearin. Lipid Fett 1992, 94, 94–100. [Google Scholar] [CrossRef]
- Metin, S.; Hartel, R.W. Thermal analysis of isothermal crystallization kinetics in blends of cocoa butter with milk fat or milk fat fractions. J. Am. Oil Chem. Soc. 1998, 75, 1617–1624. [Google Scholar] [CrossRef]
- Oh, J.-H.; Mc Curdy, A.R.; Clark, S.; Swanson, B.G. Characterization and thermal stability of polymorphic forms of synthesized tristearin. J. Food Sci. 2002, 67, 2911–2917. [Google Scholar] [CrossRef]
- Acevedo, N.C.; Marangoni, A.G. Nanostructured fat crystal systems. Annu. Rev. Food Sci. Technol. 2015, 6, 71–96. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, H.; Shiota, M.; Murakami, M.; Nakajima, I. Polymorphic behavior of palm oil and modified palm oils. Food Sci. Technol. Int. Tokyo 1997, 3, 77–81. [Google Scholar] [CrossRef]
- Lopez, C.; Lesieur, P.; Bourgaux, C.; Ollivon, M. Thermal and structural behavior of anhydrous milk fat. 3 influence of cooling rate. J. Dairy Sci. 2005, 88, 511–526. [Google Scholar] [CrossRef] [Green Version]
- Acevedo, N.C.; Peyronel, F.; Marangoni, A.G. Nanoscale structure intercrystalline interactions in fat crystal networks. Curr. Opin. Colloid Interface Sci. 2011, 16, 374–383. [Google Scholar] [CrossRef]
- Kellens, M.; Meeussen, W.; Hammersley, A.; Reynaers, H. Synchrotron radiation investigations of the polymorphic transitions in saturated monoacid triglycerides. Part 2: Polymorphism study of a 50:50 mixture of tripalmitin and tristearin during crystallization and melting. Chem. Phys. Lipids 1991, 58, 145–158. [Google Scholar] [CrossRef]
- Ueno, S.; Nishida, T.; Sato, K. Synchrotron radiation microbeam X-ray analysis of microstructures and the polymorphic transformation of spherulite crystals of trilaurin. Cryst. Growth Des. 2008, 8, 751–754. [Google Scholar] [CrossRef]
- Lutton, E.S. Review of the polymorphism of saturated even glycerides. J. Am. Oil Chem. Soc. 1950, 27, 276–281. [Google Scholar] [CrossRef]
- Ravotti, R.; Worlitschek, J.; Pulham, C.R.; Stamatiou, A. Triglycerides as novel phase-change materials: A review and assessment of their thermal properties. Molecules 2020, 25, 5572. [Google Scholar] [CrossRef]
- Acevedo, N.C.; Marangoni, A.G. Toward nanoscale engineering of triacylglycerol crystal networks. Cryst. Growth Des. 2010, 10, 3334–3339. [Google Scholar] [CrossRef]
- Acevedo, N.C.; Marangoni, A.G. Characterization of the nanoscale in triacylglycerol crystal networks. Cryst. Growth Des. 2010, 10, 3327–3333. [Google Scholar] [CrossRef]
- Bouzidi, L.; Omonov, T.S.; Garti, N.; Narine, S.S. Relationships between molecular structure and kinetic and thermodynamic controls in lipid systems. Part I: Propensity for oil loss of saturated triacylglycerols. Food Funct. 2013, 4, 130–143. [Google Scholar] [CrossRef] [PubMed]
- Silva, R.C.d.; Martini Soares, F.A.S.D.; Maruyama, J.M.; Dagostinho, N.R.; Silva, Y.A.; Ract, J.N.R.; Gioielli, L.A. Microscopic approach of the crystallization of tripalmitin and tristearin by microscopy. Chem. Phys. Lipids 2016, 198, 1–9. [Google Scholar] [CrossRef]
- Bertoni, S.; Albertini, B.; Facchini, C.; Prata, C.; Passerini, N. Glutathione-loaded solid lipid microparticles as innovative delivery system for oral antioxidant therapy. Pharmaceutics 2019, 11, 364. [Google Scholar] [CrossRef] [Green Version]
- Bertoni, S.; Albertini, B.; Dolci, L.S.; Passerini, N. Spray congealed lipid microparticles for the local delivery of β-galactosidase to the small intestine. Eur. J. Pharm. Biopharm. 2018, 132, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Albertini, B.; Di Sabatino, M.; Melegari, C.; Passerini, N. Formulating SLMs as oral pulsatile system for potential delivery of melatonin to pediatric population. Int. J. Pharm. 2014, 469, 67–79. [Google Scholar] [CrossRef]
- Bertoni, S.; Tedesco, D.; Bartolini, M.; Prata, C.; Passerini, N.; Albertini, B. Solid lipid microparticles for oral delivery of catalase: Focus on the protein structural integrity and gastric protection. Mol. Pharm. 2020, 17, 3609–3621. [Google Scholar] [CrossRef] [PubMed]
- Macridachis-González, J.; Bayés-García, L.; Calvet, T. An insight into the solid-state miscibility of triacylglycerol crystals. Molecules 2020, 25, 4562. [Google Scholar] [CrossRef]
- Mayama, H. Blooming theory of tristearin. Soft Matter 2009, 5, 856–859. [Google Scholar] [CrossRef] [Green Version]
- Siekmann, B.; Westesen, K. Thermoanalysis of the recrystallization process of melt-homogenized glyceride nanoparticles. Colloids Surf. B Biointerfaces 1994, 3, 159–175. [Google Scholar] [CrossRef]
- Jenning, V.; Thünemann, A.F.; Gohla, S.H. Characterisation of a novel solid lipid nanoparticle carrier system based on binary mixtures of liquid and solid lipids. Int. J. Pharm. 2000, 199, 167–177. [Google Scholar] [CrossRef]
- Bertoni, S.; Albertini, B.; Passerini, N. Spray congealing: An emerging technology to prepare solid dispersions with enhanced oral bioavailability of poorly water soluble drugs. Molecules 2019, 24, 3471. [Google Scholar] [CrossRef] [Green Version]
- Windbergs, M.; Strachan, C.J.; Kleinebudde, P. Influence of the composition of glycerides on the solid-state behaviour and the dissolution profiles of solid lipid extrudates. Chall. Nano Micro Macro Syst. 2009, 381, 184–191. [Google Scholar] [CrossRef]
- Kreye, F.; Siepmann, F.; Willart, J.F.; Descamps, M.; Siepmann, J. Drug release mechanisms of cast lipid implants. Eur. J. Pharm. Biopharm. 2011, 78, 394–400. [Google Scholar] [CrossRef] [PubMed]
- Jaspart, S.; Bertholet, P.; Piel, G.; Dogné, J.-M.; Delattre, L.; Evrard, B. Solid lipid microparticles as a sustained release system for pulmonary drug delivery. Eur. J. Pharm. Biopharm. 2007, 65, 47–56. [Google Scholar] [CrossRef]
- Fang, W.; Mayama, H.; Tsujii, K. Spontaneous formation of fractal structures on triglyceride surfaces with reference to their super water-repellent properties. J. Phys. Chem. B 2007, 111, 564–571. [Google Scholar] [CrossRef]
- Albertini, B.; Passerini, N.; Di Sabatino, M.; Vitali, B.; Brigidi, P.; Rodriguez, L. Polymer–lipid based mucoadhesive microspheres prepared by spray-congealing for the vaginal delivery of econazole nitrate. Eur. J. Pharm. Sci. 2009, 36, 591–601. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, H.; Zheng, A.Y.; Heng, P.W.S.; Chan, L.W. Effect of lipid additives and drug on the rheological properties of molten paraffin wax, degree of surface drug coating, and drug release in spray-congealed microparticles. Pharmaceutics 2018, 10, 75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pyka, A. Evaluation of the lipophilicity of fat-soluble vitamins. JPC Mod. TLC 2009, 22, 211–215. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| d 003 | d 001 | ||
|---|---|---|---|
| °2θ | Å | Å | |
| Dyn 118 α | 5.27 | 16.75 | 50.27 |
| Dyn 118 β | 5.94 | 14.86 | 44.60 |
| Etil oleate | 5.96 | 14.81 | 44.45 |
| IM | 5.98 | 14.76 | 44.30 |
| Oleic acid | 5.96 | 14.81 | 44.45 |
| GMO | 5.93 | 14.98 | 44.95 |
| Span 80 | 5.91 | 14.94 | 44.83 |
| Vit E | 5.91 | 14.94 | 44.83 |
| MCT | 5.89 | 14.99 | 44.98 |
| Lecithin | 5.88 | 15.01 | 45.06 |
| Formulation | Composition | Experimental CAF Content (%, w/w ± SD) | |
|---|---|---|---|
| Amount of Lipid Carrier (%, w/w) | Amount of CAF (%, w/w) | ||
| Tristearin 10% | 90 | 10 | 11.53 ± 0.39 |
| Tristearin 20% | 80 | 20 | 22.57 ± 0.29 |
| Tristearin 30% | 70 | 30 | 31.35 ± 0.55 |
| IM | 70 | 30 | 31.98 ± 1.26 |
| Ethyl oleate | 70 | 30 | 32.66 ± 1.73 |
| Oleic acid | 70 | 30 | 32.83 ± 0.29 |
| MCT | 70 | 30 | 33.27 ± 0.28 |
| Vitamin E | 70 | 30 | 32.99 ± 1.02 |
| GMO | 70 | 30 | 32.84 ± 0.11 |
| Lecithin | 70 | 30 | 32.24 ± 0.81 |
| Span 80 | 70 | 30 | 32.78 ± 0.12 |
| Liquil Lipids (LL) | Molecular Structure | Molecular Weight (g/mol) | Tm (°C) | Density (g/cm3) at 25 °C | Hydrophilic Lipophilic Balance (o/w) |
|---|---|---|---|---|---|
| Oleic acid | 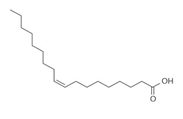 | 282.5 | 13–14 | 0.89 | |
| Ethyl oleate | 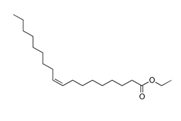 | 310.5 | −32 | 0.87 | - |
| Span 80 | 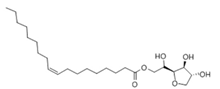 | 428.6 | 10–12 | 0.99 | 4.6 |
| Glyceryl monoleate (GMO) | 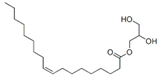 | 365.5 | 33–35 | 0.98 | 3.4–3.8 |
| Medium chain triglycerides (MCT) |  | 470–640 | −5 | 0.94–0.96 | - |
| Isopropyl myristate (IM) | 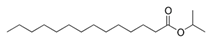 | 270.5 | 3 | 0.85 | - |
| Tocopheryl acetate (Vitamin E) | 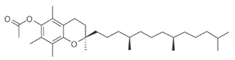 | 472.7 | −28 | 0.95 | - |
| Soyabean lecithin |  | 758.1 | * | 1.05 | ~7 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bertoni, S.; Passerini, N.; Albertini, B. Liquid Lipids Act as Polymorphic Modifiers of Tristearin-Based Formulations Produced by Melting Technologies. Pharmaceutics 2021, 13, 1089. https://doi.org/10.3390/pharmaceutics13071089
Bertoni S, Passerini N, Albertini B. Liquid Lipids Act as Polymorphic Modifiers of Tristearin-Based Formulations Produced by Melting Technologies. Pharmaceutics. 2021; 13(7):1089. https://doi.org/10.3390/pharmaceutics13071089
Chicago/Turabian StyleBertoni, Serena, Nadia Passerini, and Beatrice Albertini. 2021. "Liquid Lipids Act as Polymorphic Modifiers of Tristearin-Based Formulations Produced by Melting Technologies" Pharmaceutics 13, no. 7: 1089. https://doi.org/10.3390/pharmaceutics13071089
APA StyleBertoni, S., Passerini, N., & Albertini, B. (2021). Liquid Lipids Act as Polymorphic Modifiers of Tristearin-Based Formulations Produced by Melting Technologies. Pharmaceutics, 13(7), 1089. https://doi.org/10.3390/pharmaceutics13071089